





Abstract
Summary: Lysine succinylation orchestrates a variety of biological processes. Annotation of succinylation in proteomes is the first-crucial step to decipher physiological roles of succinylation implicated in the pathological processes. In this work, we developed a novel succinylation site online prediction tool, called SuccFind, which is constructed to predict the lysine succinylation sites based on two major categories of characteristics: sequence-derived features and evolutionary-derived information of sequence and via an enhanced feature strategy for further optimizations. The assessment results obtained from cross-validation suggest that SuccFind can provide more instructive guidance for further experimental investigation of protein succinylation.
Availability and implementation: A user-friendly server is freely available on the web at: http://bioinfo.ncu.edu.cn/SuccFind.aspx
Contact: [email protected]
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
Lysine succinylation is a novel identified PTMs pathway present in both prokaryotic and eukaryotic cells, which orchestrates a variety of biological processes. Succinylation was first discovered to occur at the active site of homoserine trans-succinylase (Rosen et al., 2004). With an antibody based enrichment strategy and MS-based proteomics, Zhang et al. (2011) identified several dozens of succinylation sites in bacteria in 2011. Subsequent work recognized a number of succinylation sites in animal tissues (Du et al., 2011) and on histones (Xie et al., 2012). Recently, plenty of lysine succinylation sites were also identified in extracellular toxoplasma tachyzoites (Li et al., 2014). As the rapid development of sequencing techniques, identification and functional study of succinylation has emerged to be an intriguing topic and attracted much attention. But it is still unknown about the mechanism of succinylation as well as the succinyl donor to lysine remains, a global analysis of lysine succinylation in diverse organisms is still lacking. Yet annotation of succinylation in proteomes is a first-crucial step toward decoding protein function and understanding of their physiological roles that have been implicated in the pathological processes. For systematically investigating the lysine succinylation and its relevant function, a prerequisite is to establish a reliable and comprehensive dataset. However, while only a small amount of known succinylation sites was detected, experimental verification of succinylated substrates is labor-intensive, time-consuming and biased toward abundant proteins and proteotypic peptides. Thus, in silicon prediction of succinylation sites can serve as an alternative strategy for whole proteome annotation. We thus developed the SuccFind to determine whether a fragment which contains lysine residues can be succinylated or not, which is constructed to predict the lysine succinylation sites based on sequence-derived features, and evolutionary-derived information of sequence and via a enhanced feature strategy for further optimizations.
Coincidentally, when we built SuccFind to predict protein succinylation sites, another protein succinylation site predictor called SucPred was reported as well (Zhao et al., 2015). But when we try to visit the web server of SucPred, the website is not accessible. So there is no way to compare with it. Since SucPred only considered unilateral features with no further optimization to build prediction models, features might contain redundant and noisy information and some other potential value information will inevitably lose. Here, we have considered different aspects of the features and via a two-step feature selection strategy to carry further optimizations when developed the model so our system can provide a more accurate and comprehensive prediction performance as well as a more stable and sustained online service.
2 Methods
2.1 Data collection and preprocessing
By searching with multiple keywords such as ‘lysine succinylation’, ‘succinylation’, we collected 2938 experimentally verified lysine succinylation sites in 1044 proteins from different database including UniProtKB/Swiss-Prot (Boutet et al., 2007), CPLM databases (Liu et al., 2014) etc. as well as the relevant literatures (Li et al., 2014; Park et al., 2013). As previously described, we regarded the known succinylation sites as positive data, while other non-succinylation lysine sites were taken as negative data. To avoid overestimated prediction because of the redundancy of homologous sites in the positive data, we used CD-HIT (Li and Godzik, 2006) to wipe off homologous fragments with a sensitive cutoff of 0.3. Finally, a non-redundant data set for training was constructed with 2713 positive sites and 23598 negative sites (Supplementary Table S1).
2.2 Sequence-derived characteristics
2.2.1 AAC & CKSAAP Encoding
Since all organisms try to minimize the ‘cost’ of protein synthesis by adjusting their amino acid content to specific growth conditions, a proteome-wide amino acid composition analysis can characterize the specific state of a given organism (Mazel and Marliére, 1989). Here, amino acid composition (AAC) and composition of k-spaced amino acid pairs (CKSAAP) (i.e. pairs that are separated by k other amino acids) were used to extract the potential sequence information of amino acid residues surrounding the succinylation sites and non-succinylation sites. More details about the encodings are described in Supplementary Materials Ep1.
2.2.2 AAindex
Amino Acid index database (AAindex) (Kawashima and Kanehisa, 2000) contains a total of 544 amino acid indices that specify the physicochemical properties of amino acids. Thus the amino acids adjacent to the succinylation sites can be encoded as an input vector to develop our prediction model. After examined all of the 544 physicochemical properties with the default parameters of SVM, isoelectric point was chosen as the best physicochemical property (with high accuracy) for succinylation prediction from AAindex.
2.3 Evolutionary-derived information
Local sequence clusters often exist around succinylation site because substrate sites of same enzyme usually share similar patterns. To take full advantage of such cluster information of local sequence fragments for predicting succinylation sites, we took the local sequence around the succinylation site in a query protein and extracted features from its similar sequences in both positive and negative datasets by a LSC score algorithm. More detail about the encoding is described in Supplementary Materials Ep2.
