





Abstract
Motivation: Molecular recognition of N-terminal targeting peptides is the most common mechanism controlling the import of nuclear-encoded proteins into mitochondria and chloroplasts. When experimental information is lacking, computational methods can annotate targeting peptides, and determine their cleavage sites for characterizing protein localization, function, and mature protein sequences. The problem of discriminating mitochondrial from chloroplastic propeptides is particularly relevant when annotating proteomes of photosynthetic Eukaryotes, endowed with both types of sequences.
Results: Here, we introduce TPpred3, a computational method that given any Eukaryotic protein sequence performs three different tasks: (i) the detection of targeting peptides; (ii) their classification as mitochondrial or chloroplastic and (iii) the precise localization of the cleavage sites in an organelle-specific framework. Our implementation is based on our TPpred previously introduced. Here, we integrate a new N-to-1 Extreme Learning Machine specifically designed for the classification task (ii). For the last task, we introduce an organelle-specific Support Vector Machine that exploits sequence motifs retrieved with an extensive motif-discovery analysis of a large set of mitochondrial and chloroplastic proteins. We show that TPpred3 outperforms the state-of-the-art methods in all the three tasks.
Availability and implementation: The method server and datasets are available at http://tppred3.biocomp.unibo.it.
Contact: [email protected]
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
The vast majority of proteins localized in mitochondria and chloroplasts are nuclear-encoded and synthesized by the cytoplasmic ribosomes. Then, they target the different organelles with the aid of specific and different molecular machineries, still partially characterized. One of most common ones requires the interaction between the import system and specific sequence signals, called targeting or transit peptides. Routinely these signals are located in the N-terminal region of the native protein sequence. Different proteolytic enzymes cleave targeting peptides upon protein translocation across organelle membranes. The recognition and annotation of targeting peptides starting from protein sequence assumes an important role in the characterization of the protein sub-cellular localization and function. For this reason, over the past years, several computational approaches have been described to address this task.
Some of the available tools are organelle-specific, such as ChloroP (Emanuelsson et al., 1999) and MitoProt (Claros and Vincens, 1996), specialized on chloroplasts and mitochondria, respectively. Both are able to detect the presence of targeting peptides and to predict the position of the cleavage site along the sequence. Other tools, such as TargetP (Emanuelsson et al., 2000), iPSORT (Bannai et al., 2002), Predotar (Small et al., 2004), PredSL (Petsalaki et al., 2006), can predict both the presence of a targeting peptide and the localization of the protein. Only PredSL and TargetP, however, perform a prediction of the cleavage site.
Recently, we introduced TPpred (Indio et al., 2013), a method based on Grammatical-Restrained Hidden Conditional Random Fields (GRHCRF) predicting the presence of targeting peptides and the localization of cleavage sites in both mitochondrial and chloroplastic proteins. TPpred, however, does not address the task of discriminating the subcellular localization on the basis of the targeting peptide. TPpred2.0 (Savojardo et al., 2014) was introduced in order to improve the prediction of cleavage sites in mitochondrial proteins. It exploits the information extracted from sequence motifs known in literature and particularly frequent in the regions containing the cleavage sites of mitochondrial targeting peptides (Mossmann et al., 2012; Savojardo et al., 2014).
In this article, we introduce TPpred3, a comprehensive and integrated pipeline to address the prediction of organelle-targeting peptides in Eukaryotic proteins, and particularly in green ones. Indeed in photosynthetic Eukaryotes, targeting peptides should be differently recognized by the import machinery in order to address both mitochondria and chloroplasts.
Our method is organized into three different modules, which collectively accomplish the following tasks: (i) the detection of the targeting signal in the N-terminal region of the protein; (ii) the classification of the identified signal as mitochondrial or chloroplastic; (iii) the precise identification of the targeting-peptide cleavage site in an organelle-specific manner.
For the first task (detection of the targeting signal), we rely on our previously developed method, called TPpred (Indio et al., 2013), which is based on a discriminative probabilistic sequence-labeling model (Fariselli et al., 2009).
The detected targeting peptide is then classified into two classes (mitochondrial or chloroplastic) by means of an N-to-1 Extreme Learning Machine (ELM) model, an architecture of artificial neural networks specifically suited to address classification problems involving sequences (Savojardo et al., 2011). Finally, a refining procedure is applied in order to better localize the cleavage site. This final task is addressed by means of two organelle-specific Support Vector Machines (SVMs) that exploit information derived from organelle-specific sequence motifs surrounding the cleavage sites. We extracted them with an extensive motif-discovery analysis carried out on two datasets of experimentally validated mitochondrial and chloroplastic targeting peptides.
We trained and tested the whole pipeline on a non-redundant dataset of proteins previously released to test TPpred and TPpred2 (Indio et al., 2013; Savojardo et al., 2014) and generated a different dataset for motif discovery.
