





Abstract
Motivation: As next generation sequencing gains a foothold in clinical genetics, there is a need for annotation tools to characterize increasing amounts of patient variant data for identifying clinically relevant mutations. While existing informatics tools provide efficient bulk variant annotations, they often generate excess information that may limit their scalability.
Results: We propose an alternative solution based on description logic inferencing to generate workflows that produce only those annotations that will contribute to the interpretation of each variant. Workflows are dynamically generated using a novel abductive reasoning framework called a basic framework for abductive workflow generation (AbFab). Criteria for identifying disease-causing variants in Mendelian blood disorders were identified and implemented as AbFab services. A web application was built allowing users to run workflows generated from the criteria to analyze genomic variants. Significant variants are flagged and explanations provided for why they match or fail to match the criteria.
Availability and implementation: The Mutadelic web application is available for use at http://krauthammerlab.med.yale.edu/mutadelic.
Contact: [email protected]
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
Distinguishing harmful from benign variants in clinical exome or whole genome sequencing is comprised of two steps: Variant annotation and interpretation. Annotation involves the labeling of variants with diverse features, including population frequencies and effect on amino acid composition, which then guide interpretation for finding variants with damaging effect on protein function (what we call variant prioritization).
There are several annotation systems that assist medical geneticists in the prioritization of a patients variants. Some of the existing systems, including Annovar and VAX (Wang etal., 2010; Yourshaw etal., 2014), can accommodate custom annotations for guiding a geneticists decision process. As most annotations can be pre-computed and efficiently retrieved, these systems can rapidly annotate most or all variants in a human genome.
While these existing systems are an invaluable tool for medical geneticists, they do have some shortcomings. First, they do not address the need to accommodate several variant filters based on disease-, and often expert-dependent interpretation rules. Second, as some annotations cannot be easily pre-computed, like complex Indels, or substrate binding properties, the computational cost of generating annotation for all observed variants is high. Third, as the number of annotations increases, storing the complete set of annotations for all variants is progressively costly. Fourth, as data resources are changing over time, there is a need to recompute the entire set of variants for retrieving the most current annotations. Fifth, there is little emphasis on using standards or provenance when generating variant annotations.
What we propose in this article is a fresh look at how we perform variant prioritization. Rather than annotating all observed variants, with subsequent interpretation, we propose to merge the two steps into a single process that reduces the necessary annotations to the minimum number necessary. To do so, we borrow from prior work in semantic workflows, and related fields such as hierarchical task network planning. In essence, we allow geneticist to formulate an objective (identify variants with a particular annotation combination), which is sufficient to generate an efficient plan for a series of annotation steps (a workflow) to which each variant is subjected. If at any point in the workflow it is determined that the objective cannot be reached, the variant under consideration is dropped from further consideration. Using smart workflow planning, computationally cheap annotation steps are front-loaded. For example, if the objective states that the geneticist is interested in variants with a specific population frequency only (a simple database lookup), many variants with higher frequency can be discarded at an early stage and do not need further annotations. If a workflow completes successfully, only the absolute necessary annotations were computed to reach the objective. Using elements from semantic workflow research, we treat each annotation step as a separate service with semantically defined input and output criteria. The use of a service-oriented architecture has distinct advantages; among them a straightforward procedure for determining whether a variant needs re-computing. For example, only variants that have been subjected to a particular annotation service need re-processing if the service is updated. Finally, the use of a coherent semantic framework, expressed in RDF/OWL, is not only helpful in workflow planning, but also for defining diverse sets of annotation objectives, for generating a provenance trail, and to link annotated variants to other resources using constrained data elements.
2 Approach
2.1 Mutadelic overview
Our system, which we call Mutadelic, is based on workflows that are created based on the user’s objectives for variant classification (i.e. what characteristics a mutation will have that an ordinary variant will not). To build these workflows, Mutadelic relies upon abductive reasoning. Abductive reasoning differs from the more familiar deductive reasoning in which logical rules are used to derive unstated inferences from an asserted KB. Abductive reasoning instead takes an initial KB (KBI) and a resulting observation (O) and determines what statements need to be added to KBI to allow O to be logically inferred. Using ontologies, we express the geneticist’s objective (identify variants with a particular annotation combination) as a variant class with a set of annotations and restrictions on annotations. We also represent observed patient variants as individuals of the variant class, with initially no annotations added. The overall goal of the abductive reasoning workflow is to progressively add annotations to the individual, until the geneticist’s defined variant class subsumes this individual. Annotation services are an important part of this process, as it is they that add the necessary annotations. Conceptually, the process starts from the objective, iterating through the defined annotations and restrictions identifying those that can be addressed by the computationally cheapest service. Once the first annotation is added to the individual (variant), and the annotation complies with the stated restriction, the process addresses the next annotations. Some defined annotations can only be addressed by a combination of annotation services. In these cases, automated workflow generation allows the chaining of services based on semantic service descriptions. For example, if the objective states that the ‘variant should be in a protein domain’, the workflow needs to chain at least two services that first map the variant to some amino acid position and then checks whether the position is located in a protein domain. As the services add annotation, they also provide provenance information that allows the reconstruction of the chain of services.
