





Abstract
Motivation: The rapidly increasing amounts of data available from new high-throughput methods have made data processing without automated pipelines infeasible. As was pointed out in several publications, integration of data and analytic resources into workflow systems provides a solution to this problem, simplifying the task of data analysis. Various applications for defining and running workflows in the field of bioinformatics have been proposed and published, e.g. Galaxy, Mobyle, Taverna, Pegasus or Kepler. One of the main aims of such workflow systems is to enable scientists to focus on analysing their datasets instead of taking care for data management, job management or monitoring the execution of computational tasks. The currently available workflow systems achieve this goal, but fundamentally differ in their way of executing workflows.
Results: We have developed the Conveyor software library, a multitiered generic workflow engine for composition, execution and monitoring of complex workflows. It features an open, extensible system architecture and concurrent program execution to exploit resources available on modern multicore CPU hardware. It offers the ability to build complex workflows with branches, loops and other control structures. Two example use cases illustrate the application of the versatile Conveyor engine to common bioinformatics problems.
Availability: The Conveyor application including client and server are available at http://conveyor.cebitec.uni-bielefeld.de.
Contact: [email protected]; [email protected].
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Workflows have become an important aspect in the field of bioinformatics during the last years (e.g. Romano, 2007 and Smedley et al., 2008). Applications like Galaxy (Goecks et al., 2010), Taverna (Hull et al., 2006), Pegasus (Deelman et al., 2005) and Kepler (Altintas et al., 2004) offer an easy way to access local and remote resources and perform automatic analyses to test hypotheses or process data. In many cases, they have become a reasonable alternative to write simple software tools like Perl scripts, especially for users without an in-depth computer science background. Libraries like Ruffus (Goodstadt, 2010) add workflow functionality to programming languages, providing methods to define and build workflow within own applications.
A workflow is built from several linked steps that consume inputs, process and convert data and produce results. The most simple workflows are linear chains of steps to convert input data to the required output. More complex setups may also include loops, branches, parallel and conditional processing, reading from various sources and writing different outputs in various formats.
For creating reusable workflow components, a processing step may be composed of nested processing steps, allowing the user to build complex pipelines for higher level analysis.
Many workflow engines act as a wrapper using existing command line utilities or enact and orchestrate existing web services as basic modules for their processing steps. As a result, adding new processing steps by wrapping existing applications often does only require little or no programming effort.
Of course, this comes at a price. Passing data between processing steps depends on a common data format, especially in the case of distributed processing nodes. Most analysis tools available as command line applications or web services are consuming simple text formats, e.g. the FASTA format for DNA and amino acid sequences. These formats are in turn used by the workflow engines to exchange data between processing nodes. Integrating other processing nodes or input sources requires explicit data conversion prior to processing. This often leads to the loss of information; e.g. gene features annotated in EMBL or GenBank entries cannot retain all qualifiers after converting them to FASTA format. An analysis done by Wassink et al. (2009) shows that most tasks used in publicly available Taverna workflows are dedicated to data conversion. To some extent, this problem is solved by meta information provided with types that allow the definition of type hierarchies and interfaces. The BioMoby (Wilkinson et al., 2008) data type management is an example for an ontology-based approach to data type handling; nonetheless, it requires extra efforts by the developer and/or maintainer. Other attempts to define common data types like BioXSD (Kalaš et al., 2010) or Seibel et al. (2006) were made, but none of them has been successfully adopted by the community yet. The situation is even worse if legacy data from applications are to be integrated into a workflow. Accessing data, e.g. stored in a relational database or available by a local application only, requires special processing steps; passing the data between distributed processing nodes may not be possible at all.
Another problem arises from the nature of web services used in processing steps. Although they offer an elegant and easy way to provide and consume useful services, users have to be aware of the pitfalls of web services if they rely on them for an analytical workflow. A service may become unavailable without prior notice of the provider. For plain web services, only the syntax of input and output data types is defined, for example by using a WSDL file. They completely lack information about semantics and whether a data type is compatible to another type used in a different service. Passing large amounts of data to or from web services adds a noticeable processing overhead. Last but not least, external web services should not be used with confidential data, since the exact service provider and the means of transferring data (unsecure/secured by https) are unknown in many workflow systems. In a scenario working with confidential data, all services used in a workflow system should be deployed in the internal network only.
Workflow systems also attempt to offer an alternative way to run more complex analyses and thus replace small software tools, e.g. Perl scripts. Controlling the flow of data in workflows is essential for these applications. This includes branching the flow and using alternative processing based on intermediate results, and enabling/disabling complete processing branches for reusability of workflows.
With high-throughput methods producing more and more data, using web service or grid-based solutions introduces additional bottlenecks, and service providers will likely get into problems providing the necessary resources. A possible solution could be the integration of local compute infrastructures into the workflow, accessing external resources for special tasks only.
A thorough analysis of the mentioned systems shows that all offer some of these requested features. Taverna provides a considerable amount of processing nodes based on various web service registries, but support for a local execution or control flow is rather limited. Mobyle (Neron et al., 2009) offers a web-based workflow system, but lacks the necessary flow control elements to create complex workflows. Pegasus is designed for large grids and includes the necessary interfaces for handling distributed data and various batch systems, but operates on file-based data only. Galaxy is also able to utilize compute clusters and grids, and also uses plain formats only. Kepler finally implements almost all of the requested features, but its type system only differentiates between primitive types and opaque objects for data tokens, without including features like type hierarchies or interfaces.
