





Abstract
Accurate pathogen identification is crucial during outbreaks, especially with the emergence of new variants requiring frequent primer updates. However, resources for maintaining up-to-date verification of primer sequences are often limited, which poses challenges for reliable diagnosis and hinders potential monitoring efforts based on genome sequencing. To address this, we introduce ViralPrimer, a web server facilitating primer design, SARS-CoV-2 and Mpox variant monitoring, and adaptation to future threats. ViralPrimer aims to enhance diagnostic accuracy with its comprehensive primer database, mutation analysis assistance, and user primer upload feature. Its adaptable design allows monitoring of other rapidly mutating pathogens, contributing to broader public health protection efforts.
ViralPrimer is freely accessible and open to all users with no login requirement at https://viralprimer.elte.hu/. The application is hosted on a DJANGO v3.2.13 web server, with a PostgreSQL database, and the frontend was implemented using jQuery v3.6.0, vanilla JavaScript vES6, and Bootstrap v5.1.
1 Introduction
The importance of accurate diagnostics in comprehending and managing rapidly mutating viral pathogens has become increasingly evident, particularly in the context of the SARS-CoV-2 pandemic, which presents a challenge to the effectiveness of diagnostic techniques (Fernandes et al. 2022). Although reverse transcription-polymerase chain reaction (RT-PCR) is currently considered the gold standard for diagnosing SARS-CoV-2, other nucleic acid amplification tests (NAATs) such as loop-mediated isothermal amplification (LAMP) offer rapid, robust, and cost-effective alternatives (Kashir and Yaqinuddin 2020). Additionally, next-generation sequencing (NGS) can be used not only for detecting viral presence (Bloom et al. 2021) but also for tracking the evolution of SARS-CoV-2 to identify emerging variants (Rosenthal et al. 2022). Consequently, these diagnostic methods as well as sequencing technologies require continuous evaluation, and the tracking of mutations in the target regions of the primers is critical for the reliable and accurate detection and sequencing of the viral genome.
It was shown that mutations in primer target regions can impact binding efficiency, potentially leading to false negative diagnostic results (Ai et al. 2020, Chen et al. 2020, Li et al. 2020a,b, Mentes et al. 2022). A recent study estimates that up to 29% of tests could result in misdiagnosis, particularly in highly affected populations (disease prevalence up to 50%), highlighting the importance of careful primer design (Pecoraro et al. 2022). While diagnostic primers are crucial for avoiding misdiagnosis, sequencing primers play a vital role in obtaining high-quality genome assemblies for tracking viral evolution. However, the potential impact of primer failure differs between these methods: diagnostic errors have more immediate epidemiological consequences, while errors in sequencing may result in “dropout” or the loss of an amplicon, leading to incomplete genome sequences and lower-quality genomic data (Sanderson and Barrett 2021, Bei et al. 2022). The latter is exemplified by the revisions of the ARTIC network and Midnight sequencing primers in response to the emergence of new variants such as Delta or Omicron (Bei et al. 2022, Lambisia et al. 2022, Ulhuq et al. 2023).
Given the importance of accurate primer design to accommodate viral evolution, the lack of readily available and current resources poses a significant challenge. To address this, we propose ViralPrimer, a website to help with primer design, monitor SARS-CoV-2 variants, and adapt to future viral threats. The web server provides an extensive database of diagnostic, sequencing, and LAMP primers and facilitates analysis through visualizations of genomic regions. The variant data is obtained from GISAID (https://gisaid.org/) (Khare et al. 2021) and the COVID-19 Data Portal (http://www.covid19dataportal.org) (Cantelli et al. 2021), which can be filtered temporally, across various countries of sample origin and by mutational profiles based on Variants of Concern (VOC) lineages. Users can also upload their own primer sequences. The potential of this tool lies in the identification of mutations that may affect primer binding efficiency. Mutations in the primer target regions are classified as high-risk mutations if they occur at the 3ʹ end of the primer or is an indel, and as moderate-risk if they have a minor effect on binding, however, the presence of three or more moderate-risk mutations in a primer's target region can also disrupt binding, although careful detection thresholds and multiple primer systems can mitigate this issue.
ViralPrimer not only enhances the ability to respond effectively to the evolving SARS-CoV-2 landscape but also prepares for future viral threats. Its adaptable design allows monitoring of other rapidly mutating viruses, as demonstrated by the integration of Mpox data.
2 Server description
ViralPrimer utilizes DJANGO's third major release (version 3.2.13) for backend functionality. Information is stored in a consistent relational database under the PostgreSQL framework. Data is either fetched directly from the database or computed in real-time from available information, eliminating the need for external APIs. The frontend combines DJANGO template language, jQuery (version 3.6.0), vanilla JavaScript (ES6), and Bootstrap (version 5.1) for its design. The visual elements are predominantly created using jQuery and vanilla JavaScript, with Plotly.js (version 2.11) utilized for chart rendering. Despite its reliance on advanced web technologies, ViralPrimer ensures compatibility across all browsers that support HTML5 and WebP.