2.2 KB representation
Mutadelic is built on a description logic (DL) model described in the Web Ontology Language (OWL). It uses and extends classes and properties from the Scientific Information Ontology (SIO) and Genomic Element Ontology (GELO). We represent genomic variants as subclasses of the GELO GenomicElement class. GELO provides data properties to represent locus_start, locus_end, (observed) sequence and strand as literal values. An object property on_chromosome points the variant to an instance of SIO’s chromosome class. To represent the reference sequence, we use SIO’s is_modelled_by property and restrict it to an individual with a string value filled by the SIO has_value data property. Annotations of variants are represented by the following pattern. A class Annotation is defined as an extension of the SIO description class. An Annotation uses the SIO cites property to connect it to its provenance (an instance of SIO’s information_content_entity or one of its subclasses). The variant is connected to the Annotation via the SIO is_described_by property. The annotated variant is connected to the actual data through the SIO refers_to property. The refers_to property is restricted to a particular information_content_entity or subclass thereof (e.g. VariationOutcome, VariationDomainColocation, etc.). Many annotation data classes extend the SIO bioinformatic_data class, itself an extension of information_content_entity. Examples include PhylopScore and SiftScore. Finally, the annotation data class is assigned its actual value through the has_value data property which is filled by a literal representation of the actual data. Figure 1 illustrates the representation of a typical variant in OWL.
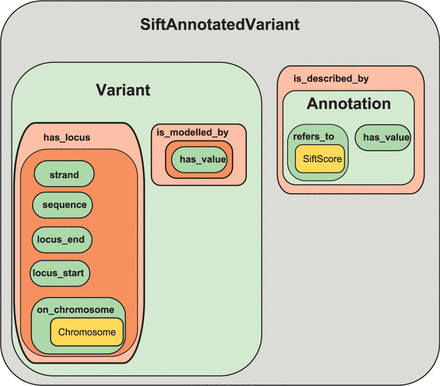
An illustration showing how variants are represented in OWL in Mutadelic. The diagram show SiftAnnotatedVariant as an example. Rectangles at the same vertical level reflect conjunction. For example, SiftAnnotatedVariant is a Variant and is_described_by some Annotation
2.3 AbFab
2.3.1 Services
An abductive reasoning framework for workflow generation is a central focus of development and a key contribution of this project. OWL does not provide semantics for abductive reasoning and most DL reasoners do not provide abductive reasoning capabilities out of the box. To fill this gap, we developed the a basic framework for abductive workflow generation (AbFab) tool. AbFab represents workflows as paths connecting services. Services are OWL classes which are constrained by the class of input that is required and the class of output that will be generated. In OWL, this is implemented very loosely as a Service class with restrictions on object properties has_output and has_input that may be filled by any OWL class expression. Other than this basic requirement, AbFab can support OWL domain models of arbitrary complexity. The service class also contains a reference to the process that should be run when the service is executed. In service execution, an instance fitting the input constraint is passed to the execution process. Services add facts to the input instance, such that the instance will be a member of the specified service output class. By separating the actual execution of services from the workflow model, AbFab allows access to procedures that must, for performance reasons or because the results are non-deterministic, operate outside the context of a DL reasoner while still maintaining a formal logical structure over the workflow as a whole. Figure 2 illustrates the structure and representation of AbFab services.