Our results show that TPpred3 outperforms previous approaches in the task of predicting organelle-targeting peptides. In particular, by exploiting organelle-specific motifs, it is possible to reduce the error in the exact identification of the cleavage site in both mitochondrial and chloroplastic protein sequences.
2 Methods
2.1 Dataset for targeting-peptide prediction
For training and testing our method we adopted the non-redundant dataset described by Indio et al. (2013) and used to benchmark TPpred2.0 (Savojardo et al., 2014). We refer to this dataset as the Cleavage Prediction Set (CPS). It contains sequences extracted from UniprotKB/SwissProt (release November 2011), sharing <30% sequence identity.
Briefly, CPS includes 8307 proteins, with a positive subset (proteins annotated with an experimental targeting peptide) and a negative subset (proteins without targeting peptide). The positive set comprises 297 proteins, 202 of which are localized into mitochondria (M set) and 95 into chloroplasts (C set). The negative set includes 8010 proteins (N set) (Indio et al., 2013). When considering only proteins from photosynthetic Eukaryotic organisms, the CPS restricted (CPSr) set includes 718 proteins, of which 18 are in M, 95 in C and the remaining 605 in N.
2.2 Datasets for motif discovery in the cleavage site of mitochondrial and chloroplastic sequences
The datasets for motif discovery were generated downloading proteins with an experimentally detected targeting peptide (and a clear cleavage site), released after November 2011 up to April 2014, and complementing CPS. All the proteins with >40% sequence identity to CPS were filtered out. We ended up with 453 mitochondrial (Mito453) and 198 chloroplastic proteins (Chloro198), respectively.
Table 1 lists the major features of the two sets. In Mito453, 34 (7%) proteins in Mito453 proteins are from plants and 12 (∼3%) are from protists [Dictyostelium discoideum (six proteins), Trypanosoma brucei (three proteins), Euglena gracilis (one protein), Leishmania tarentolae (one protein), Tetrahymena thermophila (one protein)].
Composition of Mito453 and Chloro198
| Sequences with targeting peptides (number of) | Mito453 | Chloro198 |
|---|---|---|
| Total | 453 | 198 |
| Plant proteins | 46 | 198 |
| Animal proteins | 303 | – |
| Fungi proteins | 104 | – |
| Average length of targeting peptides* | 35 | 55 |
| Standard deviations* | 16 | 20 |
| Sequences with targeting peptides (number of) | Mito453 | Chloro198 |
|---|---|---|
| Total | 453 | 198 |
| Plant proteins | 46 | 198 |
| Animal proteins | 303 | – |
| Fungi proteins | 104 | – |
| Average length of targeting peptides* | 35 | 55 |
| Standard deviations* | 16 | 20 |
*residues.
Composition of Mito453 and Chloro198
| Sequences with targeting peptides (number of) | Mito453 | Chloro198 |
|---|---|---|
| Total | 453 | 198 |
| Plant proteins | 46 | 198 |
| Animal proteins | 303 | – |
| Fungi proteins | 104 | – |
| Average length of targeting peptides* | 35 | 55 |
| Standard deviations* | 16 | 20 |
| Sequences with targeting peptides (number of) | Mito453 | Chloro198 |
|---|---|---|
| Total | 453 | 198 |
| Plant proteins | 46 | 198 |
| Animal proteins | 303 | – |
| Fungi proteins | 104 | – |
| Average length of targeting peptides* | 35 | 55 |
| Standard deviations* | 16 | 20 |
*residues.
Proteins in the Chloro198 dataset are almost entirely from Viridiplantae organisms (96% as shown in Table 1), with the exception of seven proteins that are from non-Viridiplantae organisms such as Euglena gracilis (2), Amphidinium carterae (2), Rhodomonas species (1), Odontella sinensis (1), Corallina officinalis (1).
As expected, mitochondrial targeting peptides are in general shorter than the chloroplastic ones (Table 1, bottom row). In particular, average lengths of targeting peptides are of 35 and 55 residues, respectively in mitochondrial and chloroplastic sequences.
2.3 Overview of TPpred3
Our approach for the prediction and classification of targeting peptides in Eukaryotic proteins is outlined in Figure 1. It includes our previously released TPpred predictor (Indio et al., 2013) with two additional modules newly introduced in order to analyze a given input sequence in an organelle-specific manner.
