




Abstract
Enhancers, noncoding DNA fragments, play a pivotal role in gene regulation, facilitating gene transcription. Identifying enhancers is crucial for understanding genomic regulatory mechanisms, pinpointing key elements and investigating networks governing gene expression and disease-related mechanisms. Existing enhancer identification methods exhibit limitations, prompting the development of our novel multi-input deep learning framework, termed Enhancer-MDLF. Experimental results illustrate that Enhancer-MDLF outperforms the previous method, Enhancer-IF, across eight distinct human cell lines and exhibits superior performance on generic enhancer datasets and enhancer–promoter datasets, affirming the robustness of Enhancer-MDLF. Additionally, we introduce transfer learning to provide an effective and potential solution to address the prediction challenges posed by enhancer specificity. Furthermore, we utilize model interpretation to identify transcription factor binding site motifs that may be associated with enhancer regions, with important implications for facilitating the study of enhancer regulatory mechanisms. The source code is openly accessible at https://github.com/HaoWuLab-Bioinformatics/Enhancer-MDLF.
INTRODUCTION
Enhancers, noncoding fragments within DNA sequences, play a pivotal role in regulating gene transcription [1, 2]. As a class of regulatory elements, enhancers exert control over diverse cellular activities, including tissue-specific gene expression [3], cell growth and differentiation [4] and cell carcinogenesis [5]. Mutations or abnormal expression of enhancers can disrupt gene regulatory networks, thereby affecting cellular function, tissue development and disease progression [6]. Many recent studies have found that the genetic mechanisms of complex diseases can be better revealed by understanding the role of enhancers in gene expression [7–9]. Therefore, the identification of enhancers is crucial for advancing the comprehension of gene expression and regulation.
High-throughput computational and experimental methods have been employed to predict enhancers. Several methods for identifying enhancers are as follows: (i) Computational Analysis Using Conserved Sequences and Transcription Factor Binding Site Data [10, 11]. This method effectively predicts the genomic locations where known transcription factors (TFs) with binding sequence motifs are likely to interact. However, it may yield false positives by encompassing regulatory element sequences that bind TFs but do not serve as enhancers. (ii) Utilizing ChIP-seq Data for Transcription Factors and P300. ChIP-seq data for TFs can identify enhancers bound by known TFs [12]. However, it cannot distinguish between enhancer and promoter regions because both can bind TFs. Furthermore, not all enhancers necessarily bind TFs. ChIP-seq data for p300 [13], commonly used for enhancer prediction, faces limitations in distinguishing between active and inactive enhancers. (iii) Chromatin Accessibility-Related Data (e.g. DNase-seq [14], FAIRE-seq [15], ATAC-seq [16]). This method relies on data related to chromatin accessibility, However, it may yield false positives by including other transcriptional regulatory elements, such as promoters, insulators and silencers. (iv) Histone Modification Data [17] (e.g. H3K4me1 and H3K27ac). Using both H3K4me1, which marks active and poised enhancers, and H3K27ac, which marks associated with active regulatory regions from both promoters and enhancers to identify activated enhancer regions, this method benefits from the widespread availability of histone modification data across different species, effectively supporting various research needs. However, its drawback lies in the broad nature of histone modification data across the entire genome, hindering precise enhancer prediction. (v) Prediction based on enhancer RNA (eRNA) data [18] (e.g. RNA-seq, ChAR-seq, GRO-seq and NET-seq). Enhancers transcribe eRNAs, and their locations can be predicted using eRNA data obtained through sequencing techniques. However, this method is limited in predicting enhancers that are not actively transcribed. Despite their individual merits, these experimental methods have inherent limitations, and they are both time-consuming and expensive. Therefore, it is essential to develop reliable computational tools for enhancer identification.