The diagram on the left provides a representation of AbFab services in OWL. The diagram on the right shows how services add information and provenance to input such that it ultimately fulfills the constraints of the goal criteria. This generated workflow is referred to as an AbFab path
2.3.2 Workflow generation/execution
AbFab operates in two phases: staging and execution. In the staging phase, AbFab takes as input: (i) an object called an IndividualPlus, which consists of an OWL individual and the set of OWL axioms describing that individual which would be added to the KB, and (ii) an OWL class expression which describes the goal output. It uses the KB of available services to determine a sequential path of services which, when executed would lead to an IndividualPlus consisting of the original OWL individual plus new axioms that would cause that individual to be consistent with the goal class. There are three types of steps which can be combined to form a path. Simple steps execute a single service. Branches are used where two or more services must be called to fulfill the input criteria of the next step. This corresponds with a logical AND condition. The output will be a new IndividualPlus which merges the set of axioms resulting from each service call. Conditions are used where two or more services may be each alternatively used to fulfill the input criteria of the next step. This corresponds with a logical OR condition. Conditions are useful for workflows where the next step of execution is the dependent on the result of the previous service and therefore not available until the execution phase. Branches and conditions are illustrated in Figure 3.
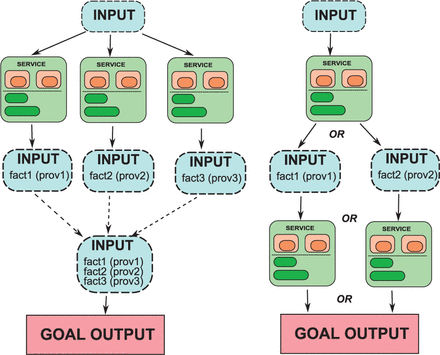
The diagram on the left illustrates a goal which is fulfilled by the output of three separate services A branch is formed which joins the three outputs. The diagram on the right illustrates a case where the input criteria of different services may be fulfilled depending on the output of a prior service. Because this result is not known until execution time, a condition is created which forms separate paths depending on the output of the prior service
Internally, the path is created in reverse, by first finding ‘terminal’ services (i.e. services whose output satisfies the goal class) and then adding services whose output satisfies the input of the next step. When the initial input satisfies the input criteria of the top service, the path is complete. Determining whether service inputs/outputs are satisfied is performed by the DL reasoner through class subsumption checking. Specifically, the output class of a service must be subsumed by the input class of the next step. For example, if some service S1 produces an output of class A and B, it would satisfy the input criteria of a service S2 which required input of class A but not that of a service S3 which required input of class A and B and C. To determine if the initial input satisfies the input criteria of the top service, an instance check is performed. This will determine if the initial input can be declared as an instance of the input criteria for the top service without rendering the model inconsistent.
Once all possible paths from initial input to goal output are determined, a cost function is used to determine the best path. The cost function is useful in that it prevents tasks that take a long time to run or consume a large amount of computational resources from being run unless absolutely necessary. The cost function is also used to determine the order in which the multiple services within a branch are executed and the sub-paths of a conditional are attempted. In both cases, the cheapest services are executed first. Our current cost function assigns a quantitative value to a service by filling the data property has_cost of a Service individual with a numerical literal expressing the relative expense of execution. Statically assigning a cost to a service is a fairly naive approach and in future versions we wish to implement cost values that are dynamically assigned based on input to the service. Another type of case is one in which multiple Services are capable of fulfilling a goal criteria. Suppose, for example, that an arbitrary ServiceA consumes more resources but yields more precise or reliable results than a less costly version, ServiceB. It may be preferable in some cases to prefer the accuracy of ServiceA and in others the speed of ServiceB. The inclusion of a quality metric to the cost function is an area of future research.
In the execution phase, the initial input IndividualPlus is passed down the chain of services, acquiring axioms from each step as it goes. At each level, the IndividualPlus is tested using consistency checking to make sure that it matches the criteria to continue and in the case of a condition whether to attempt a different fork. Since DL reasoner operations such as consistency checking are potentially time-consuming and computationally expensive, a variety of optimizations are made to keep AbFab performing efficiently. These optimizations are discussed in Supplementary Methods section.
2.4 Services for Mutadelic
A number of AbFab Services were defined in this project to correspond with tasks commonly used for variant analysis. These include Services which obtain Sift scores (measuring impact of a DNA variant on protein formation), PhyloP scores (measuring the level of evolutionary conservation of a genomic region), allele frequencies (measuring the rarity of a variant in healthy populations) and information about critical protein domains obtained from the PFAM database. Typically these services are written in Java and will acquire the data from a local database and attach it to the set of OWL axioms held by the input object. A large amount of data is required locally for these services. For example, the PhyloP database assigns a score to every base on the gene-coding regions of the chromosome. The Sift database calculates scores for all nucleotide substitution possibilities at each exonic position. We store these large collections of data in MongoDB (http://www.mongodb.org), one of the better-established of a number of ‘No-SQL’ databases that have recently come into vogue. In MongoDB, data are stored in large document-like structure rather than relational tables; schema are flexible and dynamic. Data can be retrieved rapidly as it is stored in simple indexed key-value pairs. We found the combination of performance and flexibility offered by MongoDB to be ideal for our purposes.
