





Abstract
Understanding the genetic basis of complex diseases is one of the main challenges in modern genomics. However, current tools often lack the versatility to efficiently analyze the intricate relationships between genetic variations and disease outcomes. To address this, we introduce Genopyc, a novel Python library designed for comprehensive investigation of how the variants associated to complex diseases affects downstream pathways. Genopyc offers an extensive suite of functions for heterogeneous data mining and visualization, enabling researchers to delve into and integrate biological information from large-scale genomic datasets.
In this work, we present the Genopyc library through application to real-world genome wide association studies variants. Using Genopyc to investigate the functional consequences of variants associated to intervertebral disc degeneration enabled a deeper understanding of the potential dysregulated pathways involved in the disease, which can be explored and visualized by exploiting the functionalities featured in the package. Genopyc emerges as a powerful asset for researchers, facilitating the investigation of complex diseases paving the way for more targeted therapeutic interventions.
Genopyc is available on pip https://pypi.org/project/genopyc/.The source code of Genopyc is available at https://github.com/freh-g/genopyc. A tutorial notebook is available at https://github.com/freh-g/genopyc/blob/main/tutorials/Genopyc_tutorial_notebook.ipynb. Finally, a detailed documentation is available at: https://genopyc.readthedocs.io/en/latest/.
1 Introduction
The onset of complex disorders is influenced by a multitude of components that include lifestyle, diet, environmental and genetic factors. In the last decades genome wide association studies (GWAS) have emerged as a powerful tool to investigate the genetic architecture underlying complex diseases (Bush and Moore 2012). However, now that thousands of genetic loci have been successfully associated to numerous phenotypes, we are facing another challenge: the interpretation of these associations in the biological context. We are thus entering in the so called post-GWAS Era (Gallagher and Chen-Plotkin 2018). Understanding how genetic variants are translated into biological pathways remains a complex task (Edwards et al. 2013) that brought to the development of numerous approaches to interpret GWAS results [see (Uffelmann et al. 2021) for a comprehensive review of the type of analysis and tools].
One of the main pitfalls consists in handling and interpret the extensive amount of data required to perform these studies (Edwards et al. 2013). In response, a plethora of novel methodological approaches has emerged to address this knowledge gap (Mulder and Opap 2017). These techniques rely on the large-scale omics datasets and repositories available to researchers such as Gene Expression Omnibus (Edgar et al. 2002), the genotype—tissue expression project (GTEx) (Lonsdale 2013) and the Encode project (de Souza 2012). The enormous amount of data regarding genes and variants associated to diseases is collected in knowledge bases such as the GWAS Catalog (Sollis et al. 2023), and DisGeNET (Piñero et al. 2019) that offers a standardized integration from different sources. However, 90% of the genetic variation associated to complex diseases are noncoding type and a benchmark of method to interpret how they alter genes, perturb biological pathways and ultimately lead to disease is still missing (Li and Ritchie 2021). Moreover, the application and integration of different tools to analyze GWAS data lead to discordant results, thus an unbiased assessment of the methods available is still required (Pérez-Granado et al. 2022). An advancement in associating genes to noncoding variants has been made by the Open Target Genetics platform, which implemented a pipeline consisting of a machine learning model that uses heterogeneous features such as distance from variant to the gene, expression quantitative trait loci, chromatin conformation and variant effect predictor. This method outperformed the naïve distance-based methods in the prioritization of causal genes related to complex diseases loci (Mountjoy et al. 2021).
In this context we present Genopyc, a Python library for investigating the functional effects of variants associated with complex diseases. Genopyc allows users to programmatically access multiple sources with the aim of understanding how noncoding variants could impact the biological pathways and thus infer the mechanisms underlying the development of complex diseases (Fig. 1). Moreover, being fully integrated in Python allows to perform further analysis in the same environment using the most common packages.