In recent times, several computational methods for enhancer identification have been proposed, including ienhancer-2L [19], Enhancerpred [20], ienhancer-EL [21], ienhancer-ECNN [22], BERT-enhancer [23], ienhancer-EBLSTM [24] and ienhancer-XG [25]. Notably, these methods are based on the dataset created by Liu et al. [19]. However, two notable issues arise with this dataset. Firstly, the enhancers within it are extracted as short sequences of a fixed length (200bp), raising questions about the adaptability of these methods to unequal-length sequences and their ability to maintain optimal performance under such circumstances. Secondly, the dataset is a mixed general dataset encompassing nine cell lines, notwithstanding the established understanding that enhancers exhibit cell-specificity [26, 27].
To further investigate the cell-specific nature of enhancers, Enhancer-IF [28], a framework based on integrated machine learning (ML), was proposed to identify enhancers. This framework utilizes eight cell lines with known cell-specific enhancers. Cross-cell line validation results demonstrate that a significant majority of enhancers indeed exhibit cell-type specificity. This underscores the importance of considering cell specificity in enhancer identification, a facet not fully addressed by the earlier methods relying on the general dataset approach.
Despite notable advancements in enhancer identification, there exist notable limitations. Firstly, the predictive performance of Enhancer-IF for cell-specific enhancers is not ideal, which may be due to the conventional feature encoding scheme and the relatively simplistic design of the model framework. Secondly, Enhancer-IF integrates five commonly used classifiers (Random Forest, Extremely Randomized Tree, MultiLayer Perceptron, Support Vector Machine and Extreme Gradient Boosting) and employs a grid search algorithm to optimize parameters for each classifier across every cell line. Undoubtedly, this approach is time-consuming when applied to new cell lines. Furthermore, there is a lack of in-depth exploration of potential strategies to mitigate the impact of cell specificity on the overall performance of enhancer prediction. Lastly, Enhancer-IF lacks explanations for its prediction models, which is crucial for exploring transcription factor binding sites (TFBSs) motifs in enhancer regions. The absence of such interpretability hinders a comprehensive understanding of the biological insights derived from the predictions. Addressing these limitations is crucial for advancing the accuracy, efficiency and biological interpretability of enhancer prediction models.
Therefore, we propose Enhancer-MDLF, a Multi-input Deep Learning Framework designed to predict cell-specific enhancers across multiple human cell lines. Our approach amalgamates word vector features derived from the human genome sequence and motif features extracted from the position weight matrix (PWM) of motifs. Through comprehensive evaluation on cell-specific datasets and other pertinent datasets, we demonstrate the superior performance of enhancer-MDLF. The principal contributions of our work include (i) the introduction of a novel deep learning framework, employing multi-module inputs for the identification of cell-specific enhancers; (ii) the substantiation of Enhancer-MDLF’s substantial outperformance relative to state-of-the-art predictors, accomplished without the need of parameter tuning; (iii) the incorporation of transfer learning into our model to address the challenges in cross-cell line predictions stemming from enhancer specificity and (iv) a meticulous analysis of the conservation and specificity of enhancers at the motif level, culminating in the identification of the most important TFBS motifs within enhancer regions. The overall framework of our study is depicted in Figure 1.
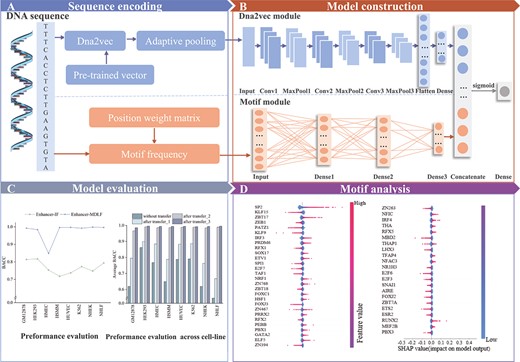
The overall flowchart of Enhancer-MDLF. (A) Sequence encoding: Enhancer-MDLF utilizes two distinct sequence encoding schemes, namely dna2vec and Motif frequency. (B) Model construction: Enhancer-MDLF is structured by integrating two pivotal modules: the dna2vec module and the motif module. (C) Model evaluation. Enhancer-MDLF undergoes thorough evaluation across multiple dimensions to assess its performance. (D) Motif analysis: Utilizing the SHAP framework, we conduct an in-depth analysis of the important features identified by Enhancer-MDLF.