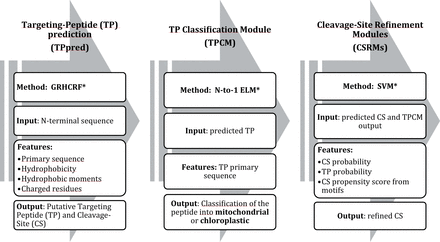
The general architecture of TPpred3. For details see Sections 2.3.1, 2.3.2 and 2.3.3, respectively. *GRHCRF, Grammatical Restrained Hidden Conditional Random Field; N-to-1 ELM, N-to-1 Extreme Learning Machines; SVM, Support Vector Machine
Starting from an input sequence, TPpred detects whether or not a targeting peptide is present in the N-terminal region of the sequence and predicts a preliminary cleavage site (Section 2.3.1). Then, the predicted targeting peptide is fed into the Targeting Peptide Classification Module (TPCM), which classifies it as mitochondrial or chloroplastic (Section 2.3.2). After this classification (TPCM output), a segment centered on the TPpred predicted cleavage site is then filtered with the organelle-specific Cleavage Site Refinement Module (CSRM) (Sections 2.3.3 and 2.3.4).
2.3.1 Prediction of organelle-targeting peptides (TPpred)
TPpred has been described before (Indio et al., 2013). Briefly, it is based on a probabilistic sequence labeling performed with a GRHCRF framework (Fariselli et al., 2009). TPpred performs the prediction using descriptors derived from protein primary sequence, which include the actual sequence, the Kyte–Doolittle hydrophobicity (Kyte and Doolittle, 1982), the hydrophobic moments and the composition in terms of charged residues. It is worth noticing that here the same model is adopted for both the mitochondrial and the chloroplastic proteins. TPpred was trained and tested on both positive and negative examples using the non-redundant CPS set described in Section 2.1. Further details are available in the reference paper (Indio et al., 2013).
2.3.2 The targeting peptide classification module
Targeting peptides detected with TPpred are then submitted to TPCM in order to be classified as mitochondrial or chloroplastic. TPCM includes a N-to-1 ELM model, a framework previously introduced to tackle classification problems (Savojardo et al., 2011). Here, predicted targeting peptides are classified either as mitochondrial or chloroplastic. The N-to-1 ELM classifier is a Neural Network with three neuron layers (the input, the hidden and the output layers).
The ELM approach gives better results than traditional gradient-based training algorithms (Huang et al., 2006). Furthermore, the ELM algorithm simplifies and speeds-up the network training by avoiding the tuning of several hyper-parameters (e.g. learning rates, number of epochs, momentum etc.) that are present in gradient-based approaches.
The two parameters of the N-to-1 method r and K were selected by performing a grid search procedure. Here, their final optimized values are 27 and 54, for r and K, respectively.
As showed in previous studies, the N-to-1 ELM framework is well-suited for structured classification involving variable-length input sequences, particularly in problems where it is not easy to extract relevant features from the input. N-to-1 ELM leverages the entire information contained in the input sequences to solve the classification task (Savojardo et al., 2011).
2.3.3 The cleavage site refinement modules
Depending on the classification TPCM output (Section 2.3.2), targeting peptides are directed to the corresponding organelle-specific CSRM (one for mitochondrial and one for chloroplastic targeting peptides, CSRMs in Fig. 1). Here, the cleavage site is refined taking advantage of the presence of organelle-specific sequence motifs in the neighborhood of the cleavage site. Motifs are found as described in Section 2.3.4 and are different for mitochondrial and chloroplastic targeting peptides. Both CSRMs include a first step that scores the presence of a given motif and a second step for refinement using SVMs. CSRMs analyze a region comprising 29 and 45 residues centered respectively on the mitochondrial and the chloroplastic TPpred predicted cleavage site. Each of the residues is scored for motif occurrence as detailed below.
2.3.3.1 Scoring a motif occurrence
2.3.3.2 SVM implementation
Two organelle-specific SVMs were trained and tested for the refinement of the cleavage site. This step was performed in a region of width 29 and 45 residues for mitochondrial and chloroplastic proteins, respectively. For each residue in this region the following descriptors were computed and used as input to SVMs:
the marginal posterior probability (computed by TPpred) of the cleavage site occurring at the given residue position;
the marginal posterior probability (computed by TPpred) that the given residue is part of a targeting peptide;
the cleavage site propensity score (Equation 8) of the given residue position computed from the organelle-specific motif set.
For the training phase, the above descriptors were computed considering only the experimentally detected cleavage sites and their closest residues regions. In testing, TPpred-predicted sites and their environments were adopted.
2.3.4 Discovery of cleavage site motifs
We carried-out an extensive motif-discovery analysis with the aim of finding characteristic sequence motifs in the regions surrounding the cleavage sites. We ran two separate analyses: one for mitochondrial and one for chloroplastic proteins, using Mito453 and Chloro198 as reference sets, respectively. We used MEME (Bailey and Elkan, 1994) and FIMO (Grant et al., 2011) of the MEME Suite (Bailey et al., 2009) as baseline motif discovery tools in protein sequences. Our procedure includes five different steps described in the following.