3 Web interface
ViralPrimer is accessible online at https://viralprimer.elte.hu/, offering an array of user-friendly options for data retrieval. The homepage presents a broad overview of the server's capabilities, including direct links to introductory guides and example use cases. Navigation is facilitated through eight primary tabs: Home, Search, Browse, Primer mapping, Mutation Analysis, Help/About, Statistics, and Tutorials. The Search tab enables users to investigate mutations in a selected reference genome region with advanced filtering options, including collection date (the date the sample was collected), variant (the viral variant present in the sample), country (the geographical location where the sample was collected), and methods (whether the primer is used for sequencing or diagnostic purposes). The Browse page provides direct links to related entries, along with their labels and unique identifiers and also presents tables grouping data based on specific attributes. The Primer Mapping tab enables users to upload their designed PCR primer sequences to check for mutations in a specific genomic region. The Mutation Analysis tab allows users to input multiple positions or mutations, either at the amino acid or nucleic acid level, and assess their potential impact on selected primers for a given pathogen. For more refined search, the advanced filtering offers similar options to those in the Primer Mapping tab. The Help/About, Statistics and Tutorials pages are designed to help users navigate the server by providing examples, data-related information and statistics.
We also present a visual analytics framework within the ViralPrimer that can help users to explore the relationship between primer design and genomic mutations. This framework comprises five key components: Header, Simple Genome Viewer, Detailed Region Viewer, Mutations in Primer Target Regions, and Summary Plots. The Header offers essential contextual information and interactive features for navigating genomic regions. The Simple Genome Viewer provides an overview with zoom and navigation functions, delivering detailed primer and gene information. The Detailed Region Viewer offers a magnified view of genomic regions, outlining genes, proteins, primers, and mutations. The Mutations in Primer Target Regions section identifies and classifies mutations within primer target regions, aiding primer design. The Summary Plots segment offers graphical summaries of mutation distributions, allowing temporal and geographic analyses of mutation trends of the selected genomic region.
4 Data source
SARS-CoV-2 mutation data was obtained from GISAID (https://gisaid.org/) (Khare et al. 2021) and the COVID-19 Data Portal (http://www.covid19dataportal.org) (Cantelli et al. 2021, Harrison et al. 2021, Rahman et al. 2024), using high-quality samples with confirmed mutations [ENA project: PRJEB43947], a process extensively detailed in our previous article (Mentes et al. 2022). The Kooplex collaboration platform (https://k8plex-veo.vo.elte.hu/hub/) (Visontai et al. 2019) was used to investigate variants and samples from systematically analysed raw reads. Briefly, to ensure high quality, human samples with an estimated N-content of no >10% were selected. Genomic positions with a sequencing depth below 100 and an alternate allele frequency below 0.5 were excluded from analysis. Mpox data was processed following a similar procedure, undergoing systematic analysis of raw read data accessible through the COVID-19 Data Portal to produce a standardized set of variant calls (in VCF format) and consensus sequences [ENA project: PRJEB55823].
The reference sequences and feature data for the virus were obtained from the GenBank file, using the NC_045512.2 reference (Wu et al. 2020) [identical to MN996528.1 used by GISAID as the reference (https://gisaid.org/wiv04/) (Zhou et al. 2020)] of SARS-CoV-2 and the NC_063383.1 reference (Mauldin et al. 2022) of the Mpox genomes. References and features data are available for download under the Genes tab of the Browse section on ViralPrimer.
In our previous investigation (Mentes et al. 2022), we collected numerous primers from reputable scientific literature, which were designed for both traditional and RT-qPCR methods to detect SARS-CoV-2. We have since expanded the repertoire of primers within the ViralPrimer web server by incorporating additional primer systems, including sequencing and LAMP techniques. Additionally, the server hosts diagnostic and sequencing primers designed for the detection of Mpox. Primer data are available for download at the Primers tab of the Browse section of ViralPrimer. Mutation data can be downloaded from the Download section on the Help/About page.
5 Result and discussion
The primary goal of our web server is to integrate genomic mutation data of prevalent global pandemic pathogens with up-to-date information, including VOC variants, geographic locations and date of collection, and sequence information of publicly available PCR, LAMP and sequencing primers (Fig. 1). Previously, various websites have been created to track SARS-CoV-2 variants and monitor mutations in primer binding regions (Primer-monitor, Campbell et al. 2021, CoV2ID, Carneiro et al. 2020, CoVrimer, Vural-Ozdeniz et al. 2021, AssayM, Naeem and Pain 2020). However, some of these platforms are no longer available a few years after the start of the COVID-19 pandemic, host outdated data or require a registration (such as PrimerChecker at GISAID.org). Therefore, a comprehensive, sustainable, and responsive platform is needed to address these challenges. This is crucial not only for efficiently tracking the evolution of pathogens like SARS-CoV-2 but also for swift adaptability to emerging pandemic pathogens to effectively monitor future viral threats in diagnostics.