MATERIALS AND METHODS
Dataset
In this study, we utilize the benchmark dataset derived from Enhancer-IF [28]. The dataset encompasses eight distinct cell lines, namely GM12878, HEK293, HMEC, HSMM, HUVEC, K562, NHEK and NHLF. They utilize Enhancer Atlas 2.0 [29] (http://www.enhanceratlas.org/indexv2.php) to extract enhancer locations for each cell line, and their corresponding sequences are obtained through Retailor [30] (http://shiva.rockefeller.edu/SeqTailor/). To ensure diversity, the CD-HIT software is applied to eliminate paired sequences with a similarity exceeding 60%. The construction of negative samples follows the methodology introduced by Dao et al. [31]. Finally, a training set and an independent test set are obtained for each of the eight cell lines. For more details regarding the dataset employed in this study, please refer to Table 1.
Statistical summary of training and independent datasets for different cell lines
| Cell lines | Training | Independent | ||
|---|---|---|---|---|
| Positives | Negatives | Positives | Negatives | |
| GM12878 | 2187 | 2187 | 1187 | 2356 |
| HEK293 | 3756 | 3756 | 2662 | 5324 |
| HMEC | 3333 | 3333 | 1795 | 3590 |
| HSMM | 2821 | 2821 | 1520 | 3040 |
| HUVEC | 4750 | 4750 | 2559 | 5118 |
| K562 | 3318 | 3318 | 1787 | 3754 |
| NHEK | 2896 | 2896 | 1559 | 3118 |
| NHLF | 1462 | 1462 | 788 | 1576 |
| Cell lines | Training | Independent | ||
|---|---|---|---|---|
| Positives | Negatives | Positives | Negatives | |
| GM12878 | 2187 | 2187 | 1187 | 2356 |
| HEK293 | 3756 | 3756 | 2662 | 5324 |
| HMEC | 3333 | 3333 | 1795 | 3590 |
| HSMM | 2821 | 2821 | 1520 | 3040 |
| HUVEC | 4750 | 4750 | 2559 | 5118 |
| K562 | 3318 | 3318 | 1787 | 3754 |
| NHEK | 2896 | 2896 | 1559 | 3118 |
| NHLF | 1462 | 1462 | 788 | 1576 |
Statistical summary of training and independent datasets for different cell lines
| Cell lines | Training | Independent | ||
|---|---|---|---|---|
| Positives | Negatives | Positives | Negatives | |
| GM12878 | 2187 | 2187 | 1187 | 2356 |
| HEK293 | 3756 | 3756 | 2662 | 5324 |
| HMEC | 3333 | 3333 | 1795 | 3590 |
| HSMM | 2821 | 2821 | 1520 | 3040 |
| HUVEC | 4750 | 4750 | 2559 | 5118 |
| K562 | 3318 | 3318 | 1787 | 3754 |
| NHEK | 2896 | 2896 | 1559 | 3118 |
| NHLF | 1462 | 1462 | 788 | 1576 |
| Cell lines | Training | Independent | ||
|---|---|---|---|---|
| Positives | Negatives | Positives | Negatives | |
| GM12878 | 2187 | 2187 | 1187 | 2356 |
| HEK293 | 3756 | 3756 | 2662 | 5324 |
| HMEC | 3333 | 3333 | 1795 | 3590 |
| HSMM | 2821 | 2821 | 1520 | 3040 |
| HUVEC | 4750 | 4750 | 2559 | 5118 |
| K562 | 3318 | 3318 | 1787 | 3754 |
| NHEK | 2896 | 2896 | 1559 | 3118 |
| NHLF | 1462 | 1462 | 788 | 1576 |
DNA sequence encoding schemes
Enhancer-MDLF is a deep learning model, including a dna2vec module and a motif module. The two modules utilize different sequence encoding schemes as input, as depicted in the following sections.
dna2vec
In recent times, word embedding techniques have gained significant popularity within the bioinformatics community, offering a promising solution to address the challenge posed by the similarity of kmer-based features in different sequences, even when their orders are reversed [32, 33]. However, a notable limitation arises when employing word vector techniques for sequence coding. Typically, the training corpus for learning word vectors encompasses only one cell line dataset. This constrained corpus imparts a limited amount of information to the learned word vectors, thereby restricting their representational capacity.