In contrast to the other workflow tools considered so far, Ruffus is a library providing a framework for defining workflow steps and executing pipelines within Python applications. Although the indiviual steps are implemented as Python functions, it does not utilize Python data types, but relies on files.
2 APPROACH AND DESIGN
To overcome these limitations, we have developed Conveyor as a novel software library offering its functionality to other applications. A client-server setup based on the library is used to separate the design of a workflow from its execution. Additionally, a command line tool allows to execute workflows in a batch manner. As a software library, Conveyor may also be included into other applications, providing an easy to update and maintain data processing layer.
Workflows in Conveyor are represented by directed graphs, composed of nodes for processing and input/output steps, and edges moving data between nodes. This design allows simple pipelines built from concatenated processing steps, and also complex graphs with branches, loops and parallel flow of data. Data are passed one by one between nodes, creating a stream of input data. This model enables the parallel processing of nodes, using multiple CPU cores if available. For a first impression, the reader may look ahead at the genome annotation workflow presented in Section 4 (Fig. 6).
Node and data types in Conveyor are constructed with a strongly typed, interface driven, object-oriented design. It supports an easy wrapping of existing classes for functionality and thus the integration of existing applications into Conveyor. The interface-driven design makes processing steps usable with any appropriate data type. Relations between data types can be easily expressed by object-oriented concepts like inheritance and do not require extra efforts.
2.1 Data types
The definition of a data type in Conveyor only requires marking a class with a special empty interface. No further restrictions are applied to data types. Relations between types are expressed in an object-oriented way by inheritance or by implementing additional interfaces.
2.2 Node types
Node types are derived from certain abstract classes that provide a default implementation of the node life cycle and execution control. The developer does not have to take care of the life cycle at all; nonetheless, the base classes are designed to allow developers to override the default behaviour if necessary.
Important information about node types is expressed by meta data (attributes in .NET). This includes a human-readable name for the node type, a description and a classification of the type based on tags. This information is also available in the design GUI to help selecting the right node type for a specific task.
A node class may contain a number of elements that are examined by reflection and it defines the way a user can work with a node:
Endpoints: an endpoint is a data exchange point of a node, either consuming data or producing data. A Conveyor instance is build by creating nodes and connecting their endpoints, thus modelling the data flow in the graph. An endpoint itself is defined as generic data type, with the type parameter being the type of data that is exchanged via the endpoint. As a result, the node class implementation does not need to use type casts to work with data elements exchanged at an endpoint. The number of endpoints is not limited by the design; the currently implemented node classes have between one and up to eight endpoints. Nonetheless, at least one endpoint is required to make a node class usable at all. Similar to the node class itself, an endpoint may be decorated with a description via attributes that are visible to the user.
Configuration fields: these fields define configuration values that may influence the processing within a node class. Table 1 shows the available configuration types. Configuration fields are visible to the user and for mandatory ones, the user is required to enter valid information prior to running the graph. A node class may have any number of configuration fields and it is the responsibility of the class itself to check that all fields contain reasonable values. Fields may be decorated with attributes, providing a user visible description, a default value and a flag to mark them as optional.
Configuration field types available in Conveyor nodes
| Type | Description |
|---|---|
| Integer | 32 bit signed integer |
| Long | 64 bit signed integer |
| Boolean | boolean value |
| Double | IEEE double precision value |
| String | UTF-8 string |
| File | A stream read as file |
| Enumeration<T> | One of the values of the enumeration type T |
| Selection<T,U<T>> | One of the values of type T, generated by type U |
| Type | Description |
|---|---|
| Integer | 32 bit signed integer |
| Long | 64 bit signed integer |
| Boolean | boolean value |
| Double | IEEE double precision value |
| String | UTF-8 string |
| File | A stream read as file |
| Enumeration<T> | One of the values of the enumeration type T |
| Selection<T,U<T>> | One of the values of type T, generated by type U |
The enumeration type uses the values from the enumeration type given as type parameter, eliminating the need for comparing string values in the node code. The selection type provides a more generic way to define a selection of values. The first parameter is the item type, and the second parameter a class generating these items. Selection types are used for example to provide a list of BLAST databases by scanning certain directories.
Configuration field types available in Conveyor nodes
| Type | Description |
|---|---|
| Integer | 32 bit signed integer |
| Long | 64 bit signed integer |
| Boolean | boolean value |
| Double | IEEE double precision value |
| String | UTF-8 string |
| File | A stream read as file |
| Enumeration<T> | One of the values of the enumeration type T |
| Selection<T,U<T>> | One of the values of type T, generated by type U |
| Type | Description |
|---|---|
| Integer | 32 bit signed integer |
| Long | 64 bit signed integer |
| Boolean | boolean value |
| Double | IEEE double precision value |
| String | UTF-8 string |
| File | A stream read as file |
| Enumeration<T> | One of the values of the enumeration type T |
| Selection<T,U<T>> | One of the values of type T, generated by type U |
The enumeration type uses the values from the enumeration type given as type parameter, eliminating the need for comparing string values in the node code. The selection type provides a more generic way to define a selection of values. The first parameter is the item type, and the second parameter a class generating these items. Selection types are used for example to provide a list of BLAST databases by scanning certain directories.