2.3.4.1 Five-fold cross-validation procedure
Both reference sets of protein sequences (either the Mito453 or the Chloro189) were divided into five cross-validation subsets. In defining these subsets, we took care of confining similar sequences into the same subset. The CD-HIT program (Fu et al., 2012) was used to calculate sequence clusters at 40% sequence identity. Then, proteins in the same cluster were assigned to the same cross-validation subset. For each cross-validation run, four of the five subsets were used to define the Motif Discovery Set (MDS), while the remaining one was used to build the Motif Validation Set (MVS). MDS was used to perform the actual motif-discovery analysis using MEME, while the MVS was used as hold-out set in order to validate the found motifs.
2.3.4.2 Building positive and negative sets
We extracted from proteins included in MDS, nine-residue long segments centered on the cleavage sites. The value 9 was set after maximizing the prediction accuracy in cross-validation. All together, these segments formed the organelle-specific positive Motif Discovering Set (MDS+). We refer to each of these segments as the Cleavage Site Environment (CSE) of a given protein sequence.
For building an organelle-specific negative MDS (MDS−), we applied the following procedure. For each sequence in the MDS set, we extracted all 9-mer centered on non-cleavage sites, excluding those that overlapped with any CSEs. All 9-mer were ranked according to their sequence identities with the CSEs of their sequence of origin. We formed the final MDS− by choosing the 1000 top-ranking sites. The same procedure was applied to generate MVSs (MVS+ and MVS−, positive and negative, respectively).
2.3.4.3 Discriminative motif discovery using MEME
Both the MDS+ and the MDS− were used to perform discriminative motif discovery using MEME. First, we estimated a Position-Specific Prior (PSP) model using the PSP-GEN program (Bailey et al., 2010) of the MEME suite. A PSP model scores the likelihood that a motif starts at a particular position in the sequences included in the MDS+ input set (Bailey et al., 2010). In this way, we identify motifs that are representative of cleavage-site regions and under-represented in the remaining part of the sequences.
The generated PSP model was then provided as input to MEME along with the MDS+ set. The MEME program was executed using different settings for the input parameters (width of motifs ranging from 4 to 9; minimum number of motif occurrences ranging from 25 to 60% of the MDS+ set). In order to identify only significant motifs, the e-value threshold of the MEME program was set to 0.05. Motifs found in all the MEME runs were finally collected into the organelle-specific Initial Motif Sets (IMSs).
2.3.4.4 Motif validation, clustering and filtering
Motifs in the IMSs were clustered and validated against the MVSs (Section 2.3.4.1). Motif clustering was performed using average-linkage hierarchical clustering. The distance measure derives from the alignment-free method described by Xu and Su (2010), originally devised to compare transcription factor binding sites. Briefly, for each motif in the IMS we compiled a 400-valued vector, called Di-peptide Frequency Vector (DFV), representing the di-peptide composition in the motif.
Hierarchical clustering by average-linkage was then computed from the motif distance matrix. Motif clusters were created by cutting the resulting distance tree using a cut-off threshold of 0.75. By this, motifs whose DFVs had a PCC higher than 0.25 ended-up into the same cluster.
For each motif in a cluster we searched for the occurrences in the MVS+ and MVS− sets, using the program FIMO (Grant et al., 2011) setting the P-value threshold to 0.001, and we selected as representative of the cluster the motif scoring with the lowest false-positive rate.
All the selected motifs were then collected into the organelle-specific Refined Motif Sets (RMSs).
2.4 Evaluation metrics
2.4.1 Evaluation of the classification procedure (TPCM)
With TPCM (in Fig. 1), for a given protein, its TPpred predicted targeting peptide is classified into one of the two mithocondrial and chloroplastic class. In the following, we describe the procedure adopted for scoring the classification.
the recall of the class i, defined as:
the precision of the class i, defined as:
the Matthews Correlation Coefficient (MCC) of class i, defined as:
2.4.2 Evaluation of CSRMs
the fraction of exactly identified cleavage sites of proteins in class i:
the mean absolute error in cleavage site determination of proteins in class i:
the fraction of predicted cleavage sites of proteins in class i whose distance from real site is below the standard deviation of the organelle-targeting peptide length distribution:
3 Results and discussion
3.1 Discriminating mitochondrial from chloroplastic targeting peptides
After the first and second modules of TPpred3 (TPpred+TCPM in Fig. 1) any protein is classified in one of the three classes: (i) without targeting peptide (N); (ii) endowed with a mitochondrial targeting peptide (M); (iii) endowed with a chloroplastic targeting peptide (C). We compared the performance of TPpred3 on the classification task with the results obtained with iPSORT (Bannai et al., 2002), TargetP (Emanuelsson et al., 2000), Predotar (Small et al., 2004) and PredSL (Petsalaki et al., 2006).
Table 2 lists the performance of the different methods on the classification task on the whole CPS set (Section 2.1). It is important to remark that we can guarantee a real cross validation procedure only for our predictor and not for all the others. Predictors are used without providing information on the source organism of the protein. All the methods in rows 2-5 have a higher recall: however TPpred3 outperforms the other state-of-the-art methods, being much less affected by false positives (higher P(M), P(C) and MCCs than other methods in Table 2).