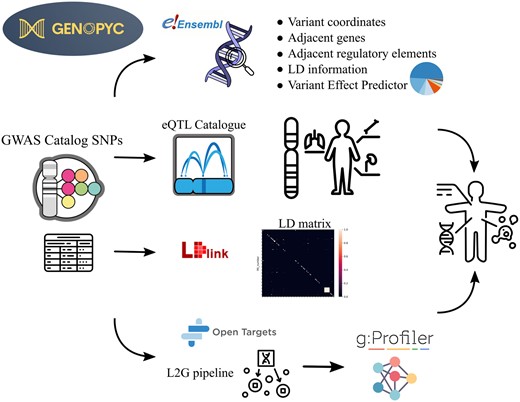
The main Genopyc features and knowledge bases accessed schematically represented. Variants associated with a specific trait are initially obtained from GWAS catalog and then subjected to various analyses, including examination of genomic context, LD features, eQTLs, VEP, and Locus2gene pipeline. Subsequently, as the variants are linked to genes through these analyses, the functions enriched within the gene set can be explored to identify potential dysregulated pathways relevant to the disease.
2 Implementation and features
Genopyc is a Python package integrating information from several knowledge bases. The tool can receive as an input a trait, coded with Experimental Factor Ontology (EFO) identifiers (Malone et al. 2010), or the results of a GWA study. If an EFO code is used as an input, the variants associated to the trait are retrieved from the GWAS Catalog. Information such as the β coefficient, standard error, risk allele frequency and the mapped genes related to the SNPs are also retrieved. Additionally, other features such as genomic coordinates, linkage disequilibrium (LD) correlated SNPs and neighboring functional elements can be obtained from Ensembl Genome Browser (Martin et al. 2023). Genopyc also queries the variant effect predictor (VEP) to obtain the consequences of the SNPs on the transcript and its effect on neighboring genes and functional elements (McLaren et al. 2016). Often SNPs associated to complex phenotypes fall in noncoding regions of the genome and are more likely to have regulatory effects (Prokunina and Alarcón-Riquelme 2004). Therefore, it is possible to retrieve the expression quantitative trait loci (eQTL) related to variants through the eQTL Catalogue (Kerimov et al. 2021). Finally, Genopyc integrates the locus to gene (L2G) pipeline from Open Target Genetics to uncover the target gene or genes of variants located in noncoding regions. Once a variant is associated to a gene or genes, the significantly enriched pathways are retrieved through G: Profiler (Raudvere et al. 2019). In this way the user can elucidate the functions whose perturbation could ultimately lead to the disease. Genopyc package also offers functionality to visualize the results of the functional enrichment as an interactive network (see Supplementary material). In this network, genes of interest are mapped to a protein-protein interaction network derived from the HIPPIE database (Alanis-Lobato et al. 2017) in which nodes represent the gene products and edges correspond to the physical interactions between proteins. A dropdown menu allows the user to select the function enriched in the gene set and, when a function is selected, the gene-products belonging to that function are highlighted.
Genopyc can also retrieve a linkage-disequilibrium (LD) matrix for a set of SNPs by using LDlink (Machiela and Chanock 2015), convert genome coordinates between genome versions and retrieve genes coordinates in the genome. LDlink calculates the LD matrix through the population-specific 1000 genomes haplotype panels (Auton et al. 2015). Retrieving genome coordinates and mapping between genome builds are made possible by accessing Ensembl genome browser. A comparison between the main functionalities of Genopyc and other tools for post-GWAS analysis is shown in Table 1. Genopyc is the only library that integrates multiple analysis to connect variants to genes (conditional, colocalization, fine mapping) through L2G pipeline, gather functional information to annotate variants (eQTLs, HI-C, linkage disequilibrium, VEP, functional genomic elements), maps between different vocabularies of gene and variant identifiers and perform functional enrichment to detect possible pathways perturbed by genetic variations. The visualization capabilities of the library help the user to directly unveil biological associations and can be fully exploited in an interactive computational environment such as Jupyter Notebook. In summary, we provide an all-in-one tool to retrieve and interpret the effect of genomic variants on the development of complex disease. Genopyc is easily installable via pip and can be integrated into Python environments being built upon main Python libraries.