Settings: in contrast to configuration fields mentioned before, settings are static objects that refer to a node class instead of a single node instance. They can be used to define setup and policy-dependent values like the paths to executables or default values. Table 2 shows the various types available as settings in node classes. The value of a setting cannot be manipulated by the user. Instead, the system may either deduce the value itself if possible (e.g. in the case of an executable, Conveyor uses the application search path to locate executables) or the administrator may use a simple graphical user interface after installing Conveyor for adjusting the settings.
Setting types available in Conveyor
| Type | Description |
|---|---|
| Boolean | A boolean value |
| Directory | A directory in the file system, with optional check for existence of the directory and entries in the directory. |
| Double | A floating point value |
| Executable | A binary in the file system, with optional scanning of the default search path for the binary. |
| Integer | A 32 bit integer |
| String | A UTF-8 string |
| StringList | A list of UTF-8 strings |
| Type | A class implementing a given interface |
| TypeList | A list of classes implementing an interface |
| Type | Description |
|---|---|
| Boolean | A boolean value |
| Directory | A directory in the file system, with optional check for existence of the directory and entries in the directory. |
| Double | A floating point value |
| Executable | A binary in the file system, with optional scanning of the default search path for the binary. |
| Integer | A 32 bit integer |
| String | A UTF-8 string |
| StringList | A list of UTF-8 strings |
| Type | A class implementing a given interface |
| TypeList | A list of classes implementing an interface |
Static fields using these types allow for an easy configuration of plugins and the core system by external configuration files.
Setting types available in Conveyor
| Type | Description |
|---|---|
| Boolean | A boolean value |
| Directory | A directory in the file system, with optional check for existence of the directory and entries in the directory. |
| Double | A floating point value |
| Executable | A binary in the file system, with optional scanning of the default search path for the binary. |
| Integer | A 32 bit integer |
| String | A UTF-8 string |
| StringList | A list of UTF-8 strings |
| Type | A class implementing a given interface |
| TypeList | A list of classes implementing an interface |
| Type | Description |
|---|---|
| Boolean | A boolean value |
| Directory | A directory in the file system, with optional check for existence of the directory and entries in the directory. |
| Double | A floating point value |
| Executable | A binary in the file system, with optional scanning of the default search path for the binary. |
| Integer | A 32 bit integer |
| String | A UTF-8 string |
| StringList | A list of UTF-8 strings |
| Type | A class implementing a given interface |
| TypeList | A list of classes implementing an interface |
Static fields using these types allow for an easy configuration of plugins and the core system by external configuration files.
To avoid an unnecessary growth of the number of node and data types and to reduce redundancy, Conveyor supports generic types for both data and node elements. Generics add another abstraction layer to types, allowing developers to implement functionality shared between similar, but not related types. The best known example are the list types in Java or C#. The generic type contains all functionality necessary for the list, the concrete type only determines the element type of the list. This feature simplifies the implementation of generic node types, and allows their type safe use. Certain functionality only needs to be implemented once as a generic node type. It also allows the creation of higher order functions as known from functional programming languages. For example, the core library already contains nodes acting similar to the well-known map and fold operations for list types. An example node class implementation is shown in Figure 1. It defines a generic node type for building a dictionary from key–value pairs.

Example of a node class for creating a dictionary from key-value pairs. The code shows the complete class, including documentation, all methods and fields. The dictionary is built by collecting key-value pairs read from the inputs. The node terminates if either input is depleted, and the dictionary is sent to the output. Lines 10 + 11 define the node class, using two generic type parameters for the key and value types. Line 8 contains a user-visible name for the class, and line 9 a description of the function. Lines 13–20 declare the fields for the two input endpoints and the output endpoint, including a description for each endpoint. The remaining lines overwrite the default life cycle methods to initialize the internal dictionary for collecting the key-value pairs, and sending it to the output upon termination. The Process() method in lines 34–42 also checks whether a key is already stored in the dictionary and prints a warning to a configurable logging facility.
The Conveyor client-server system does not solely address the needs of an end user; by allowing complex control structures like branches and loops, it also focuses on experienced users with a computer science or data analysis background, and it is intended as a replacement for analyses written in Perl scripts or other programming languages.
2.3 Life cycle
Figure 2 depicts the life cycle of a node in a Conveyor instance. The methods used to change from state to state are as follows:
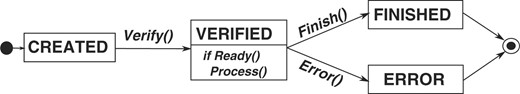
Life cycle of Conveyor nodes: after creation and configuring the node (e.g. setting configuration values and links to other nodes), the Verify() method validates the configuration. Upon successful validation, the node enters the VERIFIED state and is able to process data using the Ready() and Process() methods. The life cycle is terminated either by calling Finish() to indicate normal termination and changing to the FINISHED state, or by calling Error() in case of an exception during processing.
Verify(): after reading the definition of a node instance in a Conveyor instance from a XML formatted file, the processing engine creates an object of the node type given in the definition, sets configuration values and creates connections between endpoints of nodes as defined in the XML file. It then invokes this method to check that (i) all endpoints of the node are connected, (ii) the data types defined for the endpoints are compatible and (iii) all mandatory configuration fields are set. Derived node types may overwrite and extend this method, e.g. to check their configuration fields for reasonable values, open files, connect to a database or to perform any operation necessary to setup a node.