Benchmarking different methods in the targeting-peptide classification task
| Method | R(M) | P(M) | R(C) | P(C) | MCC(M) | MCC(C) | MCC(N) |
|---|---|---|---|---|---|---|---|
| TPpred3 | 0.70 | 0.44 | 0.64 | 0.46 | 0.54 | 0.54 | 0.58 |
| iPSORTa | 0.88 | 0.12 | 0.61 | 0.07 | 0.27 | 0.18 | 0.28 |
| TargetPa | 0.91 | 0.15 | 0.87 | 0.11 | 0.33 | 0.29 | 0.32 |
| Predotara | 0.89 | 0.25 | 0.83 | 0.23 | 0.45 | 0.43 | 0.44 |
| PredSLa | 0.99 | 0.06 | 0.81 | 0.05 | 0.20 | 0.18 | 0.17 |
| Method | R(M) | P(M) | R(C) | P(C) | MCC(M) | MCC(C) | MCC(N) |
|---|---|---|---|---|---|---|---|
| TPpred3 | 0.70 | 0.44 | 0.64 | 0.46 | 0.54 | 0.54 | 0.58 |
| iPSORTa | 0.88 | 0.12 | 0.61 | 0.07 | 0.27 | 0.18 | 0.28 |
| TargetPa | 0.91 | 0.15 | 0.87 | 0.11 | 0.33 | 0.29 | 0.32 |
| Predotara | 0.89 | 0.25 | 0.83 | 0.23 | 0.45 | 0.43 | 0.44 |
| PredSLa | 0.99 | 0.06 | 0.81 | 0.05 | 0.20 | 0.18 | 0.17 |
Benchmark is done on the entire CPS (Section 2.1). In bold the highest score achieved for each index.
aOur testing set is included in the training set adopted by all the methods, respectively. Scoring indexes are defined in Section 2.4. Only TPpred3 is used in cross-validation.
Benchmarking different methods in the targeting-peptide classification task
| Method | R(M) | P(M) | R(C) | P(C) | MCC(M) | MCC(C) | MCC(N) |
|---|---|---|---|---|---|---|---|
| TPpred3 | 0.70 | 0.44 | 0.64 | 0.46 | 0.54 | 0.54 | 0.58 |
| iPSORTa | 0.88 | 0.12 | 0.61 | 0.07 | 0.27 | 0.18 | 0.28 |
| TargetPa | 0.91 | 0.15 | 0.87 | 0.11 | 0.33 | 0.29 | 0.32 |
| Predotara | 0.89 | 0.25 | 0.83 | 0.23 | 0.45 | 0.43 | 0.44 |
| PredSLa | 0.99 | 0.06 | 0.81 | 0.05 | 0.20 | 0.18 | 0.17 |
| Method | R(M) | P(M) | R(C) | P(C) | MCC(M) | MCC(C) | MCC(N) |
|---|---|---|---|---|---|---|---|
| TPpred3 | 0.70 | 0.44 | 0.64 | 0.46 | 0.54 | 0.54 | 0.58 |
| iPSORTa | 0.88 | 0.12 | 0.61 | 0.07 | 0.27 | 0.18 | 0.28 |
| TargetPa | 0.91 | 0.15 | 0.87 | 0.11 | 0.33 | 0.29 | 0.32 |
| Predotara | 0.89 | 0.25 | 0.83 | 0.23 | 0.45 | 0.43 | 0.44 |
| PredSLa | 0.99 | 0.06 | 0.81 | 0.05 | 0.20 | 0.18 | 0.17 |
Benchmark is done on the entire CPS (Section 2.1). In bold the highest score achieved for each index.
aOur testing set is included in the training set adopted by all the methods, respectively. Scoring indexes are defined in Section 2.4. Only TPpred3 is used in cross-validation.
In Table 3, we benchmark all the methods towards proteins of photosynthetic Eukaryotes (CPS reduced), where targeting peptides of both mitochondrial and chloroplastic types coexist in the same cytoplasm. Again, considering that only TPpred3 is tested adopting a rigorous cross validation procedure, it appears that it outperforms the other methods, being much less affected by false positives.