To address the above limitation, we utilize the pre-trained DNA vectors provided in dna2vec as a sequence encoding index. dna2vec, a method grounded in the word2vec word embedding model, is employed to compute distributed representations of variable-length k-mers within DNA sequences. Earlier investigations have substantiated that the mathematical operations applied to dna2vec vectors exhibit similarity to nucleotide concatenation [34]. dna2vec employs the human genome sequence as a learning corpus for unsupervised training, employing the continuous skip-gram (Skip-gram) model in word2vec. This process results in embedding k-mers into a continuous vector space with 100 dimensions.
In this study, we conducted experiments within the range of [3,8] (see Supplementary Table S1) to determine that the optimal value for k is 3. Subsequently, we utilize the pre-trained dna2vec model to obtain a 100-dimensional feature vector for each word. These feature vectors are obtained by concatenating the vectors of short sequences with a step size of 1 in the overall sequence. They are then input to the dna2vec module as sequence features. Assuming that the sequence length is denoted as L, the input dimension of the dna2vec module becomes 100|$\times $| (L - k + 1). It is noteworthy that due to varying feature dimensions for unequal sequences in the dataset, adaptive pooling operations are employed to flatten the feature dimensions to 10 000 dimensions. This pre-processing step is essential for subsequent input into the deep learning model.
Motif frequency
In dna2vec, the parameter k in k-mer is set to 3, signifying that DNA fragments are divided into short sequences with a nucleotide length of 3. Given the relatively brief nature of these sequences, addressing this limitation necessitates the utilization of longer features for capturing intricate sequence patterns. Notably, TFs play an important role in gene transcription by directly binding motifs in the genome. A previous study has successfully identified some potential TF binding within DNA sequences, particularly those inclined to bind in the enhancer regions [35]. Leveraging this biological characteristic, we extract the count of TFBS motifs within each DNA sequence and convert it into a frequency representation for input to the motif module.
We extract the PWM of motifs from the HOCOMOCO Human v11 database [36] for sliding-scale matching to the sequence data in this dataset. Assuming that the length of the motif is denoted as |$L_{m}$|, the PWM is structured as a matrix with |$L_{m}$| rows and four columns, representing the values corresponding to each base (A, C, G and T). The process involves dividing each sequence of length |$L_{s}$| into subsequences of length |$L_{m}$| with a stride of 1. Consequently, we obtain |$L_{s}$| - |$L_{m}$| +1 subsequence segments, each of length |$L_{m}$|. For each subsequence segment, we calculate the sum of the corresponding value for each base, and this sum serves as the final matching score. This score is then compared with the predetermined threshold score to determine whether this subsequence segment matches a motif. The criteria for subsequence segment comparison are defined by the following equation:
where the variable j takes on values 0, 1, 2 and 3, corresponding to the nucleotides A, C, G and T in the subsequence segments, and Q represents the matching score. Assuming P represents the P-value threshold score (set at |$10^{-4}$|) for respective motifs, the subsequence segment is deemed to match this motif when |$Q> P$|.
Upon traversing each sequence, we gather information on the count of each TFBS motif, resulting in a 401-dimensional feature vector denoted as |$V_{count}$|. Additionally, to accommodate varying sequence lengths, we utilize the feature vector |$V_{frequency}$| as the final model input, calculated through the following equation:
where |$L_{s}$| represents the length of each sequence.
The framework of Enhancer-MDLF
Model architecture
The DNA fragments extracted from the dataset undergo encoding through two schemes, namely dna2vec and motif. Subsequently, these encoded fragments are input into the model to predict whether the fragment contains enhancer regions. Through systematic experimentation involving various combinations of convolutional layers, max-pooling layers and dense layers, along with meticulous parameter tuning for optimal balance between accuracy, efficiency and generalization capabilities, we have defined the comprehensive framework of Enhancer-MDLF, as illustrated in Figure 2A. The details are elaborated as follows.