Ready(): nodes within a Conveyor instance are executed independently of other nodes in a thread of their own; the Ready() method is used by the thread to determine whether the node is ready to process data. The default implementation signals the ability to process data if all endpoints defined as input to the node can provide at least one data element. It also checks the state of connected nodes and terminates processing if one of the connected nodes has terminated. Most derived classes should work fine with the default implementation; nonetheless, the method may be overwritten to implement a different logic.
Finish()/Error(): being the last steps in the life cycle, these methods are used to release resources claimed by a node instance. Derived classes should overwrite this method and release resources allocated during the Verify() method at this stage.
Process(): finally, this method is intended for derived classes to implement whatever functionality the node class provides.
The base classes already provide default implementations for all of these methods except Process(). The core library also contains a number of generic class definitions for special purposes like simple unary operations, binary operations, generation of data elements and more. A complete overview of these classes is found in the manual available at the Conveyor web site.
2.4 Graph execution
The number of cores available in modern CPUs has been growing over the recent years from single core CPUs to modern multi-core CPUs with 8 or more cores available on a single chip. Thus, parallelization of processing is a design goal of Conveyor and every processing step is running independent of other steps. The processing step itself may implement its own logic, use an external command line application or invoke web services. Conveyor does not restrict the functionality in any way, but expects a node type to respect the order and cardinality of the data flow. Nodes that rely on these constraints may be marked with annotations that result in an extra check during runtime. The map, for example, transforms a list of data objects into a list of processed objects, maintaining their order and cardinality. The transformation loop may not contain any node type violating these rules. If a node type has to deviate from the default behaviour, it has to be marked with a corresponding annotation. An example is the PassFilter node, that filters elements based on an external condition. If elements are dropped, it violates the cardinality rule, and may not be used in most loops. All annotations are available in the meta data at runtime, allowing the processing engine to validate the rules and their exceptions.
3 IMPLEMENTATION
To reconcile the design defined in the previous paragraph, the implementation of Conveyor is done using a 2-fold strategy: Java for the GUI design client and the .NET framework for the processing engine. Being deployed as a web start application, the GUI design client is available on every platform supported by Java. Updates to the client are automatically installed if an internet connection is available. Information about Conveyor services and their corresponding processing steps are cached by the client to allow users to work offline; submitting a Conveyor instance and monitoring it naturally requires access to the central service.
The .NET framework was chosen for the implementation of the core library and the processing engine. It offers a variety of different programming languages for implementation. Among designated languages like C# or F#, using libraries from many other languages including C, Java and Python is supported. The core library and the processing engine are implemented in C#, without any dependency on external libraries that are not part of the .NET framework. This ensures that Conveyor itself is portable across platforms supported by the .NET framework or by Mono,1 the free implementation of .NET available for Unix-like systems and Mac OS X.
The overall architecture is split into three components (see Fig. 3).
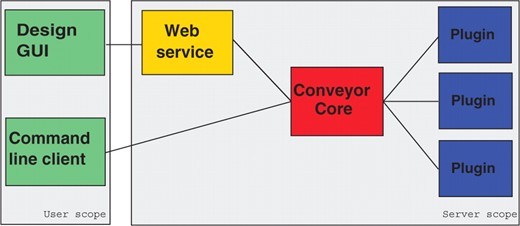
Architecture of the Conveyor system, showing the three main components and component's scopes.
3.1 Core library and processing engine
The core library contains the basic building blocks of Conveyor. This includes main classes for data import, data export and processing steps and the interface to mark a class as a data type usable by Conveyor. It also contains wrappers for primitive data types like numbers and strings, and definitions of control flow processing steps.
The processing engine manages the plugins and controls the execution of a Conveyor instance. It provides a flexible configuration mechanism for processing steps and includes a command line utility for executing a Conveyor instance, and a web service-based interface for the design GUI. The complete core library is independent of the application domain; every functionality and all data types specified for a certain field can be bundled in plugins. Similar to Ruffus, the Conveyor core and processing libraries may be included in applications, enhancing them with workflow capabilities.
3.2 Plugins
Conveyor is bundled with a number of default plugins for functionality used in many fields of application. It includes handling of tables, lists and parallelization of command line tools on compute clusters. These are independent of the field of application. To simplify developing a plugin, all information about available processing steps and data types is extracted automatically from the plugin by reflection.
Most plugins available for Conveyor handle bioinformatics tasks. Two simple rules were used while developing the bioinformatics node types in Conveyor. These rules ensure that node types are usable independent of the context.
Data interfaces instead of classes: all important data types are declared as interfaces, with a default implementation available for constructing them in Conveyor instances or other node types.
Processing nodes depend on interfaces only: node types never refer to a concrete class. The only exception of this rule are processing steps building data types from primitives, e.g. creating a DNA sequence object from a name and the sequence itself. This rule ensures that the node types work independently of the concrete data type and avoids the need for semantically duplicated node types. As an example, the BLAST wrapping node types only rely on the Sequence interface, allowing to use them with any data type that implements this interface. The node type does not have to take care whether the data are read from a FASTA file, is defined as a feature in an EMBL file or provided as an opaque object by a linked application. Node and data types for handling various sequence formats and invoking tools like BLAST (Altschul et al., 1997), ClustalW (Thompson et al., 1994), Muscle (Edgar, 2004), etc. are already implemented. A full list is available at the Conveyor web server.