Benchmarking different methods in the targeting-peptide classification task in photosynthetic Eukaryotes
| Method | R(M) | P(M) | R(C) | P(C) | MCC(M) | MCC(C) | MCC(N) |
|---|---|---|---|---|---|---|---|
| TPpred3 | 0.67 | 0.39 | 0.64 | 0.91 | 0.49 | 0.74 | 0.74 |
| iPSORTa | 0.89 | 0.14 | 0.61 | 0.54 | 0.31 | 0.50 | 0.53 |
| TargetPa | 0.89 | 0.19 | 0.87 | 0.59 | 0.31 | 0.50 | 0.61 |
| Predotara | 0.94 | 0.23 | 0.83 | 0.72 | 0.44 | 0.73 | 0.65 |
| PredSLa | 0.94 | 0.11 | 0.81 | 0.32 | 0.29 | 0.39 | 0.36 |
| Method | R(M) | P(M) | R(C) | P(C) | MCC(M) | MCC(C) | MCC(N) |
|---|---|---|---|---|---|---|---|
| TPpred3 | 0.67 | 0.39 | 0.64 | 0.91 | 0.49 | 0.74 | 0.74 |
| iPSORTa | 0.89 | 0.14 | 0.61 | 0.54 | 0.31 | 0.50 | 0.53 |
| TargetPa | 0.89 | 0.19 | 0.87 | 0.59 | 0.31 | 0.50 | 0.61 |
| Predotara | 0.94 | 0.23 | 0.83 | 0.72 | 0.44 | 0.73 | 0.65 |
| PredSLa | 0.94 | 0.11 | 0.81 | 0.32 | 0.29 | 0.39 | 0.36 |
Benchmark is done on the reduced CPS restricted to the green Eukaryotic proteins (Section 2.1). In bold the highest score achieved for each index.
aOur testing set is included in the training set adopted by all the other methods, respectively. Scoring indexes are defined in Section 2.4.
Benchmarking different methods in the targeting-peptide classification task in photosynthetic Eukaryotes
| Method | R(M) | P(M) | R(C) | P(C) | MCC(M) | MCC(C) | MCC(N) |
|---|---|---|---|---|---|---|---|
| TPpred3 | 0.67 | 0.39 | 0.64 | 0.91 | 0.49 | 0.74 | 0.74 |
| iPSORTa | 0.89 | 0.14 | 0.61 | 0.54 | 0.31 | 0.50 | 0.53 |
| TargetPa | 0.89 | 0.19 | 0.87 | 0.59 | 0.31 | 0.50 | 0.61 |
| Predotara | 0.94 | 0.23 | 0.83 | 0.72 | 0.44 | 0.73 | 0.65 |
| PredSLa | 0.94 | 0.11 | 0.81 | 0.32 | 0.29 | 0.39 | 0.36 |
| Method | R(M) | P(M) | R(C) | P(C) | MCC(M) | MCC(C) | MCC(N) |
|---|---|---|---|---|---|---|---|
| TPpred3 | 0.67 | 0.39 | 0.64 | 0.91 | 0.49 | 0.74 | 0.74 |
| iPSORTa | 0.89 | 0.14 | 0.61 | 0.54 | 0.31 | 0.50 | 0.53 |
| TargetPa | 0.89 | 0.19 | 0.87 | 0.59 | 0.31 | 0.50 | 0.61 |
| Predotara | 0.94 | 0.23 | 0.83 | 0.72 | 0.44 | 0.73 | 0.65 |
| PredSLa | 0.94 | 0.11 | 0.81 | 0.32 | 0.29 | 0.39 | 0.36 |
Benchmark is done on the reduced CPS restricted to the green Eukaryotic proteins (Section 2.1). In bold the highest score achieved for each index.
aOur testing set is included in the training set adopted by all the other methods, respectively. Scoring indexes are defined in Section 2.4.
3.2 Motif discovery results
The motif discovery procedure retrieved two and three motifs around the cleavage site for mitochondrial and chloroplastic proteins, respectively. These motifs describe sequence patterns that are frequently present in the regions surrounding the cleavage sites. Motifs found by the MEME program are in the form of Position-Specific Scoring Matrices (PSSMs) whose regular expressions, obtained by extracting the most frequent residues in each position, are listed in Table 4. Sequence logos generated with the Seq2Logo program (Thomsen and Nielsen, 2012), show the relative frequency of the different residues in the motifs and are available in Supplementary Materials (Supplementary Figs S1, Supplementary Data, Supplementary Data, Supplementary Data and Supplementary Data).
Mitochondrial and chloroplastic motifs around the cleavage site of targeting peptides
| Motif label | Regular expression |
|---|---|
| M1 | RC|[YF][AS] |
| M2 | SVRx|Y[SA][TS]G |
| C1 | [VI][RA]|[AC]AAE |
| C2 | S[VI][RSV]|[CA]A |
| C3 | [AV]N|A[AM]AG[ED] |
| Motif label | Regular expression |
|---|---|
| M1 | RC|[YF][AS] |
| M2 | SVRx|Y[SA][TS]G |
| C1 | [VI][RA]|[AC]AAE |
| C2 | S[VI][RSV]|[CA]A |
| C3 | [AV]N|A[AM]AG[ED] |
M, Mitochondrial and C, Chloroplastic motifs. We indicate in brackets ‘[]’ alternative choices and with the symbol ‘|’ the most frequent position for the cleavage site when the PSSM are searched on the Mito453 and Chloro198 sets (Section 2.2).