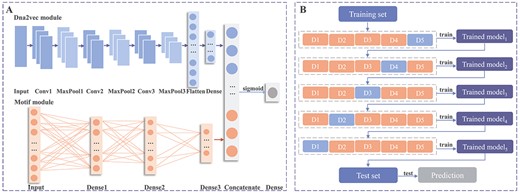
The model architecture and training procedure of Enhancer-MDLF. (A) Enhancer-MDLF comprises a dna2vec module predominantly composed of convolutional layers and a motif module primarily consisting of Dense layers. It takes dna2vec and motif frequency, the two sequence encoding results, as inputs. The fused features are derived by concatenating after feature extraction and subsequently passed through a sigmoid function for enhancer detection. (B) The training procedure of the Enhancer-MDLF framework. The process involves iterations through the training set five times to iteratively refine the predictive model. Subsequently, this model undergoes testing on the test set to yield the final prediction results.
Feature Extraction: We employ a combination of three 1D convolutional layers with corresponding 1D max-pooling layers in the dna2vec module, and three dense layers in the motif module. The convolutional layers are instrumental in capturing complex features from the inputs through convolutional computations. Simultaneously, the max-pooling layers implement a down-sampling approach, selecting the maximum value for each sub-region. This not only improves the robustness of the model but also mitigates the risk of overfitting. Specifically, we define three convolutional layers with 64 filters, a kernel size of 7 and a stride of 3. Additionally, three max-pooling layers are constructed with a pool size of 2, respectively.
To further enhance the model’s generalization and prevent overfitting, we introduce a dropout layer with a probability of 0.6. This layer randomly removes certain neural network units during the training phase, contributing to the model’s overall robustness and preventing excessive adaptation to the training data.
Prediction: The dna2vec module undergoes computation through a dense layer comprising 500 neurons after flattening. Simultaneously, the motif module is designed to culminate in 16 neurons, with a choice of the ‘relu’ activation function for both modules. Within the motif module, we incorporate a dropout layer with a probability of 0.6 following the three dense layers. Finally, Enhancer-MDLF combines the results from the dna2vec and motif modules and generates predictive values using a dense layer with a single neuron and a ‘sigmoid’ activation function. To further enhance model generalization, a dropout layer with a probability of 0.5 is applied after concatenating the two sets of results. The classification criterion is set such that a sample is considered positive if the predicted value exceeds 0.5; otherwise, it is deemed negative.
Hyperparameters: The hyperparameters of Enhancer-MDLF include learning rate, batch size and maximum epoch. Following a comprehensive comparison of performance across multiple hyperparameter combinations through the grid search method, we establish the optimal settings as follows: learning rate = 0.0001, batch size = 100 and max epoch = 200. The specific details of the grid search method and the details of the search ranges for hyperparameters can be found in the Supplementary Information.
Loss function
The dataset we utilize exhibits an imbalance, a characteristic that poses challenges for traditional loss functions. Traditional approaches tend to disproportionately penalize dominant classes, often neglecting the informative contributions of minority classes in imbalanced datasets, thereby resulting in suboptimal prediction performance. Recognizing this, the focal loss [37] initially employed in computer vision [38, 39] has emerged as a solution. Consequently, we use the focal loss as the primary loss function for Enhancer-MDLF, aiming to mitigate the drawbacks associated with the traditional cross-entropy loss function in managing imbalanced datasets. The focal loss is formally defined as follows:
where |$P_{t}$| represents the predicted probability, |$\alpha $| denotes the balance parameter and |$\gamma $| is the focus parameter. To determine the optimal configuration for Enhancer-MDLF, we conduct a thorough performance evaluation using the grid search method over a range of |$\alpha $| values [0.25, 0.5, 0.75] and |$\gamma $| values [1, 2, 3]. Our findings reveal that the model achieves optimal performance when |$\alpha $|=0.5 and |$\gamma $|=3.