3.3 Design GUI
Being the main interface to the Conveyor system, the graphical user interface client is designed with a focus on usability and simplicity. It allows users to create Conveyor instances, monitor graphs running on Conveyor servers, and it can be used to retrieve and view the results of computations. The client is written in the Java programming language, using a Webstart setup for easy installation and upgrading of the client. Given a web start environment on a computer (which comes with the Java runtime), clicking a simple button on a web page is all that is needed to install the client; furthermore, the client is able to check for updates at every start, keeping itself in sync without any effort on behalf of the user. A list of servers is stored by the client, together with a cache of nodes and data types available on the servers. The cache allows the user to work offline with the system, without the need of an active internet connection. A new Conveyor pipeline is designed by drag‘n’drop. Figure 4 shows the main design screen, with the list of available node types on the left and the design area on the right. The list of node types may be searched for terms in node type names and descriptions. Dragging a node type from the list on the left side to the design area on the right side will create an instance of the node. Connections are simply created by clicking on the source and the target node, configuration values are set by a double click on the node. One of the key features built into the design interface is the ability to create composite node types. These are node types build from other nodes, allowing the user to create complex functionality by combining lower level functions. Composite node types may also be annotated and send to the server, allowing them to be shared with other users. After designing the graph, it may be sent to the associated Conveyor server for execution. The designer GUI keeps track of running graphs and stores information about them on the local computer. The user thus may design a graph, submit it to the server, shut down his computer and return the next day and retrieve the results. A permanent connection to the server is not needed for Conveyor. During execution, the designer GUI displays information about the progress, including the status of each node and the number of data elements passed along each connection in the graph. Figure 5 shows a screenshot of the Conveyor design GUI while monitoring a Conveyor instance.

Screenshot of the Conveyor graph designer—the screen is split into three parts: the list of available node types on the left side, an outline of the current graph in the lower left corner and the main working area for manipulating the graph instance. New nodes are added by dragging them from the node type list and dropping them on the work area. Connections between nodes are also created by dragging a line from one node to another. Configuration items are accessible by a double click on a node in the work canvas. Node colours in the work space indicate whether the node is an input node (blue), a processing node (green) or an output node (red).
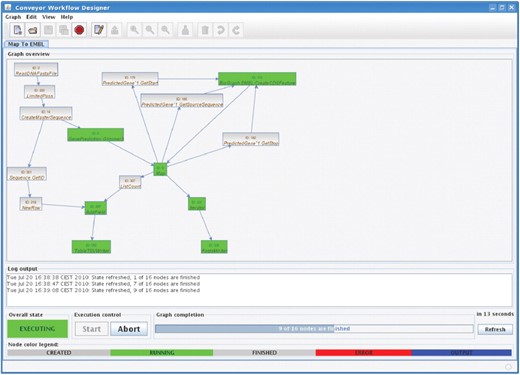
Execution of a workflow in the Conveyor graph designer: the integrated execution monitor displays a graph currently being executed on a Conveyor service. Finished nodes are shown in gray, and nodes currently executing or waiting for data are coloured green. The monitor also provides access to results of output nodes after execution has finished.
4 USE CASES
Conveyor offers a new platform for implementing analytical workflows. Two example use cases demonstrate the unique features like the type system and control flow nodes.
4.1 Workflow 1: annotation by reference genome
The workflow presented in this use case study implements a pipeline for annotating a genome using an annotated reference genome. The new ultrafast sequencing technologies introduced in the recent years result among other benefits in a flood of new draft genomes. Instead of concentrating on a single organism or a single strain, a complete species including several subspecies can be sequenced in a single run. Due to the amount of data generated in these cases, manually finishing or annotating these genomes is not feasible with respect to financial or man power constraints. The research objectives thus focus on the identification of differences among the draft genomes by mapping them onto a reference and comparing the results for the different strains. In many cases, manual annotation is only done for the genes of interest, if done at all. The overall workflow is built from three different parts: gene prediction on plain DNA genomic sequences, homology search for the predicted genes versus an annotated reference genome and handling of predicted genes without homologues. The result of the workflow is exported as EMBL-formatted file containing the predicted genes and their annotations. The workflow in this use case uses a BLAST search to map genes to the reference genome. It also uses another BLAST search for genes without mapped reference genes. The workflow as shown in Figure 6 consists of several parts: (a) the preparation of the reference genome. A BLAST database is built from all CDS features of the given EMBL or GenBank file; (b) gene prediction for the novel genomes. Putative coding regions are predicted using the Glimmer3 (Delcher et al., 1999) prokaryotic gene prediction software with default parameters, and CDS regions are created; (c) search for homologues in the reference genome. Using BLAST, all CDS of the novel genome are compared with the CDS of the reference genome. The results are filtered based on e-value and per cent coverage and put into a list; (d) copy of annotation fields from matching CDS. If a homologous CDS was found during the BLAST search, its annotation fields like gene product, gene name and function are transferred to the novel CDS; (e) search for homologues in a public database. For novel CDS without known similar genes in the reference genome, a public database like the SwissProt database as part of the UniProt database [Consortium (2010) or the GenBank database, Benson et al. (2010)] is used with BLAST to search for homologues; (f) copy of annotation fields from matching database hits. Similar to (a) the content of annotation fields of the best hits against the public database are transferred to the novel CDS; (g) mark still unassigned CDS. All CDS without assigned annotation during steps d) and f) are marked as being putative; (h) Export of genome. The complete genome including the predicted CDS is exported as an EMBL or GenBank file.
