





Abstract
Summary: Kalign2 is one of the fastest and most accurate methods for multiple alignments. However, in contrast to other methods Kalign2 does not allow externally supplied position specific gap penalties. Here, we present a modification to Kalign2, KalignP, so that it accepts such penalties. Further, we show that KalignP using position specific gap penalties obtained from predicted secondary structures makes steady improvement over Kalign2 when tested on Balibase 3.0 as well as on a dataset derived from Pfam-A seed alignments.
Availability and Implementation: KalignP is freely available at http://kalignp.cbr.su.se. The source code of KalignP is available under the GNU General Public License, Version 2 or later from the same website.
Contact: [email protected]
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
The alignment of multiple sequences is essential for many tasks in sequence analysis, e.g. protein structure and function prediction, similarity search and phylogenetic analysis (Pei, 2008). Due to the importance of multiple sequence alignment (MSA) in a variety of biological applications, a number of programs have been developed. Three aspects are usually considered when developing an MSA program, the alignment accuracy, the computational speed and the memory usage. Kalign2 (Lassmann et al., 2009) is one of the fastest MSA methods, and still the alignment accuracy is comparable to many of the slower methods. Moreover, it produces high quality alignments consistently when aligning large number of sequences, i.e. Kalign2 is well suited for MSA tasks in large-scale genome analysis. However, in contrast to other methods such as ClustalW (Thompson et al., 1994), Kalign2 does not allow externally supplied position specific gap penalties (ESPSGP). Thompson et al. (1994) showed that gaps occur far more often between major secondary structure elements than within. Also gaps are much more frequent in disordered regions of proteins. For integral transmembrane (TM) proteins, residues are more conserved at TM regions and few gaps are allowed within (Kauko et al., 2008). Further, the inclusion of ESPSGP allows some inclusion of expertise knowledge into the alignment procedure.
Here, we introduce KalignP as a modified version of Kalign2 that accepts ESPSGP. Unlike ClustalW, where ESPSGP is only applicable to profile alignments, KalignP incorporates ESPSGP into sequences and forward them to the full progressive alignment.
2 METHODS
2.1 Implementation of KalignP
KalignP inherits the syntax of Kalign2 but extends its functionality for protein sequence alignment. The ESPSGP can be supplied in the Enhanced Fasta format (see Supplementary Material) so that gap penalties are set individually for each position. The default gap penalties will be replaced by ESPSGP when aligning two single sequences. Gap penalties of profiles (consensus of sequences) are calculated in the same way as Kalign2 but ESPSGP will be used instead of the constant gap penalty. KalignP also inherits one of the best merits of Kalign2: low memory consumption. Since KalignP uses the same progressive method as Kalign2, it consumes almost the same amount of memory as Kalign2. KalignP takes about 50% more CPU time than Kalign2 for the alignment itself. The KalignP package, including automatic secondary structure prediction and ESPSGP calculation from secondary structures predicted by PSIPRED_single (Jones, 1999) (version 3.2), is about 15 times slower than Kalign2, but this speed is still comparable to many other MSA programs such as ClustalW and Muscle (Edgar, 2004), see Table 1 and Supplementary Material.