3 Results
3.1 Mutadelic implementation
3.1.1 Motivation
The most notable disorders of red cell membranes in humans are hereditary spherocytosis (HS), hereditary elliptocytosis (HE) and hereditary pyropoikilocytosis (HPP). They are inherited disorders, marked by genetic heterogeneity, that are linked to hemolytic anemia (Mohandas and Gallagher, 2008). HS is characterized by erythrocytes that are spherical in shape rather than the expected olive-shaped conformation (Perrotta etal., 2008). HS is a fairly common disorder, especially among Northern European populations where it occurs in 1:1000–2500 individuals. It most frequently manifests itself in the form of mild to moderate anemia, although severe anemia can occur in some cases. HS is characterized by mutations in the following genes: ANK1 (ankyrin 1, erythrocytic), SLC4A1 (solute carrier family 4, anion exchanger, member 1 [erythrocyte membrane protein band 3, Diego blood group)], SPTB (spectrin, beta, erythrocytic), SPTA1 [spectrin, alpha, erythrocytic 1 (elliptocytosis)] and EPB42 (erythrocyte membrane protein band 4.2). Of these five, mutations in ANK1 are most frequent, followed by SLC4A1 and SPTB (Gallagher, 2004a). HE results in elongated, cigar-shaped erythrocytes. It is most frequently asymptomatic but will occasionally lead to mild or even severe anemia (Gallagher, 2004b). It is also fairly common (1:2000–4000), with higher incidence in African populations (estimated as high as 1:100). HPP is related to HE but is much more severe. It is characterized by large numbers of fragmented and misshapen erythrocytes and results in severe hemolytic anemia. Mutations in SPTA1, SPTB and EPB42 are associated with HE and HPP, with SPT1 being the most frequent. Most documented mutations modify the self-association regions of spectrin proteins which causes damage to cell membrane structure (Zhang etal., 2001).
3.1.2 Criteria
An initial test workflow was created which would prove useful for the analysis of variants on a set of five genes associated with Mendelian blood cell disorders. The workflow defines a potential mutation as one that either (i) has been recognized as a disease-causing mutation by the Red Cell Membrane Disorder Mutations Database (http:// research.nhgri.nih.gov/RBCmembrane) or (ii) is extremely rare in healthy populations (<0.01 MAF) and either (a) occurs at a splice site, (b) is considered severely damaging to the resulting protein (<0.05 SIFT score), (c) occurs in a highly conserved region (>1.0 PhyloP score) or (d) occurs in a region that codes for a critical domain of the protein.
Specific services to perform these checks are implemented as (i) KnownMutationService, (ii) VariantFrequencyRareService, (iia) TranscriptLocalSpliceService, (iib) PhylopConservedService, (iic) SiftSevereService and (iid) InCriticalDomainService. In addition, a handful of services are required which perform prerequisite tasks. For example, the Red Cell Mutations database identifies variants by position relative to the transcript rather than genomic position so a preliminary conversion from genomic to transcript-based position is required. This conversion is handled by the AlignVariantService. AlignVariantService is also a prerequisite for TranscriptLocalSpliceService. Each of PhylopConservedService, SiftSevereService and InCriticalDomainService need consider only non-synonymous variants. Hence, a prerequisite service (AAChangeNonSynonymous) is required. This in turn requires the determination that a variant occurs in the protein-coding region of a transcript. Again a prerequisite service (TranscriptLocaleProteinCodingService) is required, which in turn requires AlignVariantService as a prerequisite. Finally as part of the optimization process, two marking services are generated by AbFab. This process is discussed in detail in the Supplementary Methods section. The first (MarkedUniqueVariantService) marks the disjunction of TranscriptLocaleSpliceService, PhylopConservedService, SiftSevereService and InCriticalDomainService; the second (MarkedRareAndUniqueVariantService) marks the conjunction of VariantFrequencyRareService and MarkedUniqueVariantService. The full criteria for interesting variants is illustrated in Figure 4.