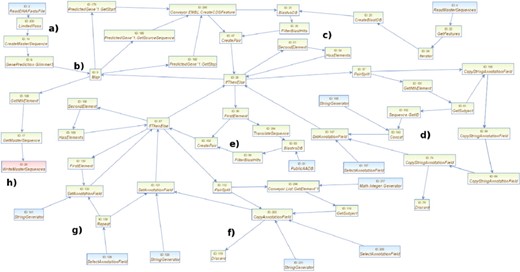
Example workflow for annotating a genome using a reference genome. (a) The preparation of the reference genome. (b) Gene prediction for the novel genomes. (c) Search for homologues in the reference genome. (d) Copy of annotation fields from matching CDS. (e) Search for homologues in a public database. (f) Copy of annotation fields from matching database entry. (g) Mark still unassigned CDS. (h) Export of genome.
The main benefits of implementing the above workflow in the Conveyor system are flexibility and extensibility. The use case only shows an exemplary design of the workflow. Changing the reference organism or the gene prediction tool is only a matter of changing a node in the workflow. Thresholds e.g. for the e-value of BLAST results or settings like the database to use in step (e) are configuration fields directly accessible in the designer GUI. The complete workflow is implemented without writing a single line of code or invoking an application except Conveyor and the designer GUI. Moreover, the command line execution utility that comes with Conveyor allows executing the workflow completely without user interaction. It also offers the ability to change the configuration for individual runs. The BLAST steps used in this use case are good examples for generic types. The BLAST result data type is a generic type, using a type parameter for the BLAST query and another type parameter for the database sequence. Retrieving the query or the subject from a BLAST hit thus results in the original feature from the EMBL or GenBank file, with full access to all feature keys as annotation fields. This also allows transferring keys usually not exported to BLAST databases like notes or EC numbers in a consistent way.
4.2 Workflow 2: core genome calculation
With several genomes of a selected species available, dividing them into groups according to their phenotype and determining the specific gene content of these groups is the next important step in comparative genomics. Conveyor provides all necessary functions for this kind of analysis. A figure showing the complete workflow is available in the Supplementary Material. The core genome consists of sets of all genes present in all organisms, taking paralogs into account. Based on annotated genomes, the homologues of all genes of one organism are computed. If the list of homologues of a single gene only contains one homologue in each organisms, it is considered a member of the core genome.
The workflow thus consists of several parts: (i) import of genomes including their annotation from EMBL or GenBank formatted files. Sequences shorter than a given length are filtered out, e.g. plasmids. (ii) A list of all genome IDs is created. (iii) A blastable database is created from the CDS of all organisms. (iv) The genome with the lowest number of coding sequences is selected. (v) A second blastable database containing all coding sequences of the smallest genome is created. (vi) Homologous sequences for all coding sequences of the smallest genomes are determined using BLAST. (vii) The list of BLAST hits is reduced to significant hits. Their bit scores have to exceed a certain ratio if compared to the bit score of the most significant hit (first hit in BLAST result). (viii) The set of sequences referred to by the remaining BLAST hits has to contain a single gene of each organism to be considered to be a part of the core genome. (ix) Each sequence detected as a possible homologue is compared against all coding sequences of the smallest genome using BLAST. The step filters out possible paralogs in the genome and ensures that all genes in all candidate sets are indeed homologues. (x) Alignments are created from the sequences of all candidate core gene sets using Muscle and written as result using the Phylip alignment format.
The workflow is only an exemplary implementation of a core genome calculator. Other restrictions may also apply, e.g. filtering out genes related to mobile elements. The number of genomes processed by the workflow is not restricted. Many parameters like thresholds for BLAST results, the required score ratio in step (vii) or the required length of the genome sequence used in step (i) can easily be adapted to the input sequences. The workflow may also be extended by additional processing steps based on the alignment of the core gene sets. Nodes for alignment processing and phylogenetic analyses are already available for Conveyor. This workflow demonstrates the list processing and branching flow features of Conveyor.
5 RESULTS
The use cases shown in the previous section demonstrate the usefullness of the Conveyor system to common problems. Both use cases were processed with the set of available Escherichia coli genomes2 (Accession numbers NC_011602, NC_011603, NC_008253, NC_011748, NC_008563, NC_009837, NC_009838, NC_012947, NC_012759, NC_012967, NC_004431, NC_010468, NC_009786, NC_009787, NC_009788, NC_009789, NC_009790, NC_009791, NC_009801, NC_011745, NC_009800, NC_011741, NC_011750, NC_010473, NC_000913, AC_000091, NC_002127, NC_002128, NC_002695, NC_002655, NC_007414, NC_011350, NC_011351, NC_011353, NC_013008, NC_013010, NC_011742, NC_011747, NC_011407, NC_011408, NC_011411, NC_011413, NC_011415, NC_011416, NC_011419, NC_010485, NC_010486, NC_010487, NC_010488, NC_010498, NC_011739, NC_011749, NC_011751, NC_007941, NC_007946, NC_011740, NC_011743) by concatenating their GenBank formatted files available at the NCBI web site. The workflows in both use cases were executed on a single host with four Intel Xeon E7540 CPUs running at 2 GHz. Each CPU provides six physical cores and six additional virtual cores by hyper threading, summing up to 48 cores managed by the operation system. The system is equipped with 256 GB system RAM and 200 GB swap. Conveyor was configured to use a thread-based processing model with one thread per workflow node, and a process-based processing model for external applications. The number of parallel running processes were set to one, two, four, eight and sixteen. Each setup was run five times. Execution time was measured using the time command, reporting the overall time (real), the accumulated CPU time of all processes (user) and the time spend in the kernel (sys). The complete benchmark data is available as Supplementary Material.