Performance of multiple sequence alignment methods on Balibase 3.0 (using core blocks) in SP score. All SP scores are multiplied with 100 for easy reading
| Method | CPUa (s) | RV11b | RV12c | RV20d | RV30e | RV40f | RV50g | All |
|---|---|---|---|---|---|---|---|---|
| Kalign2 | 57 | 64.4 | 91.3 | 91.9 | 82.5 | 88.4 | 81.9 | 83.6 |
| KalignP | 95 (974h) | 65.6 | 91.8 | 92.4 | 83.4 | 89.8 | 83.9 | 84.6 |
| Mafft | 223 | 56.0 | 89.6 | 91.1 | 84.0 | 87.8 | 84.9 | 81.8 |
| ClustalW | 1281 | 58.2 | 88.4 | 88.8 | 77.1 | 78.9 | 76.9 | 78.6 |
| Muscle | 1281 | 65.7 | 92.3 | 91.5 | 84.2 | 86.5 | 85.3 | 84.4 |
| T_coffee | 25 293 | 73.0 | 94.4 | 93.4 | 87.1 | 89.2 | 90.2 | 87.8 |
| Probcons | 26 085 | 74.0 | 94.6 | 93.7 | 87.5 | 90.3 | 90.1 | 88.3 |
| Method | CPUa (s) | RV11b | RV12c | RV20d | RV30e | RV40f | RV50g | All |
|---|---|---|---|---|---|---|---|---|
| Kalign2 | 57 | 64.4 | 91.3 | 91.9 | 82.5 | 88.4 | 81.9 | 83.6 |
| KalignP | 95 (974h) | 65.6 | 91.8 | 92.4 | 83.4 | 89.8 | 83.9 | 84.6 |
| Mafft | 223 | 56.0 | 89.6 | 91.1 | 84.0 | 87.8 | 84.9 | 81.8 |
| ClustalW | 1281 | 58.2 | 88.4 | 88.8 | 77.1 | 78.9 | 76.9 | 78.6 |
| Muscle | 1281 | 65.7 | 92.3 | 91.5 | 84.2 | 86.5 | 85.3 | 84.4 |
| T_coffee | 25 293 | 73.0 | 94.4 | 93.4 | 87.1 | 89.2 | 90.2 | 87.8 |
| Probcons | 26 085 | 74.0 | 94.6 | 93.7 | 87.5 | 90.3 | 90.1 | 88.3 |
aThe CPU time refers to single threaded process running on a Linux box with Intel quad-core 2.50 GHz CPU and 4 GB memory.
bReference 1: equi-distant sequences with <20% identity.
cReference 1: equi-distant sequences with 20–40% identity.
dReference 2: families aligned with highly divergent ‘orphan’ sequences.
eReference 3: subgroups with <25% residue identity between groups.
fReference 4: sequences with N/C-terminal extensions.
gReference 5: sequences with internal insertions.
hKalignP running time including secondary structure prediction and ESPSGP calculation.
Performance of multiple sequence alignment methods on Balibase 3.0 (using core blocks) in SP score. All SP scores are multiplied with 100 for easy reading
| Method | CPUa (s) | RV11b | RV12c | RV20d | RV30e | RV40f | RV50g | All |
|---|---|---|---|---|---|---|---|---|
| Kalign2 | 57 | 64.4 | 91.3 | 91.9 | 82.5 | 88.4 | 81.9 | 83.6 |
| KalignP | 95 (974h) | 65.6 | 91.8 | 92.4 | 83.4 | 89.8 | 83.9 | 84.6 |
| Mafft | 223 | 56.0 | 89.6 | 91.1 | 84.0 | 87.8 | 84.9 | 81.8 |
| ClustalW | 1281 | 58.2 | 88.4 | 88.8 | 77.1 | 78.9 | 76.9 | 78.6 |
| Muscle | 1281 | 65.7 | 92.3 | 91.5 | 84.2 | 86.5 | 85.3 | 84.4 |
| T_coffee | 25 293 | 73.0 | 94.4 | 93.4 | 87.1 | 89.2 | 90.2 | 87.8 |
| Probcons | 26 085 | 74.0 | 94.6 | 93.7 | 87.5 | 90.3 | 90.1 | 88.3 |
| Method | CPUa (s) | RV11b | RV12c | RV20d | RV30e | RV40f | RV50g | All |
|---|---|---|---|---|---|---|---|---|
| Kalign2 | 57 | 64.4 | 91.3 | 91.9 | 82.5 | 88.4 | 81.9 | 83.6 |
| KalignP | 95 (974h) | 65.6 | 91.8 | 92.4 | 83.4 | 89.8 | 83.9 | 84.6 |
| Mafft | 223 | 56.0 | 89.6 | 91.1 | 84.0 | 87.8 | 84.9 | 81.8 |
| ClustalW | 1281 | 58.2 | 88.4 | 88.8 | 77.1 | 78.9 | 76.9 | 78.6 |
| Muscle | 1281 | 65.7 | 92.3 | 91.5 | 84.2 | 86.5 | 85.3 | 84.4 |
| T_coffee | 25 293 | 73.0 | 94.4 | 93.4 | 87.1 | 89.2 | 90.2 | 87.8 |
| Probcons | 26 085 | 74.0 | 94.6 | 93.7 | 87.5 | 90.3 | 90.1 | 88.3 |
aThe CPU time refers to single threaded process running on a Linux box with Intel quad-core 2.50 GHz CPU and 4 GB memory.
