





Abstract
Summary: SiGN-SSM is an open-source gene network estimation software able to run in parallel on PCs and massively parallel supercomputers. The software estimates a state space model (SSM), that is a statistical dynamic model suitable for analyzing short time and/or replicated time series gene expression profiles. SiGN-SSM implements a novel parameter constraint effective to stabilize the estimated models. Also, by using a supercomputer, it is able to determine the gene network structure by a statistical permutation test in a practical time. SiGN-SSM is applicable not only to analyzing temporal regulatory dependencies between genes, but also to extracting the differentially regulated genes from time series expression profiles.
Availability: SiGN-SSM is distributed under GNU Affero General Public Licence (GNU AGPL) version 3 and can be downloaded at http://sign.hgc.jp/signssm/. The pre-compiled binaries for some architectures are available in addition to the source code. The pre-installed binaries are also available on the Human Genome Center supercomputer system. The online manual and the supplementary information of SiGN-SSM is available on our web site.
Contact: [email protected]
1 INTRODUCTION
Analyzing the dynamical regulatory mechanisms of gene expressions in a cellular system is a challenging problem in systems biology. To this end, many computational methods have been proposed to estimate dynamical systems of regulatory dependencies between gene expressions from temporal gene expression profiles. The major difficulty of these studies comes from insufficient data time points as opposed to the number of variables (genes) in a computational model. A state space model (SSM) (Hirose et al., 2008; Kitagawa and Gersch, 1996; West and Harrison, 1997) is a statistical model that is applicable to small time-point temporal datasets because it can reduce the number of parameters to be estimated. The SSM decomposes the temporal gene expressions into a dynamical system of modules called the system model (or state space) and a mapping from the modules to the particular genes called the observation model. There are a number of gene network studies using the SSM (Beal et al., 2005; Hirose et al., 2008; Rangel et al., 2004).
SiGN-SSM is a re-implemented, new version of the previously released one called TRANS-MNET (Hirose et al., 2008). In addition to TRANS-MNET, SiGN-SSM has the following improvements: (i) it implements a novel constraint on the model parameters effective to stabilize the estimated models for the short time series data with irregular time intervals; (ii) runs in parallel as a multithreaded program exploiting multicore CPUs, as a bulk (array) job through a job dispatching system such as Sun (Oracle) Grid Engine (SGE) on PC cluster systems, and a multi-process MPI (Message Passing Interface) application on massively parallel supercomputers; (iii) is an open-source software so that everyone can freely access the source code and improve, modify and distribute it, and (iv) implements a statistical permutation test to determine a gene network structure as proposed in Hirose et al. (2008) and differentially regulated gene extraction presented in Yamaguchi et al. (2008) on the Human Genome Center (HGC) supercomputer system.
2 STATE SPACE MODEL
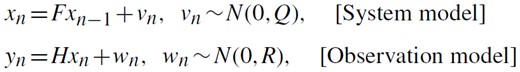
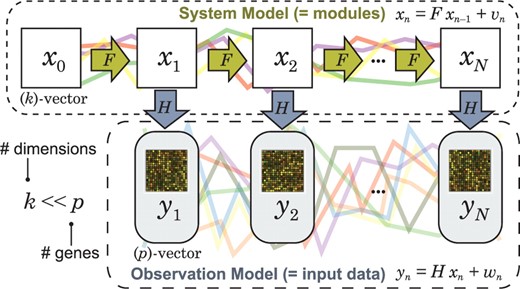
Conceptual view of the state space model.
3 IMPLEMENTATION AND PARALLELIZATION
SiGN-SSM is written in C, using the BLAS/LAPACK library. Since the EM algorithm finds only locally optimal parameters, it is required to run the algorithm many times to obtain better estimate for a single k. To speed up the parameter estimation and the optimal k determination, SiGN-SSM supports multiprocess parallelization using MPI, multithread parallelization using OpenMP and parallelization by bulk jobs on PC clusters. By using MPI, we confirmed that SiGN-SSM can optimize multiple k in parallel very efficiently with up to 256 CPU cores. See Supplementary information on our web site for detailed results. The permutation test determines the gene network structure from the estimated model parameters. However, it requires much more computational time than the parameter estimation. To solve this problem, we parallelized it on the HGC supercomputer using SGE.
4 NEW CONSTRAINT ON PARAMETERS
When the algorithm estimates the model parameters from short time series data measured for irregular time intervals, they often oscillate undesirably (Fig. 2). To suppress such spurious patterns, we propose a novel constraint on the system transition matrix F along with the smoothness prior approach (Kitagawa and Gersch, 1996). With the constraint, we assume that the value of the state vector at time n is similar to that at time n − 1. The constrained version of F, denoted by  , has its diagonal elements
, has its diagonal elements  for i = 1,…, k where 0 ≤ g ≤ 1 is a constant to control the smoothness, which user can choose (we set 0.8 as the default value). The off-diagonal components of
for i = 1,…, k where 0 ≤ g ≤ 1 is a constant to control the smoothness, which user can choose (we set 0.8 as the default value). The off-diagonal components of  are estimated by the EM algorithm with the constraint of the diagonal components, in which we utilize the general framework of Wu et al. (1996) to constrain parameters of an SSM in the EM algorithm. The value of g can be determined by such as comparison of BIC values, cross-validation, etc.
are estimated by the EM algorithm with the constraint of the diagonal components, in which we utilize the general framework of Wu et al. (1996) to constrain parameters of an SSM in the EM algorithm. The value of g can be determined by such as comparison of BIC values, cross-validation, etc.
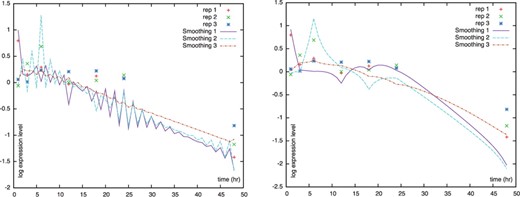
Comparison without (left) and with (right) the proposed constraint on F. The plotted lines are the estimated smoothing observation variables for the very short, triplicate sample data.
5 CONCLUSION
SiGN-SSM is a highly-scalable, open-source implementation of an SSM estimation algorithm. The newly proposed constraint on the parameters can significantly stabilize the estimation of the parameters for the very short time point temporal data with irregular time intervals. Users can analyze their time series expression datasets using SiGN-SSM, and the estimated model can be applied to other data to extract differentially expressed genes.
ACKNOWLEDGEMENT
Computational resources required for the development of SiGN-SSM was provided by the HGC Supercomputer System, Human Genome Center, Institute of Medical Science, The University of Tokyo and RIKEN Supercomputer system RICC.
Funding: ISLiM (Next-generation integrated simulation of living matter) project in RIKEN Computational Science Research Program.
Conflict of Interest: none declared.
REFERENCES
Author notes
Associate Editor: Martin Bishop


{kind=link}
{kind=link}