Training procedure
We utilize a novel training strategy for Enhancer-MDLF to enhance its capacity to effectively learn information from the input feature vectors of the dna2vec and motif modules. First, we implement a 5-fold cross-validation algorithm, randomly partitioning the training set into five folds with a 4:1 ratio between the training and validation sets. To prevent overfitting, an early stopping mechanism is applied to the validation set. The model undergoes training five times, with the initial training parameters being the model’s starting parameters, determined by TensorFlow’s default network initialization methods [40]. Xavier initialization is frequently used in TensorFlow for a variety of neural network layers, such as dense and convolutional layers. This method initializes weights randomly from a uniform or normal distribution, and the calculation of the standard deviation depends on the number of input and output units in the layer. Subsequent trainings build upon the most recent model, utilizing different folds of the training and validation sets. Following the completion of the five training sessions, the model is applied to the testing set for prediction.
As shown in Figure 2B, the workflow of Enhancer-MDLF is as follows:
(i) Divide the training dataset |$D_{train}$| into five folds: D1, D2, D3, D4 and D5 utilizing a 5-fold cross-validation strategy. For the first training iteration, choose |$D_{train}$| - D1 (The remaining samples after removing D1 in |$D_{train}$|) as the current training set, utilizing ‘D1’ as the validation set to train the model and obtain |$model_{1}$|.
(ii) Subsequent to model1, for the second training iteration, employ |$D_{train}$| - D2 as the current training set and D2 as the validation set to continue training |$model_{1}$|, obtaining |$model_{2}$|. By analogy, repeat the training process five times to obtain the final model, denoted as |$model_{5}$|.
(iii) Evaluate the performance of the final model on the test set |$D_{test}$|.
Evaluation metrics
We utilize seven evaluation metrics to assess the performance of Enhancer-MDLF and compare it with that of other methods. These metrics encompass accuracy (ACC), balanced accuracy (BACC), area under the receiver operating characteristics (AUROC), Matthews correlation coefficient (MCC), sensitivity (Sn), specificity (Sp) and F1 score. The details of these evaluation metrics are provided in the Supplementary Information. In general, a higher value for these metrics indicates superior model performance.
RESULTS
Performance evaluation of combinatorial module in comparison with individual modules
Recent studies have underscored the superiority of leveraging multiple features over individual ones in sequence-based prediction tasks [41–44]. Consequently, Enhancer-MDLF, proposed in this study, incorporates a dna2vec module and a motif module to comprehensively extract information on enhancer sequences. To assess the effectiveness of this combinatorial module in improving the model’s performance for predicting cell-specific enhancers, we individually train the dna2vec module and the motif module utilizing training sets for each cell line. Subsequently, we evaluate the performance of these modules on independent test sets and draw comparisons with the performance of the combinatorial module.
Given that the datasets used in this study are imbalanced, we utilize the BACC metric rather than ACC to evaluate the performance of models. What surprised us is that Enhancer-MDLF can effectively improve the performance of the model by fusing the two modules while maintaining a reasonable computational cost (refer to Figure 3, Supplementary Tables S2 and S3). All six metrics exhibit improvement across the eight cell lines. Especially, the performance of two individual modules on the HMEC cell line is unsatisfactory, while the combinatorial module greatly improves the prediction performance. Furthermore, the training time for the combined module is shorter than that for the dna2vec module alone in the HSMM and NHEK cell lines (Supplementary Table S3), potentially due to an accelerated convergence speed facilitated by our early stopping strategy. Overall, these results indicate that leveraging multiple features indeed improves prediction performance, establishing Enhancer-MDLF as a powerful and robust tool for predicting cell-specific enhancers.
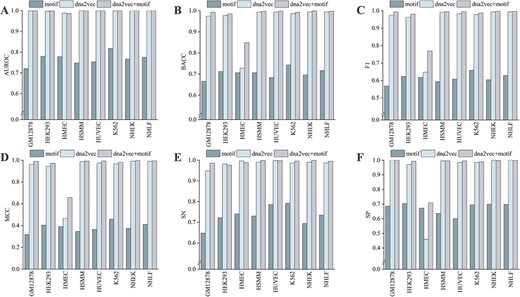
Performance evaluation of combinatorial module in comparison with individual modules. Panels (A–F) depict the evaluation of model performance across eight cell lines using metrics such as AUROC, BACC, F1 score, MCC, SN and SP.