bReference 1: equi-distant sequences with <20% identity.
cReference 1: equi-distant sequences with 20–40% identity.
dReference 2: families aligned with highly divergent ‘orphan’ sequences.
eReference 3: subgroups with <25% residue identity between groups.
fReference 4: sequences with N/C-terminal extensions.
gReference 5: sequences with internal insertions.
hKalignP running time including secondary structure prediction and ESPSGP calculation.
2.2 Estimation of ESPSGP from predicted secondary structure
The principle to estimate ESPSGP from the predicted secondary structure is simple: increase the gap penalty at helices and sheets while lowering the gap penalty at coils. ESPSGP for position i is set as
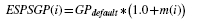
where m(i) is a variable to adjust the gap penalty at residue i based on the predicted structural information, see Supplementary Material for details.
3 RESULTS
We benchmarked KalignP using gap penalties from predicted secondary structure with other methods on Balibase 3.0. KalignP outperformed Kalign2 on all five reference-sets (Table 1). The results were evaluated by SP (Sum-of-Pairs) score and TC (Total Columns) score (Thompson et al., 2005). SP score is the sum of scores of the projected pairwise alignments and TC score is the sum of scores of each column for a multiple alignment. The P-values of paired t-test of all 386 alignments in Balibase 3.0 are 4.51e-5 and 0.0167 for SP and TC scores, respectively, with the alternative hypothesis that true difference in the means of Kalign2 and KalignP is less than 0. The paired t-test was performed by R. Here, KalignP were optimized on Balibase 3.0 using a 3-fold cross-validation (see Supplementary Material). The largest improvement was made on RV50 (for proteins with insertions) where KalignP made a 2.0% improvement for SP score and 2.4% for TC score (see Supplementary Table S1). Only two methods in the benchmark outperformed KalignP, T_coffee (Notredame et al., 2000) and Probcons (Do et al., 2005) but these methods are much slower. Note that the speed of KalignP for alignment alone is comparable to Kalign2. However, including the time for secondary structure prediction and ESPSGP calculation it is slower but still comparable to ClustalW and Muscle (Edgar, 2004).
KalignP made a steady improvement on the alignment of TM proteins as well. As shown in the benchmark on ref7 (for TM proteins) of Balibase 3.0, KalignP outperformed Kalign2 by 1.6% for SP score and 1.8% for TC score (Table 2). However, the significance of the improvement as shown by the t-test is not strong (P-values are 0.0623 and 0.278 for SP and TC scores, respectively). This is most likely because the sample size is too small.
Benchmark on ref7 (transmembrane proteins) of Balibase 3.0 (left) and PfamA-mem using 51 alignments of transmembrane proteins derived from Pfam-A seed alignments (right)
| Method | CPUa (s) | SP | TC | CPUa (s) | SP | TC |
|---|---|---|---|---|---|---|
| Kalign2 | 5 | 89.2 | 43.6 | 3 | 84.6 | 59.3 |
| KalignP | 9 (64b) | 90.8 | 45.4 | 4 (88b) | 86.2 | 62.6 |
| Mafft | 12 | 87.0 | 35.0 | 20 | 83.6 | 56.6 |
| ClustalW | 155 | 79.2 | 40.4 | 81 | 84.0 | 59.3 |
| Muscle | 93 | 88.7 | 43.1 | 59 | 85.7 | 61.3 |
| T_coffee | 2946 | 90.8 | 50.2 | 3093 | 88.4 | 66.8 |
| Probcons | 3217 | 92.4 | 53.0 | 1441 | 88.6 | 67.1 |
| Method | CPUa (s) | SP | TC | CPUa (s) | SP | TC |
|---|---|---|---|---|---|---|
| Kalign2 | 5 | 89.2 | 43.6 | 3 | 84.6 | 59.3 |
| KalignP | 9 (64b) | 90.8 | 45.4 | 4 (88b) | 86.2 | 62.6 |
| Mafft | 12 | 87.0 | 35.0 | 20 | 83.6 | 56.6 |
| ClustalW | 155 | 79.2 | 40.4 | 81 | 84.0 | 59.3 |
| Muscle | 93 | 88.7 | 43.1 | 59 | 85.7 | 61.3 |
| T_coffee | 2946 | 90.8 | 50.2 | 3093 | 88.4 | 66.8 |
| Probcons | 3217 | 92.4 | 53.0 | 1441 | 88.6 | 67.1 |
aThe CPU time refers to single threaded process running on a Linux box with Intel quad-core 2.50 GHz CPU and 4 GB memory.