2.4 Two-step feature selection strategy via support vector machine
As heterogeneous features are often redundant and noisy, we performed feature selection to pick up most important features by a feature selection method known as F-score (Sokolova et al., 2006). In particular, a two-step feature selection manner was applied in our study. First, averaged F-score values of the each ten training sets were calculated with the purpose of ranking the features vector. We used a wrapper-based feature selection with the forward best first search. More specifically, for a given list of feature sorted in the descending order by their average F-score value and an empty list R that store the selected features. We add the top-ranked feature from F to R and run SVM using feature set R in the cross validation strategy. If the addition of the top ranked features improves the average accuracy value over the ten test folds, then this features vector is retained in R; otherwise it will be removed. We repeat that until F is empty. More details about the methods are described in Supplementary Materials Ep3.
3 Results and discussion
Based on the succinylation data sets, we firstly generated the graphical sequence logo (P < 0.01; t-test) and detected a statistically significant differences in position-specific symbol compositions and biochemical environment (Fig. 1a, Supplementary Fig. S1). We then calculated the amino acid frequencies (Fig. 1b, Supplementary Fig. S2) and probed the weight distribution of amino acid pairs (Fig. 1c, Supplementary Fig. S3) in the sequence surrounding the succinylation sites. Information gain (IG) values of different residues also were calculated in Figure 1b, which was used to distinguish the importance of different residues for succinylation sequence. More detail about the IG is described in Supplementary Materials Ep4. Note that the larger the IG values of residues are, the more important the residues are. These results suggested not only the compositions of amino acids, amino acid pairs but also the weight of residues is vary widely in succinylation and non-succinylation segments. After examined all of the 544 physicochemical properties with the default parameters of SVM, isoelectric point was chosen as the best physicochemical property for succinylation prediction (Figs 1d, 2a). In addition, as for biological analysis, one of the most important aspects of concern is the evolutionary conservation. Figure 2b and 2c compares the LSC scores of succinylation sites with those of non-succinylation sites. The result declared the LSC scores could capture evolutionary similarity information in the local sequence around succinylation sites and hence distinguish them from the background. To assess the prediction performance of SuccFind, the LOO validation and 4-, 6-, 8-, 10-fold cross-validations were applied and the receiver operating characteristic (ROC) curves were drawn in Figure 2d and the corresponding value of average area under the curve (AAUC) values were calculated as 0.8070 (LOO), 0.8201 (4-fold), 0.8182 (6-fold), 0.8226 (8-fold) and 0.8162 (10-fold), respectively. Since the prediction performance of the 4-, 6-, 8- and 10-fold cross-validations were closely similar to the LOO validation for the prediction of succinylation sites, it is evident that the method is a robust predictor.
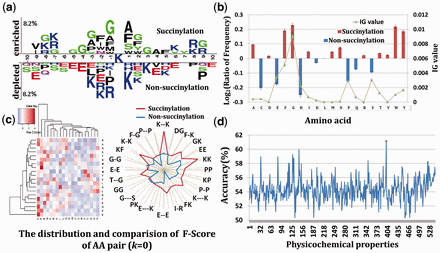
(a) A two-sample logo of the compositional biases. (b) Comparisons of AAC in positive and negative datasets. (c) Weight distribution and comparisons of k-space scores of amino acid pairs (k = 0). (d) The accuracy of 544 physicochemical properties with the default parameters of SVM
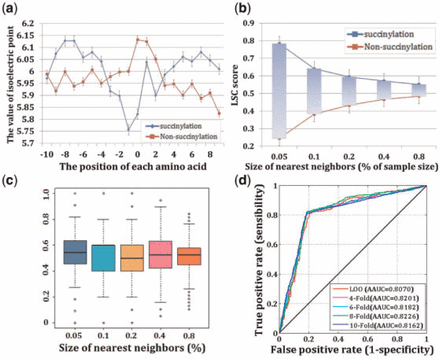
(a) Comparison of isoelectric point between succinylation sites and non-succinylation sites. (b), (c) Comparison of LSC scores between succinylation sites and non-succinylation sites. (d) The ROC curves and AAUC values for the LOO validation and 4-, 6-, 8-, 10-fold cross-validations
With the high stringency value, we adopted SuccFind to predict potential succinylation sites in lysine acetylated substrates. Totally, it was observed that 10128 (17.80%) known acetylation sites might be modified by succinylation. We are also surprised to detect that 2422 succinylation sites in our study could co-occur at the same lysine residues with acetylation. These observations show that succinylation prefer to co-occupy with acetylation at the same lysine residues. And in situ crosstalk between succinylation and acetylation might exhibit a tissue-specific manner (Supplementary Table S1 and Supplementary Data). Gene functional analysis suggested that succinylation prefers to in situ crosstalk with acetylation in a variety of biological processes and pathway (Supplementary Figs S5 and Supplementary Data), and potential impacts of lysine succinylation on enzymes involve in mitochondrial metabolism and other cellular processes that metabolism related (Supplementary Materials Ep5 and Supplementary Data).
It is our desire to build an open platform which could provide more useful guidance for experimental workers of identification of succinylation sites. The web service of SuccFind is freely available at: http://bioinfo.ncu.edu.cn/SuccFind.aspx. The improved succinylation prediction system will be done when the new succinylation sites data become available. We anticipate that the SuccFind will be a powerful and complementary tool for further experimental investigation of protein succinylation.
Funding
This work was supported by the National Natural Science Foundation of China (21175064, 21305062); and Program for New Century Excellent Talents in University (NCET-11-1002).
Conflict of Interest: none declared.
References
Author notes
Associate Editor: John Hancock


{kind=link}
{kind=link}