5.1 Reference annotation benchmark
The first use case was executed with the plain genome sequences of all 58 Escherichia coli replicons as multiple FASTA input file and the official GenBank annotation of E.coli K-12 DH10B (NC_010473) as reference genome. Figure 7 shows the average runtime of the workflow for different numbers of parallel running processes. The relative runtime is presented in Figure 8. While the user time scales well with the number of processes, the system overhead significantly increases, up to 80% in the 16 process benchmark. This is due to the increasing number of file handles and threads used to communicate with the processes. Nonetheless, the relative runtime of the workflow only increases by 3–4%. Conveyor thus scales well with the number of external processes.
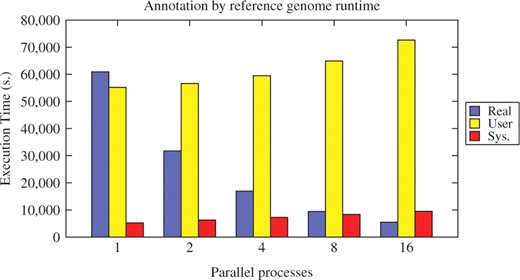
Runtime benchmark for the reference annotation use case. The plots shows the average runtime of the workflow, splitted into the user, system and overall time (measured by the UNIX time command).
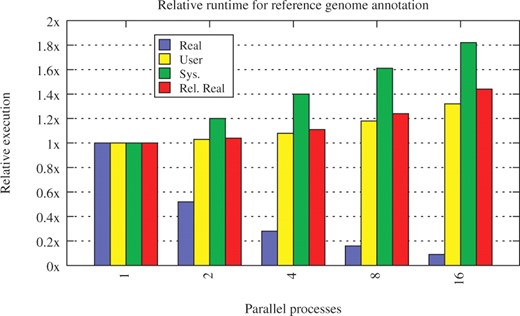
Relative runtime of the reference annotation use case. Values of the single process run are used as base. The relative real time is the ratio of the real runtime times the number of processes used by the real runtime in the single process case.
5.2 Core genome benchmark
For the second use case, the official GenBank annotations of the mentioned E.coli strains were used as input. The genome size filter was set to 1 Mb, resulting in 26 full-sized entries to be processed. A BLAST score ratio cutoff of 0.8 was used to filter the BLAST hits in step (viii). Figures showing the absolute and relative runtimes are available in the Supplementary Material. Due to the different nature of the external tools used in the workflow, e.g. different database sizes in case of the blast tools, running parallel processes adds a noticeable overhead in the case of few processes, but scales well with many instances in parallel. Compared to the former benchmark, the system overhead increases in a less dramatic way, and the overall runtime also scales well.
6 DISCUSSION
A comparison of Conveyor and the workflow systems mentioned in Section 1 is shown in Table 3. The Conveyor system offers many of the features found in workflow systems. This includes encapsulating functionality in processing steps, composite steps built from other steps and an engine to run a workflow. Similar to Taverna and Kepler, Conveyor uses a data-oriented view, modelling data flow from input to output. In contrast to Taverna, the Conveyor plugin system does not depend on external providers for most of its functionality. The current set of plugins implements locally executed processing steps, with an option to use a [DRMAA, distributed resource management application API Troeger et al. (2007)] compliant scheduling system for executing external applications like BLAST. New plugins for integrating web services are planned nonetheless. Similar to the concept of directors in Kepler, Conveyor uses a configurable system for executing workflows. Depending on the machine running the workflow, it may be executed with multiple threads, single-threaded or distributed on a compute cluster. The unique advantage of the Conveyor system with respect to the other workflow engines is the object-oriented type system. Features like inheritance, interfaces and generic data and node types greatly reduce the complexity of workflows and the effort for developing new plugins. Especially the generic types reduce redundancy and allow preserving the original data type in almost all processing nodes. In contrast, Taverna data types are based on textual representation of data, using WSDL and a hand-curated ontology to describe the relations between types and their structure. Kepler allows to restrict data types to primitive types or opaque objects only; object-oriented concepts like inheritance or interfaces are not supported by either system. Pegasus in turn is the most advanced of the compared systems, designed for large workflows with huge amounts of data, and large compute grids with distributed file systems. With respect to its intended use cases, it is clearly superior to Conveyor. Nonetheless, it neither provides a strong type system nor does it offer an easy way for local execution of workflows. As shown in the use cases studies, Conveyor offers complete control over the data flow, allowing loops and conditional branches. With experienced users in mind, the Conveyor system offers a viable alternative to writing scripts in Perl and other languages. The ability to extend the system by plugins and the generic type system allows developers to provide node types and data types based on third party applications, making their data and functionality available to Conveyor. Finally, the results of the use cases shows that Conveyor performs well on off-the-shelf hardware with a reasonable price.