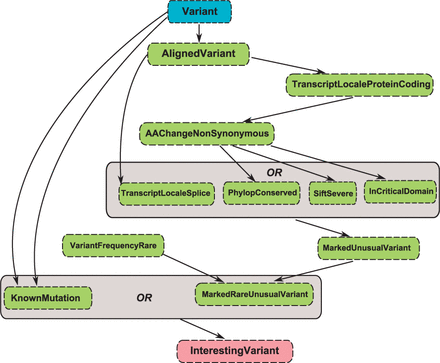
An illustration showing the criteria used to identify significant variants in Mutadelic. The map provides an overview of services which are called by the workflow path. Auto-generated marker services are highlighted in dashes
3.1.3 Web application
Mutadelic has a web application front end. A RESTful Web Service (Fielding and Taylor, 2002) was implemented using (http://jersey.java.net), a library implementing the Java JAX-RS standard. The following resources are defined: Users, Workflows, Inputs, Variants, Outputs and AnnotatedVariants. Workflows define the criteria that is used to determine whether a variant is significant. They are owned by Users. Inputs are bound by a Workflow and hold the collection of Variants which the user is analyzing. Outputs are tied to a particular Input and hold a collection of AnnotatedVariants which are annotated with the results of performing the Workflow analysis. Resources are stored in MongoDB collections and mapped to Java classes via the Morphia Object-Relational Mapping (ORM) tool. The web application runs on the Grizzly container for highly scalable Java server applications (https://grizzly.java.net).
The front end is a simple Ajax-based single-page application. The user is allowed to input genomic variants individually or in a file. Once variants are submitted, an instance of AbFab running on the Mutadelic server will generate and execute workflow paths for each variant. Once AbFab is finished running, the front end will show all variants, highlighting those determined to be interested. The user can click on a variant to view the annotations that were added by the Mutadelic server. Annotations contributing to an interesting status are highlighted. The Mutadelic web application uses Bootstrap as a UI design framework (http://getbootstrap.com). We used knockout.js as a data binding library (http://knockoutjs.com) and jQuery for Ajax interaction with the server and additional Javascript functionality (http://jquery.com).
3.2 Validation
To test the effectiveness of Mutadelic in identifying harmful variants, we attempted to validate the curated set of disease-associated variants at the Red Cell Membrane Disorder Mutations Database. (This is the same database we use in Mutadelic to flag variants previously determined to be disease causing. For obvious reasons, we disabled that portion of the criteria for this validation.) Eliminating a handful of intergenic variants, we tested 119 variants associated with HS and 43 variants associated with HE/HPP (Fig. 5). Mutadelic performed well for both sets of variants, flagging 106 of 119 HS variants (89.0%) and 38 of 43 HE/HPP variants (88.4%). Overall, performance was somewhat better for point variants (103 of 112; 92.0%) than for indels (41 of 50; 82.0%). Identification of deleterious indels could be improved by adding a service which determines if critical domains of a protein prevented from forming by a frameshift caused by a mutation. Such a service is further described in the Future section. Of the 18 variants not flagged by Mutadelic, only two were not at least found to be rare variants and those two were borderline cases (0.02 and 0.05 versus the 0.01 threshold). A significant number not identified (5 of 18) were intronic variants and it should be noted that few effective metrics for variant prioritization of introns exist.