Performance comparison with state-of-the-art method
To comprehensively assess the predictive capabilities of Enhancer-MDLF for cell-specific enhancers, we conduct a thorough comparison with Enhancer-IF, a model dedicated to cell-specific enhancer prediction, across eight cell lines. As mentioned in the introduction section, existing enhancer prediction methods predominantly rely on generic datasets, neglecting the crucial aspect of enhancer’s cell specificity. To validate the robustness of our model, we conduct performance comparisons for both methods on independent test sets and through 10-fold cross-validation (refer to Supplementary Information).
As can be seen in Figure 4 and detailed in Supplementary Table S5, Enhancer-MDLF consistently outperforms Enhancer-IF across all five metrics and all cell lines, demonstrating its significant superiority. The robust predictive performance of Enhancer-MDLF could be attributed to its unique multi-module fusion approach and a novel training strategy that incorporates comprehensive and in-depth sequence information. It is crucial to highlight that, in contrast to the training strategy of Enhancer-IF, we design a unique training strategy for Enhancer-MDLF. This strategy involves learning more distributions iteratively within a limited dataset. Our approach and Enhancer-IF employ identical input data and generate results on the same test dataset, ensuring a fair and meaningful comparison within the training process from an end-to-end perspective.
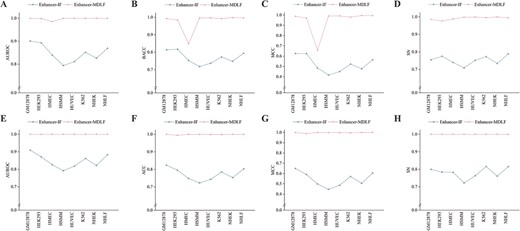
Performance comparison between Enhance-MDLF and Enhancer-IF on independent test sets. Panels (A–D) present the evaluation of model performance across eight cell lines using metrics such as AUROC, BACC, MCC and SN. Panels (E–H) present the evaluation of model performance across eight cell lines using metrics such as AUROC, BACC, MCC and SN.
Notably, a pronounced performance gap is observed between the 10-fold cross-validation and independent test sets for Enhancer-MDLF on the HMEC cell line. This discrepancy may arise from the distinct data distributions between the training and test sets, posing a challenge for the model to effectively generalize information learned from the training set to the test set. However, even under these challenging conditions, our method outperforms Enhancer-IF by 9.54% on the independent test set in terms of BACC.
However, to the best of our knowledge, there is no specific pattern observed in Enhancer-IF for the HMEC dataset. This lack of observation could be attributed to Enhancer-IF consistently performing within the range of 70–80% across multiple cell lines, which may imply limited knowledge acquisition, possibly concealing specific characteristics within the HMEC dataset. It is precisely due to the robust predictive capabilities of Enhancer-MDLF that these discrepant results may indicate that the enhancers within the HMEC cell line have more complex gene regulatory mechanisms. This suggests an intriguing direction for further exploration to help uncover the reasons behind enhancer cell specificity. Overall, these results demonstrate the robustness and effectiveness of Enhancer-MDLF in accurately predicting cell-specific enhancers.