Comparison of the features provided by the different workflow engines
| Mobyle | Taverna (2.x) | Pegasus | Kepler | Conveyor | Galaxy | Ruffus | |
|---|---|---|---|---|---|---|---|
| User interface | |||||||
| Web based | + | − | − | − | − | + | − |
| Offline client | − | + | + | + | + | − | − |
| Processing | |||||||
| Local execution | − | − | + | + | + | − | + |
| Batch support | − | + | + | + | + | + | − |
| Grid/cloud support | − | − | + | + | + | + | − |
| Threading support | − | + | + | + | + | − | − |
| Workflow features | |||||||
| File based | + | − | + | − | − | + | + |
| Type based | − | +a | − | +b | +c | − | − |
| Composed steps | − | + | + | + | + | − | + |
| Flow control | − | − | − | + | + | − | − |
| Mobyle | Taverna (2.x) | Pegasus | Kepler | Conveyor | Galaxy | Ruffus | |
|---|---|---|---|---|---|---|---|
| User interface | |||||||
| Web based | + | − | − | − | − | + | − |
| Offline client | − | + | + | + | + | − | − |
| Processing | |||||||
| Local execution | − | − | + | + | + | − | + |
| Batch support | − | + | + | + | + | + | − |
| Grid/cloud support | − | − | + | + | + | + | − |
| Threading support | − | + | + | + | + | − | − |
| Workflow features | |||||||
| File based | + | − | + | − | − | + | + |
| Type based | − | +a | − | +b | +c | − | − |
| Composed steps | − | + | + | + | + | − | + |
| Flow control | − | − | − | + | + | − | − |
Since AnaBench only wraps command line applications, it does not support any of the advanced features. Taverna 2.x is based on a new processing model that executes nodes in separate threads, with a benefit for locally executed services. Most services of the default installation rely on web services nonetheless. The grid support of Conveyor currently only supports certain nodes executing command line applications; a future extension will also distribute nodes on a compute grid. Galaxy may use a Torque or DRMAA compute cluster for distributing processing steps.
aWSDL/ontology based.
bScalar types and opaque objects without inheritance.
cGeneric object-oriented type system.
Comparison of the features provided by the different workflow engines
| Mobyle | Taverna (2.x) | Pegasus | Kepler | Conveyor | Galaxy | Ruffus | |
|---|---|---|---|---|---|---|---|
| User interface | |||||||
| Web based | + | − | − | − | − | + | − |
| Offline client | − | + | + | + | + | − | − |
| Processing | |||||||
| Local execution | − | − | + | + | + | − | + |
| Batch support | − | + | + | + | + | + | − |
| Grid/cloud support | − | − | + | + | + | + | − |
| Threading support | − | + | + | + | + | − | − |
| Workflow features | |||||||
| File based | + | − | + | − | − | + | + |
| Type based | − | +a | − | +b | +c | − | − |
| Composed steps | − | + | + | + | + | − | + |
| Flow control | − | − | − | + | + | − | − |
| Mobyle | Taverna (2.x) | Pegasus | Kepler | Conveyor | Galaxy | Ruffus | |
|---|---|---|---|---|---|---|---|
| User interface | |||||||
| Web based | + | − | − | − | − | + | − |
| Offline client | − | + | + | + | + | − | − |
| Processing | |||||||
| Local execution | − | − | + | + | + | − | + |
| Batch support | − | + | + | + | + | + | − |
| Grid/cloud support | − | − | + | + | + | + | − |
| Threading support | − | + | + | + | + | − | − |
| Workflow features | |||||||
| File based | + | − | + | − | − | + | + |
| Type based | − | +a | − | +b | +c | − | − |
| Composed steps | − | + | + | + | + | − | + |
| Flow control | − | − | − | + | + | − | − |
Since AnaBench only wraps command line applications, it does not support any of the advanced features. Taverna 2.x is based on a new processing model that executes nodes in separate threads, with a benefit for locally executed services. Most services of the default installation rely on web services nonetheless. The grid support of Conveyor currently only supports certain nodes executing command line applications; a future extension will also distribute nodes on a compute grid. Galaxy may use a Torque or DRMAA compute cluster for distributing processing steps.
aWSDL/ontology based.
bScalar types and opaque objects without inheritance.
cGeneric object-oriented type system.
6.1 Future work
While the current implementation of Conveyor has been focussed on the core library and processing engine, the further development will centre around the functions provided and the user interface. A number of additional plugins are planned and are under development. This includes taxonomy information handling, HMMER 3 support,3 deep sequencing analysis including support for SAM and BAM formats (Li et al., 2009). The interface driven object-oriented approach will also simplify integration of additional sequence formats like the mentioned BioXSD definition into Conveyor. The user interface and the Conveyor web site will be extended to support the exchange of workflows and subworkflows between users, including a cookbook of proven approaches to common analyses. New components will transform a workflow into a standalone application and web service, allowing the rapid development and deployment of workflows.
7 CONCLUSION
As presented in the preceding sections, the Conveyor system offers a comprehensive and versatile system for data analysis. Although it is designed to work in any field of application, the use cases presented in the former sections clearly prove its fidelity to the field of bioinformatics. The unique design, especially the powerful object model for both data and nodes, allows the system to fill the gap between web service-based approaches and writing custom software.
1Cross platform, open-source .NET development framework, http://www.mono-project.com
2Available at end of April 2010.
3No publication available for HMMER 3 yet.
ACKNOWLEDGEMENTS
The authors further thank the BRF system administrators for technical support.
Funding: Bundesministerium für Bildung und Forschung (BMBF) grants (0315599B and 0315599A) ‘GenoMik-Transfer’ to B.L.
Conflict of Interest: none declared.
REFERENCES
Author notes
Associate Editor: Alex Bateman


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}