Mitochondrial and chloroplastic motifs around the cleavage site of targeting peptides
| Motif label | Regular expression |
|---|---|
| M1 | RC|[YF][AS] |
| M2 | SVRx|Y[SA][TS]G |
| C1 | [VI][RA]|[AC]AAE |
| C2 | S[VI][RSV]|[CA]A |
| C3 | [AV]N|A[AM]AG[ED] |
| Motif label | Regular expression |
|---|---|
| M1 | RC|[YF][AS] |
| M2 | SVRx|Y[SA][TS]G |
| C1 | [VI][RA]|[AC]AAE |
| C2 | S[VI][RSV]|[CA]A |
| C3 | [AV]N|A[AM]AG[ED] |
M, Mitochondrial and C, Chloroplastic motifs. We indicate in brackets ‘[]’ alternative choices and with the symbol ‘|’ the most frequent position for the cleavage site when the PSSM are searched on the Mito453 and Chloro198 sets (Section 2.2).
On the overall, the motifs are present in the neighborhood of 415 (92%) and 171 (86%) mitochondrial and chloroplastic cleavage sites, respectively. Moreover, the cleavage sites indicated in the regular expressions exactly correspond to the experimental cleavage sites in 214 (47%) and 118 (60%) mitochondrial and chloroplastic targeting peptides, respectively. In other cases, the difference between the real site and that marked in the regular expressions is very small (Supplementary Figs S6 and Supplementary Data).
The two mitochondrial motifs are both characterized by the presence of Arg residues in the region immediately surrounding the site. Previous works reported distinctive sequence motifs of cleavage sites in mitochondrial proteins (Mossmann et al., 2012 for a review). They identified Arg-rich short motifs which are all included into the longer M1 and M2 motifs found in this study.
Chloroplastic cleavage sites are characterized by high concentrations of Ala, Ile, Cys and Val residues. These findings corroborate and add to the conclusions of an early study carried on a limited set of 32 chloroplastic cleavage sites (Gavel and von Heijne, 1990).
3.3 Performance of the cleavage-site prediction
Performances of the different methods on cleavage-site prediction are listed in Table 5. We compare TPpred3 to our TPpred (Indio et al., 2013 and TPpred2 (Savojardo et al., 2014), a predictor specialized for only mitochondrial targeting peptides that also includes mitochondrial sequence specific motifs. We also include in the benchmark PredSL (Petsalaki et al., 2006), TargetP (Emanuelsson et al., 2000), MitoProt (Claros and Vincens, 1996) and the recently released MitoFates (Fukasawa et al., 2015).
Benchmark results of the different methods on cleavage-site prediction
| Method | Qc(M) | ME(M) | Eσ(M) | Qc(C) | ME(C) | Eσ(C) |
|---|---|---|---|---|---|---|
| TPpred3 | 0.33 | 6 | 0.90 | 0.26 | 12 | 0.75 |
| TPpred | 0.17 | 7 | 0.89 | 0.15 | 15 | 0.74 |
| TPpred2.0 | 0.32 | 6 | 0.89 | – | – | – |
| TargetPa | 0.39 | 13 | 0.67 | 0.09 | 15 | 0.70 |
| PredSLa | 0.17 | 12 | 0.75 | 0.05 | 17 | 0.73 |
| MitoProta | 0.16 | 13 | 0.75 | – | – | – |
| MitoFatesa | 0.25 | 11 | 0.77 | – | – | – |
| Method | Qc(M) | ME(M) | Eσ(M) | Qc(C) | ME(C) | Eσ(C) |
|---|---|---|---|---|---|---|
| TPpred3 | 0.33 | 6 | 0.90 | 0.26 | 12 | 0.75 |
| TPpred | 0.17 | 7 | 0.89 | 0.15 | 15 | 0.74 |
| TPpred2.0 | 0.32 | 6 | 0.89 | – | – | – |
| TargetPa | 0.39 | 13 | 0.67 | 0.09 | 15 | 0.70 |
| PredSLa | 0.17 | 12 | 0.75 | 0.05 | 17 | 0.73 |
| MitoProta | 0.16 | 13 | 0.75 | – | – | – |
| MitoFatesa | 0.25 | 11 | 0.77 | – | – | – |
aThe benchmark dataset partially overlaps with the training set. Scoring indexes are defined in Section 2.4.2. Briefly: Qc, the fraction of exactly identified cleavage sites of proteins in class M or C; ME, the mean absolute error in cleavage site determination of proteins in class M or C; Eσ, the fraction of predicted cleavage sites of proteins in class i whose distance from real site is below the standard deviation of the organelle-targeting peptide length distribution.