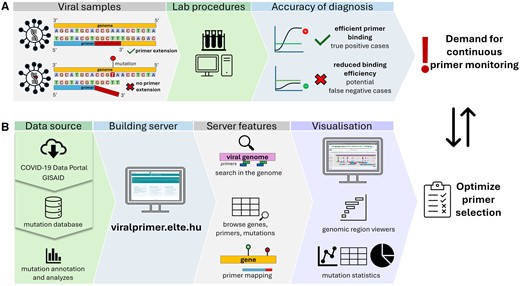
Overview of the ViralPrimer project. (A) The efficiency of PCR tests is affected by the variability in viral genome. Mutations near the 3ʹ end of the primer target region (designated as high-risk) can lead to false-negative results, highlighting the significance of continuous primer assessment. (B) Our project uses data from a mutation database incorporating the analysis results of various public sample sets (GISAID, COVID-19 Data Portal), enabling the assessment of mutations and samples concerning primer binding sites. Classification relies on the genomic location, length, and co-occurrence of mutations in each primer target region. The main features of the server are the options of searching in the genome, browsing primers, and primer mapping. The ViralPrimer webserver offers visualization tools, such as region viewers and mutation statistics, to aid in obtaining results and selecting optimal primers for specific regions. This tool further addresses the ongoing need for rapid primer testing and ensures the accuracy of diagnostic results.
The ViralPrimer web server has a comprehensive collection of NAATs primers, along with their associated mutations. We collected a total of 1372 primer oligos from 50 primer sets (10 LAMP sets, 4 sequencing sets, 35 diagnostic PCR sets). For SARS-CoV-2, a total of 19 007 636 samples were processed from two databases (GISAID—16 160 967 and COVID-19 Data Portal—2 846 669), while for Mpox, 1574 samples were included. The features allow for quick verification of mutations that affect both public and custom primers. Case studies demonstrating the use of the ViralPrimer website are available on the Tutorials page: one investigates the impact of Omicron XBB.1.5 variant mutations (Use Case 1), another focuses on advances in SARS-CoV-2 whole genome sequencing and evaluates the effectiveness of long-range PCR primers (Kandel et al. 2024) (Use Case 2), and a third provides a guide to using the Mutation Analysis page with Mpox amino acid mutations as an example (Wang et al. 2023) (Use Case 3). The data in the web server is regularly updated to ensure that it contains the most current information. In the future, we plan to expand the ViralPrimer to include viruses that are of global concern.
The ViralPrimer could fill a critical gap in the ability to respond rapidly to emerging viral threats by providing up-to-date and user-friendly resources for primer design and tracking viral evolution.
Acknowledgements
The authors thank Ádám Dán for fruitful discussions on the topic and valuable suggestions for research objectives. We gratefully acknowledge all data contributors, i.e. the Authors and their Originating laboratories responsible for obtaining the specimens, and their Submitting laboratories for generating the genetic sequence and metadata and sharing via the GISAID Initiative, on which this research is based. The authors data available at this link: https://doi.org/10.55876/gis8.231212uv. We are also grateful to all those who uploaded and continue to upload raw read data to the EMBL-EBI European Nucleotide Archive. Support from ELIXIR Hungary (www.elixir-hungary.org) also acknowledged.
Author contributions
N.D. contributed to the development of the web server, the conception and design of the study, and the methodology. Zs.D. contributed to the conception of the study and the writing of the manuscript. I.Cs. contributed to the conception of the study, the writing of the manuscript, and funding. A.M.-H. contributed to the writing of the manuscript, data acquisition, methodology, data curation, and web server testing. O.A.P. contributed to the writing of the manuscript, data curation, and web server testing. J.S., K.P., D.V., and G.E. contributed to the acquisition of the data and methodology. A.M. contributed to the coordination of the study, the development of the webserver, data analysis, conception, and writing of the manuscript. All authors read and approved the final version of the manuscript.
Conflict of interest: None declared.
Funding
This work was supported by the European Union's Horizon 2020 research and innovation programme under grant agreements No. 874735 (VEO) and No. 101046203 (BY-COVID). The work of N.D. was supported by the ÚNKP-23-3 New National Excellence Program of the Ministry for Culture and Innovation from the source of the National Research, Development and Innovation Fund and DKOP-23 Doctoral Excellence Program of the Ministry for Culture and Innovation from the source of the National Research, Development and Innovation Fund.
Data availability
The data underlying this article are freely available on the ViralPrimer website at https://viralprimer.elte.hu/. Mutation data used on the website can be downloaded via https://viralprimer.elte.hu/help#download. Detailed NAATs' primer information is accessible at https://viralprimer.elte.hu/browse/primers. Primary data are sourced from the GISAID repository (https://gisaid.org/, with sample authors acknowledged) and the COVID-19 Data Portal (http://www.covid19dataportal.org, ENA projects: PRJEB43947, PRJEB55823). Reference sequences and feature data for SARS-CoV-2 and Mpox were obtained from GenBank (NC_045512.2 and NC_063383.1).


{kind=link}