





Abstract
Summary: The DOMIRE web server implements a novel, automatic, protein structural domain assignment procedure based on 3D substructures of the query protein which are also found within structures of a non-redundant protein database. These common 3D substructures are transformed into a co-occurrence matrix that offers a global view of the protein domain organization. Three different algorithms are employed to define structural domain boundaries from this co-occurrence matrix. For each query, a list of structural neighbors and their alignments are provided. DOMIRE, by displaying the protein structural domain organization, can be a useful tool for defining protein common cores and for unravelling the evolutionary relationship between different proteins.
Availability: http://genome.jouy.inra.fr/domire
Contact: [email protected]
1 INTRODUCTION
The modular nature of proteins is well established. First described at the level of their structure, based on their compactness character and folding properties (Wetlaufer, 1973), their evolutionary role has been substantiated by comparing amino acid sequences, following the pioneering works of Doolittle (1985). Databases such as ProDom (http://prodom.prabi.fr/prodom/current/html/home.php) or Pfam (http://pfam.sanger.ac.uk/) provide domain definitions at the level of amino acid sequences. Databases such as CATH (http://www.cathdb.info/) and SCOP (http://scop.mrc-lmb.cam.ac.uk/scop/index.html) define domain at the level of the 3D structures. Domains are exchangeable segments of amino acids that retain their 3D structure and molecular function. Domain identification is thus an important tool for a number of studies about protein 3D structures or evolution.
Recently, we applied the protein structure comparison program VAST (Gibrat et al., 1996; Madej et al., 1995) and we found that the recurrence of small common 3D substructures (typically four secondary structures) between the query protein and proteins of a non-redundant dataset were sufficient to define the boundaries of the domains (Tai et al., 2011). The methodology described in the latter paper is now available as an online server named DOMIRE for DOMain Identification from REcurrence. The server also provides, for each query, a list of structural neighbors with their alignments.
2 METHODS
Domain definitions: VAST was used with very liberal cut-offs (Pcli ≥−10 and rmsd ≤5 Å) to collect a maximum number of common 3D substructures between the query protein and target proteins of the non-redundant dataset. Unaligned regions of <40 residues between two aligned secondary structures were also included, giving rise to padded Locally Similar Structural Pieces (pLSSPs). These pLSSPs were mapped onto the query protein, resulting in an alignment matrix which was then transformed into a co-occurrence N matrix (Fig. 1a) from which the domains were parsed by three different methods: PCM, SMF and SVD (see Tai et al., 2011 for details).
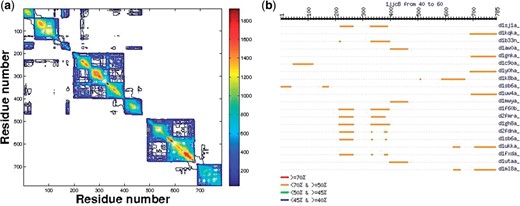
(a) N matrix of the phenylalanyl-tRNA syhthetase (1jjc) chain B (contour map) where Nij is the number of pLSSP in which residues i and j of the query protein are found together. This chain consists of six domains. For instance, SMF algorithm finds the following domains: D1: (1–41, 151–196), D2: (42–150), D3: (197–398), D4: (399–484), D5: (485–676), D6: (677–785). Note that D1 is a segmented domain as defined in CATH and SCOP. This matrix made of several ten of thousands pLSSPs, gives a global view of the recurrence and of the domain organization. (b) An example of aligned structural neighbors for 1jjc chain B, sorted by decreasing numbers of percent of aligned residues, here a snapshot of 20 out of a few hundreds with their corresponding PDB names.
Structural neighbors: from the list of pLSSPs used to build the N matrix, some targets are highlighted when they fulfil the following two criteria: (i) the length of the target pLSSP amounts to ≥80% of the target length and (ii) >40% of the target length are aligned by VAST to the query in the corresponding common 3D substructure. In other words, this region in the query protein can be extensively aligned with most of the target 3D structure in the PDB. These targets define the structural neighbors. Besides the list of these targets, a graphic representation of their alignments along the query is provided (Fig. 1b).
DOMIRE input/output: DOMIRE takes as input a single protein chain with its PDB accession code. Alternately, user can upload a file of coordinates with a PDB format. When the job is completed, the user receives an email to reach a web page displaying three interactive 3D representations of the query with colored domains using the Jmol applet (one for each domain assignment method). It shows also the N matrix as a heat map and a contour map (Fig. 1a) as well as the alignments of the structural neighbors on the query protein (Fig. 1b). These results are available online for 1 month and can be downloaded as a tarball.
3 DISCUSSION
With a benchmark of 128 chains, using SCOP or/and CATH classifications as gold standards, SMF and SVD algorithms provided results similar to those of the PUU server but less close to those of Domain Parser or PDP (Tai et al., 2011). PCM performs better than SMF and SVD for chains having three or more domains. For that reason, we provide the results of the three algorithms on the web site with their, possibly, different boundaries.
The criterion for selecting the structural neighbors of 40% or more aligned residues is based on the fact that VAST alignments involve essentially the secondary structures and that on average secondary structures amount to ∼50% of the residues of a protein. The criterion of at least 80% of the target aligned with the query tends to select regions of the query that can be aligned with whole structures in the PDB. Some of them are homologues of the query or of its domains (to be published).
4 CONCLUSION
In addition to offering an automatic partitioning of protein structures into domains with performances comparable with the best existing programs, DOMIRE provides the identification and alignments of structural neighbors. This can be useful for identifying remote homologues. It provides a tool to analyze the common 3D substructures in polypeptide chains shedding light on their evolution.
ACKNOWLEDGEMENT
We are grateful to the INRA MIGALE platform for providing computational resources.
Funding: Intramural Research Program of the Center for Cancer Research, National Cancer Institute and of the Division of Computational Bioscience, Center for Information Technology, NIH in USA and financially supported by the Institut National de la Recherche Agronomique in France (in part).
Conflict of Interest: none declared.
REFERENCES
Author notes
Associate Editor: Anna Tramontano


{kind=link}