Benchmark results of the different methods on cleavage-site prediction
| Method | Qc(M) | ME(M) | Eσ(M) | Qc(C) | ME(C) | Eσ(C) |
|---|---|---|---|---|---|---|
| TPpred3 | 0.33 | 6 | 0.90 | 0.26 | 12 | 0.75 |
| TPpred | 0.17 | 7 | 0.89 | 0.15 | 15 | 0.74 |
| TPpred2.0 | 0.32 | 6 | 0.89 | – | – | – |
| TargetPa | 0.39 | 13 | 0.67 | 0.09 | 15 | 0.70 |
| PredSLa | 0.17 | 12 | 0.75 | 0.05 | 17 | 0.73 |
| MitoProta | 0.16 | 13 | 0.75 | – | – | – |
| MitoFatesa | 0.25 | 11 | 0.77 | – | – | – |
| Method | Qc(M) | ME(M) | Eσ(M) | Qc(C) | ME(C) | Eσ(C) |
|---|---|---|---|---|---|---|
| TPpred3 | 0.33 | 6 | 0.90 | 0.26 | 12 | 0.75 |
| TPpred | 0.17 | 7 | 0.89 | 0.15 | 15 | 0.74 |
| TPpred2.0 | 0.32 | 6 | 0.89 | – | – | – |
| TargetPa | 0.39 | 13 | 0.67 | 0.09 | 15 | 0.70 |
| PredSLa | 0.17 | 12 | 0.75 | 0.05 | 17 | 0.73 |
| MitoProta | 0.16 | 13 | 0.75 | – | – | – |
| MitoFatesa | 0.25 | 11 | 0.77 | – | – | – |
aThe benchmark dataset partially overlaps with the training set. Scoring indexes are defined in Section 2.4.2. Briefly: Qc, the fraction of exactly identified cleavage sites of proteins in class M or C; ME, the mean absolute error in cleavage site determination of proteins in class M or C; Eσ, the fraction of predicted cleavage sites of proteins in class i whose distance from real site is below the standard deviation of the organelle-targeting peptide length distribution.
TPpred3 (Table 5) exactly identifies 66 mitochondrial [Qc(M) = 0.33] and 25 chloroplastic cleavage sites [Qc(C) = 0.26]. Only TargetP, and only for mitochondrial proteins, is performing better [Qc(M) = 0.39], possibly due to the fact that we are controlling homology only in our predictor. TPpred3 is outperforming all methods when considering the average prediction error (ME) and the fraction of predicted cleavage sites whose distance from real site is below the standard deviation of the organelle-targeting peptide length distribution (Eσ).
For mitochondrial targeting peptides, TPpred3 slightly outperforms even TPpred2.0, which also adopted known sequence motifs in order to improve the localization of mitochondrial cleavage site. In the case of chloroplastic targeting peptides, the motifs we extract, greatly improve the prediction.
4 Conclusions
In this article, we describe the performance of TPpred3, a new integrated method that predicts and discriminates targeting peptides of mitochondrial and chloroplastic eukaryotic proteins. The method is characterized by a low rate of false positives and high performance scores when compared with the state-of-the-art methods addressing the same task. Furthermore, TPpred3 is also improving the cleavage site prediction by introducing and exploiting different cleavage site motifs of the different import systems of mitochondria and chloroplasts.
TPpred3 is organized as a pipeline where the different software modules are devised to address targeting-peptide detection, classification and cleavage-site refinement. In principle, a single GRHCRF model can tackle all the three tasks: targeting peptide prediction and peptide classification (organelle-specific annotation), and detection of motif-aware cleavage-sites. Despite the inherent elegance and compactness of the solution, preliminary tests (data not shown) lead to poor performance compared with the ones of the pipeline presented in this article.
For the problem at hand, we still have a limited amount of experimental data and handling the problem with a pipeline allows us to exploit effectively relevant information in a separate way at each prediction step.
TPpred3 is a pipeline that integrates different machine-learning predictors. For this reason, errors propagate throughout the different prediction steps. An empirical evaluation of error propagation (Supplementary Table S1) indicates that much of the error is at the TPCM level and it is due to erroneous predictions performed by TPpred (both false negative and incorrect cleavage site predictions).
In summary, TPpred3 is particularly suited for eukaryotic proteins annotation in sequence experiments, producing large amount of data. Our method starting from sequence classifies proteins in relation to the presence or absence of targeting peptides in sequences that are targeted to two distinct types of organelles: mitochondria and chloroplasts. We propose its application particularly in annotation processes of green Eukaryotic (Archaeplastida) genomes, where proteins with targeting peptides after synthesis are delivered to either organelle.
Funding
Funding PRIN 201011 project 20108XYHJS (to P.L.M.) (Italian MIUR); Funding COST BMBS Action TD1101 and Action BM1405 (European Union RTD Framework Program, to R.C.); Funding PON projects PON01_02249 and PAN Lab PONa3_00166 (Italian Miur to R.C. and P.L.M.); Funding FARB-UNIBO 2012 (to R.C.).
Conflict of Interest: none declared.
References
Author notes
Associate Editor: John Hancock


{kind=link}