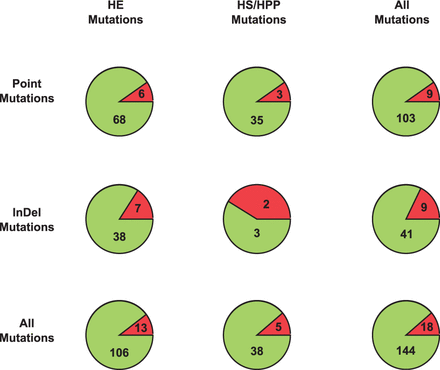
Mutadelic was used to validate the HS and HE/HPP associated variants curated by the Red Cell Membrane Disorder Mutations Database. In the pie charts, the green regions indicate disease-associated variants successfully flagged by Mutadelic and the red regions indicate those missed
4 Discussion
This work discusses clinical variant prioritization using DL reasoning, and bears similarities to existing ideas from the fields of semantic workflow generation, and related fields such as hierarchical task network planning. The key elements of our approach, in short, are the ability to express a goal or objective, and a system that decides whether an observed individual (variant) complies with the objective. To do so, we need, in a staging phase, means to automatically chain together existing variant annotation services, using semantic descriptions of these services. There is extensive prior work in this area (Zhao and Paschke, 2012), with several formalisms for web service description available, such as SWASDL (Kopecky etal., 2007). There exists integrated systems that perform workflow generation based on such descriptions, including the WINGS system (Gil etal., 2010, 2011) or TAVERNA (Oinn etal., 2004). These existing formalisms and systems are generic, and can be applied to diverse domains and research areas. Unlike WINGS, which supports automated workflow generation, but delegates service execution to external systems such as PEGASUS (Deelman etal., 2005), our system integrates workflow planning and execution in one formalism. While TAVERNA’s emphasis is less on automated workflow generation, it integrates workflow planning and execution in one platform. Both systems do not provide the second requirement of our system, which uses instance checking to determine whether the output of a workflow (once executed), corresponds to some stated objective. There is some prior work (Sirin etal., 2004) that uses hierarchical task network (HTN) planning on top of semantic web service descriptions, to achieve a similar goal. However, the described work is based on an existing task planning system, called SHOP2 (Nau etal., 2003), which is not easily applicable to the RDF/OWL formalism used in our project. In the bioinformatics domain, our approach is similar to the Semantic Automated Discovery and Integration (SADI) framework (Wilkinson etal., 2010). SADI describes web services for processing biomedical data in OWL. As in AbFab, OWL classes with restrictions on input and output are used to model services. The Semantic Health And Research Environment (SHARE) SPARQL query client also functions by connecting services to form a path of execution. Mutadelic/AbFab and SADI/SHARE differ however in terms of their focus and their means of implementing service paths. The focus of the SHARE is on answering queries where data is spread out across multiple locations. AbFab focuses instead on generation of workflows to process data where services are defined locally. SHARE matches services based upon whether execution of them adds the predicate requested by the query to the KB (Vandervalk etal., 2009). AbFab uses consistency checking to determine fulfillment of input and output constraints. This approach allows for more flexible matching and it does not require the user to know the specifics of what triples services add to the KB. The SHARE method requires that a step be executed before the next step may be determined. AbFab generates the full path before execution, potentially saving considerable expense if a path to the goal criteria cannot be reached. AbFab also offers the additional features of a cost-based determination of optimal path and structures (Branches and Conditions) to support complex paths. The ability to change execution flow based upon runtime output allows AbFab to support use cases beyond workflow generation, including decision trees and the sort of query answering targeted by SADI/SHARE. Due to their similar structure, it would be relatively straightforward to create AbFab services that act as wrappers for SADI services.
The criteria used to highlight mutations of interest in Mutadelic represents a single expert view on mutation prioritization within a particular context, in this case Mendelian blood disorders. Different criteria could be applied to the same context or criteria could be established for different contexts, e.g. a different disease or family of diseases. It is easy to imagine a scenario in which multiple criteria co-exist. In such a scenario, researchers could compare different methodologies for the same context or analyze patient data from multiple contexts. To do so, we hope, in the near future, to provide a UI framework to allow users to define their own criteria to identify significant variations. This would facilitate comparison of approaches to variant prioritization. The challenge would be creating a user-friendly interface with the OWL representation of the logical constraints. This would be achieved using the current set of defined services allowing for (i) modification of the faceted restrictions (e.g. changing significant SIFT value from 0.05 to 0.01) and (ii) different combinations of conjunction, disjunction and negation (AND, OR and NOT). Another optimization would be a framework, either a UI tool or an API for definition of new service classes. Again, there is the challenge of hiding the complexity of OWL representation from the user/developer.
5 Conclusion
This article describes a new variant annotation paradigm that is based on the idea of generating the minimum necessary annotations for reaching a diagnostically relevant conclusion. It is a departure from existing solutions, which seek to annotate all observed variants. It is also, to our knowledge, the first attempt to use semantic workflows and abductive reasoning for this purpose. We believe that our solution will contribute to a more scalable informatics infrastructure that is appropriate for next-generation sequencing-based clinical genetics.
Acknowledgements
We thank Drs Jon Morrow and Pei Hui (Department of Pathology) and Drs Patrick Gallagher and Vincent Schulz (Department of Pediatrics) for their valuable input and guidance in designing Mutadelic. We are especially grateful for their input into the crafting of genetic rules for identifying disease variants.
Funding
Dr. Krauthammer was partially supported by the Yale SPORE in skin cancer (NCI/5P50CA121974) and Dr. Holford was supported by the NLM biomedical informatics training grant at Yale (NLM/T15LM007056)
Conflict of Interest: none declared.
References
Author notes
Associate Editor: John Hancock


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}