





Abstract
Summary: Over the last decade, immunoinformatics has made significant progress. Computational approaches, in particular the prediction of T-cell epitopes using machine learning methods, are at the core of modern vaccine design. Large-scale analyses and the integration or comparison of different methods become increasingly important. We have developed FRED, an extendable, open source software framework for key tasks in immunoinformatics. In this, its first version, FRED offers easily accessible prediction methods for MHC binding and antigen processing as well as general infrastructure for the handling of antigen sequence data and epitopes. FRED is implemented in Python in a modular way and allows the integration of external methods.
Availability: FRED is freely available for download at http://www-bs.informatik.uni-tuebingen.de/Software/FRED.
Contact: [email protected]
1 INTRODUCTION
The detection of T-cell epitopes is a critical step in vaccine design and a key problem in immunoinformatics. Experimental studies to detect epitopes are expensive and time consuming. Computational methods are able to reduce this experimental effort and thereby facilitate the process of epitope detection (DeLuca and Blasczyk, 2007). Many computational methods (based on, e.g. position-specific scoring matrices, various machine learning methods or structural information) have been developed for this task. Many of these methods are freely available through the internet, however, few standalone implementations are available. While web-based predictions are easy to use on the small scale, it severely hampers large-scale predictions and makes a direct comparison of individual methods difficult. The development of flexible prediction and analysis pipelines that can handle large amounts of data and combine prediction methods becomes increasingly important. These pipelines include extensive and flexible pre- and post-processing in addition to the application of a prediction method. The web-based methods available today do not offer tools for flexible data processing. Since there is no uniform interface to access these methods it is difficult to include them into automated prediction pipelines. One option to provide convenient and coherent interfaces to immunoinformatics tools is through web services (Halling-Brown et al., 2009); however, speed and availability tend to limit this approach particularly for large-scale studies.
Here, we present FRED, a software framework for computational immunomics, that provides a uniform interface for a variety of prediction methods and support for the implementation of custom-tailored prediction pipelines. FRED offers methods for extensive data processing as well as methods to assess and compare the performance of the prediction methods. This makes it a powerful platform for the rapid development of new algorithms and the analysis of large datasets.
2 METHODS
2.1 Implementation
FRED provides methods for sequence input, sequence preprocessing, filtering and display of the results. The general organization of FRED is depicted in Figure 1. The single prediction methods are accessed internally via a consistent interface. FRED can handle polymorphic sequences (e.g. for the study of single nucleotide polymorphisms (SNPs) in an epitope context) and offers the possibility of accessing different methods simultaneously and of combining, comparing or benchmarking the methods. FRED is easily extendable by user-defined prediction methods or methods for filtering of results. FRED is implemented in Python (release 2.6) (www.python.org). All additional software required for FRED is freely available and installation packages are included in the FRED package. The prediction methods currently available in FRED are listed in Table 1.
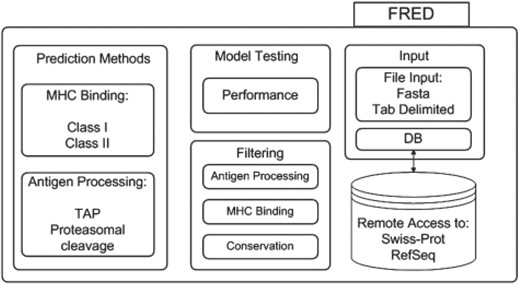
FRED is organized into four major parts: sequence input, application of prediction methods, filtering of the results and model testing.
Prediction methods currently integrated in FRED
| Method | References |
|---|---|
| MHC binding: | |
| SYFPEITHI | Rammensee et al. (1999) |
| SVMHC | Dönnes and Kohlbacher (2006) |
| BIMAS | Parker et al. (1994) |
| NetMHCpana | Nielsen et al. (2007) |
| NetMHCa | Buus et al. (2003) |
| Hammer | Sturniolo et al. (1999) |
| NetMHCIIpana | Nielsen et al. (2008) |
| Proteasomal Cleavage: | |
| PCM method from WAPP | Dönnes and Kohlbacher (2005) |
| TAP Transport: | |
| SVMTAP | Dönnes and Kohlbacher (2005) |
| Additive matrix method | Doytchinova et al. (2004) |
| Method | References |
|---|---|
| MHC binding: | |
| SYFPEITHI | Rammensee et al. (1999) |
| SVMHC | Dönnes and Kohlbacher (2006) |
| BIMAS | Parker et al. (1994) |
| NetMHCpana | Nielsen et al. (2007) |
| NetMHCa | Buus et al. (2003) |
| Hammer | Sturniolo et al. (1999) |
| NetMHCIIpana | Nielsen et al. (2008) |
| Proteasomal Cleavage: | |
| PCM method from WAPP | Dönnes and Kohlbacher (2005) |
| TAP Transport: | |
| SVMTAP | Dönnes and Kohlbacher (2005) |
| Additive matrix method | Doytchinova et al. (2004) |
aInstallation of external software is required. Due to licensing issues, we could not include the standalone versions of these methods in the FRED package.