Comparison between Genopyc and the main tools for post GWAS analysis.a
| Tool | Mapping Ids | Retrieve trait associated variants | Conditional analysis | Fine mapping | eQTL Co-localization | HI-C | Functional enrichment | Linkage disequilibrium | Genomic features | Variant annotation |
|---|---|---|---|---|---|---|---|---|---|---|
| Genopyc | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Coloc | × | × | × | × | ✓ | × | × | × | × | ✓ |
| FUMA | × | × | ✓ | × | ✓ | ✓ | × | ✓ | × | × |
| Finemap | × | × | × | ✓ | × | × | × | ✓ | × | × |
| Ensemble API | ✓ | × | × | × | × | × | × | ✓ | ✓ | ✓ |
| Open targets: genetics | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | × | ✓ | × | ✓ |
| Tool | Mapping Ids | Retrieve trait associated variants | Conditional analysis | Fine mapping | eQTL Co-localization | HI-C | Functional enrichment | Linkage disequilibrium | Genomic features | Variant annotation |
|---|---|---|---|---|---|---|---|---|---|---|
| Genopyc | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Coloc | × | × | × | × | ✓ | × | × | × | × | ✓ |
| FUMA | × | × | ✓ | × | ✓ | ✓ | × | ✓ | × | × |
| Finemap | × | × | × | ✓ | × | × | × | ✓ | × | × |
| Ensemble API | ✓ | × | × | × | × | × | × | ✓ | ✓ | ✓ |
| Open targets: genetics | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | × | ✓ | × | ✓ |
Genopyc integrates diverse functionalities allowing a more flexible investigation of variants related to diseases.
Comparison between Genopyc and the main tools for post GWAS analysis.a
| Tool | Mapping Ids | Retrieve trait associated variants | Conditional analysis | Fine mapping | eQTL Co-localization | HI-C | Functional enrichment | Linkage disequilibrium | Genomic features | Variant annotation |
|---|---|---|---|---|---|---|---|---|---|---|
| Genopyc | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Coloc | × | × | × | × | ✓ | × | × | × | × | ✓ |
| FUMA | × | × | ✓ | × | ✓ | ✓ | × | ✓ | × | × |
| Finemap | × | × | × | ✓ | × | × | × | ✓ | × | × |
| Ensemble API | ✓ | × | × | × | × | × | × | ✓ | ✓ | ✓ |
| Open targets: genetics | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | × | ✓ | × | ✓ |
| Tool | Mapping Ids | Retrieve trait associated variants | Conditional analysis | Fine mapping | eQTL Co-localization | HI-C | Functional enrichment | Linkage disequilibrium | Genomic features | Variant annotation |
|---|---|---|---|---|---|---|---|---|---|---|
| Genopyc | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Coloc | × | × | × | × | ✓ | × | × | × | × | ✓ |
| FUMA | × | × | ✓ | × | ✓ | ✓ | × | ✓ | × | × |
| Finemap | × | × | × | ✓ | × | × | × | ✓ | × | × |
| Ensemble API | ✓ | × | × | × | × | × | × | ✓ | ✓ | ✓ |
| Open targets: genetics | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | × | ✓ | × | ✓ |
Genopyc integrates diverse functionalities allowing a more flexible investigation of variants related to diseases.
3 Use case
To illustrate the utility of Genopyc, we applied it to the variants associated to lumbar disc degeneration that are available in the GWAS catalog [intervertebral disc degeneration (IDD), EFO:0004994]. IDD is a complex multifactorial condition for which the molecular mechanisms are poorly understood. Thanks to multiple data integration via Genopyc we highlight the involvement of variants associated downstream to pathways that may be relevant to the IDD, such as SP1 (Xu et al. 2016), HIF1-α (Meng et al. 2018) and AP-2α (Li et al. 2020), that according to the literature are tightly associated with IDD. Conversely, the functional enrichment did not bring any result or valuable information on the pathways that could be dysregulated in the disease. This example highlights that thanks to Genopyc a user can unveil a greater understanding of human complex traits.
Supplementary data
Supplementary data are available at Bioinformatics online.
Conflict of interest
J.P. is an employee of Medbioinformatics Solutions SL. J.P. is co-founder and holds shares of Medbioinformatics Solutions SL.
Funding
This project was supported by the Marie Sklodowska-Curie International Training Network “disc4all” [#955735]. B.O. acknowledges support from MCIN and the AEI (DOI: 10.13039/501100011033) by grants PID2020-113203RB-I00 and “Unidad de Excelencia María de Maeztu” [ref: CEX2018-000792-M].
Data availability
The library can be installed via pip: https://pypi.org/project/genopyc/. The source code is available at: https://github.com/freh-g/genopyc. The notebook with the use case is available at: https://github.com/freh-g/genopyc/blob/main/tutorials/Genopyc_tutorial_notebook.ipynb. The documentation of the package is available at: https://genopyc.readthedocs.io/en/latest/.


{kind=link}