bKalignP running time including secondary structure prediction and ESPSGP calculation.
Benchmark on ref7 (transmembrane proteins) of Balibase 3.0 (left) and PfamA-mem using 51 alignments of transmembrane proteins derived from Pfam-A seed alignments (right)
| Method | CPUa (s) | SP | TC | CPUa (s) | SP | TC |
|---|---|---|---|---|---|---|
| Kalign2 | 5 | 89.2 | 43.6 | 3 | 84.6 | 59.3 |
| KalignP | 9 (64b) | 90.8 | 45.4 | 4 (88b) | 86.2 | 62.6 |
| Mafft | 12 | 87.0 | 35.0 | 20 | 83.6 | 56.6 |
| ClustalW | 155 | 79.2 | 40.4 | 81 | 84.0 | 59.3 |
| Muscle | 93 | 88.7 | 43.1 | 59 | 85.7 | 61.3 |
| T_coffee | 2946 | 90.8 | 50.2 | 3093 | 88.4 | 66.8 |
| Probcons | 3217 | 92.4 | 53.0 | 1441 | 88.6 | 67.1 |
| Method | CPUa (s) | SP | TC | CPUa (s) | SP | TC |
|---|---|---|---|---|---|---|
| Kalign2 | 5 | 89.2 | 43.6 | 3 | 84.6 | 59.3 |
| KalignP | 9 (64b) | 90.8 | 45.4 | 4 (88b) | 86.2 | 62.6 |
| Mafft | 12 | 87.0 | 35.0 | 20 | 83.6 | 56.6 |
| ClustalW | 155 | 79.2 | 40.4 | 81 | 84.0 | 59.3 |
| Muscle | 93 | 88.7 | 43.1 | 59 | 85.7 | 61.3 |
| T_coffee | 2946 | 90.8 | 50.2 | 3093 | 88.4 | 66.8 |
| Probcons | 3217 | 92.4 | 53.0 | 1441 | 88.6 | 67.1 |
aThe CPU time refers to single threaded process running on a Linux box with Intel quad-core 2.50 GHz CPU and 4 GB memory.
bKalignP running time including secondary structure prediction and ESPSGP calculation.
The Balibase 3.0 ref7 contains only eight alignments and the results for KalignP were obtained by optimizing parameters on all these alignments. Therefore, over-training might be an issue here. To test the robustness of KalignP on the alignment of TM proteins, we created another reference dataset PfamA-mem which was derived from Pfam-A (version 24.0) seed alignments and filtered by matching the key word ‘transmembrane’ in the ‘DE’ record. The same parameters optimized from ref7 were used to test these 51 alignments in PfamA-mem (see Supplementary Table S3 for a list of these 51 Pfam-A alignments). A similar improvement of KalignP over Kalign2 was obtained (Table 2). The P-values for paired t-test are 2.75e-4 and 3.85e-3 for SP and TC scores, respectively.
4 CONCLUSIONS
Here, we present KalignP, a method that allows externally supplied position specific gap penalty, and maintains the high speed and accuracy of Kalign2. Using gap-penalties from predicted secondary structures a steady improvement is seen. Further, KalignP is more flexible than Kalign2, as researchers can modify the behavior of alignment using other knowledge.
Funding: Swedish Research Council (VR-NT 2009-5072, VR-M 2007-3065); SSF (the Foundation for Strategic Research); the EU 7'th Framework Program by support to the EDICT project (contract No: FP7-HEALTH-F4-2007-201924).
Conflict of Interest: none declared.
REFERENCES
Author notes
Associate Editor: Martin Bishop