Prediction methods currently integrated in FRED
| Method | References |
|---|---|
| MHC binding: | |
| SYFPEITHI | Rammensee et al. (1999) |
| SVMHC | Dönnes and Kohlbacher (2006) |
| BIMAS | Parker et al. (1994) |
| NetMHCpana | Nielsen et al. (2007) |
| NetMHCa | Buus et al. (2003) |
| Hammer | Sturniolo et al. (1999) |
| NetMHCIIpana | Nielsen et al. (2008) |
| Proteasomal Cleavage: | |
| PCM method from WAPP | Dönnes and Kohlbacher (2005) |
| TAP Transport: | |
| SVMTAP | Dönnes and Kohlbacher (2005) |
| Additive matrix method | Doytchinova et al. (2004) |
| Method | References |
|---|---|
| MHC binding: | |
| SYFPEITHI | Rammensee et al. (1999) |
| SVMHC | Dönnes and Kohlbacher (2006) |
| BIMAS | Parker et al. (1994) |
| NetMHCpana | Nielsen et al. (2007) |
| NetMHCa | Buus et al. (2003) |
| Hammer | Sturniolo et al. (1999) |
| NetMHCIIpana | Nielsen et al. (2008) |
| Proteasomal Cleavage: | |
| PCM method from WAPP | Dönnes and Kohlbacher (2005) |
| TAP Transport: | |
| SVMTAP | Dönnes and Kohlbacher (2005) |
| Additive matrix method | Doytchinova et al. (2004) |
aInstallation of external software is required. Due to licensing issues, we could not include the standalone versions of these methods in the FRED package.
3 APPLICATIONS
Tutorial and documentation: with the FRED package, we provide examples that demonstrate how FRED can be used to solve typical tasks in computational immunomics with short and simple scripts. A detailed tutorial is available on the project's web site. It explains how to implement prediction pipelines, offers more detailed information on the functionality of FRED and addresses problems like choosing the right prediction method or threshold. We additionally provide a detailed documentation of the code.
Vaccine design: the selection of peptides for epitope-based vaccines is a typical application for large-scale predictions of MHC binding peptides. The following short and simple program implements a typical scenario for the selection of conserved peptide candidates for a vaccine against a virus. The scenario is based on the paper by Toussaint et al. (2008): a set of sequences of the hepatitis C virus core protein from four different subtypes is used. All peptides that occur in at least 90% of the input sequences are considered candidates for conserved epitopes. Predictions are made for 29 HLA alleles using the BIMAS method (Parker et al., 1994). 
Integration of new methods and performance evaluation: epitope prediction is still a very active field, with new methods continuously being developed. Such methods not implemented in Python can be plugged in using command-line calls. FRED provides a number of standard measures to compare different prediction methods and to evaluate the performance w.r.t. experimental values (Matthews Correlation Coefficient, accuracy, sensitivity, specificity, area under the ROC curve, correlation and rank correlation). Different prediction methods can thus be compared with ease.
Web server development: using FRED as the basis for new applications in computational immunomics leads to a significant reduction of development time and allows the convenient combination of new methods with existing ones. An example of an application based on FRED is EpiToolKit (www.epitoolkit.org, Feldhahn et al., 2008; Toussaint and Kohlbacher, 2009). Only the web-based user interface and the data management in the web server had to be newly implemented. Prediction functionality of EpiToolKit is completely provided by FRED. Through the use of Python, FRED can be integrated seamlessly in web servers/content management systems like Plone (http://www.plone.org/).
4 CONCLUSIONS
FRED is a valuable tool for performing large-scale analyses in immunoinformatics with different prediction methods and is also a software framework for the development of novel immunoinformatics methods. Ease of use, extendability and openness make it an ideal tool for addressing complex immuno-informatics problems in an uncomplicated manner.
Funding: Deutsche Forschungsgemeinschaft (SFB 685/B1).
Conflict of Interest: none declared.
REFERENCES
Author notes
Associate Editor: John Quackenbush


{kind=link}