Performance evaluation across cell lines
Previous studies have established that enhancers exhibit cell-specific functionalities [27, 45]. To explore the potential relationships among enhancers across different cell lines, we conduct a comprehensive cross-cell line performance evaluation to investigate the transferability of cell-specific models. Our approach involves training models on one cell line and evaluating the performance of Enhancer-MDLF and Enhancer-IF on the test sets of seven other cell lines. The results demonstrate that Enhancer-MDLF achieves optimal performance in terms of average BACC across all eight cell lines (Figure 5A, Supplementary Tables S6 and S7). Notably, the HEK293 model achieves an average BACC of 86.18% when predicting outcomes in other cell lines, indicating its capacity to transfer to other cell lines. Despite the prevalence of cell-specific enhancers observed in most instances, such as those identified in NHLF cell lines, it is important to acknowledge the existence of enhancers that exhibit significant similarities across certain cell lines. For instance, Enhancer-MDLF demonstrates satisfactory mutual predictive performance on the HUVEC and K562 cell lines, as well as on the NHEK and HMEC cell lines (Figure 5B). These results highlight a commonality of enhancers between some cell lines. Overall, enhancers generally display specificity among most cell lines, but there exist similarities between enhancers across specific cell lines. Therefore, investigating both cell-specific and non-cell-specific enhancers emerges as a critical avenue for unveiling insights into cell specificity and differentiation.
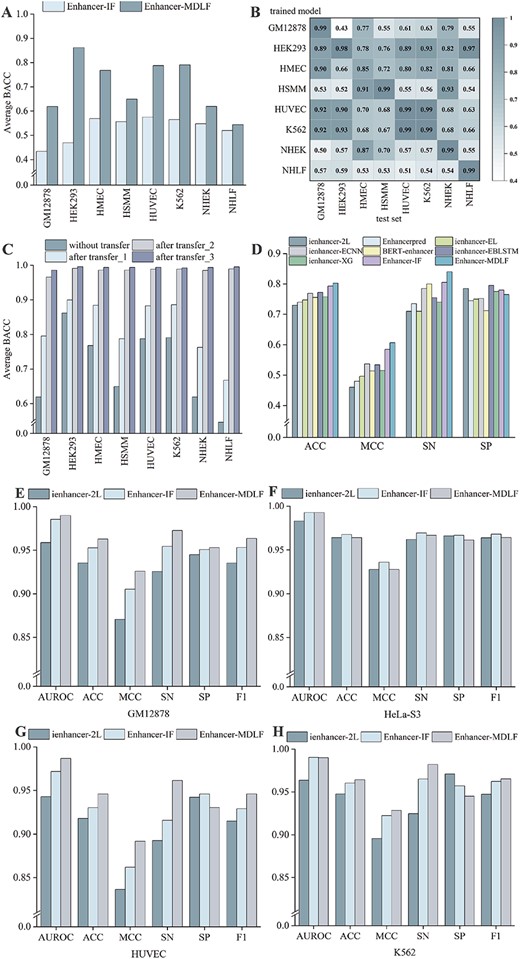
Comprehensive performance evaluation across multiple aspects. (A) Average BACC for cross-cell line prediction on eight cell lines. (B) Heat map of BACC in cross-cell line evaluation. Columns represent pre-trained models trained on different training sets, and rows represent testing on their own or other cell lines’ test sets. (C) Exploration of Transfer Learning Strategies: Three transfer learning strategies are presented to address the challenge of poor direct prediction performance in cross-cell line evaluation. (D) Performance comparison of Enhancer-MDLF with other methods on a universal dataset. (E–H) Performance comparison of Enhancer-MDLF with other methods on enhancer–promoter datasets.
Performance evaluation across cell lines with transfer learning
The challenge of predicting enhancers across different cell lines, attributed to the inherent cell specificity of enhancers, significantly hinders the exploration of gene regulatory mechanisms. To overcome this obstacle and advance the field, we further explore potential approaches. An emerging ML technique, transfer learning, proves promising as it leverages knowledge acquired from a source domain to enhance learning performance in a target domain. Notably, this technique has demonstrated success in predicting cell-specific enhancer–promoter interactions [46, 47]. To comprehensively evaluate the potential of transfer learning in the context of enhancer prediction, we adopt three strategies: transfer_1, transfer_2 and transfer_3. These strategies align with both traditional transfer learning methods and those previously utilized in a relevant study [48]. Detailed descriptions of these strategies are provided in the Supplementary Information, aiming to shed light on their effectiveness in mitigating the challenges posed by the cell specificity of enhancers.
We apply these three distinct transfer strategies separately to Enhancer-MDLF to validate their effectiveness and explore their practical applicability (Figure 5C). The results, as detailed in Supplementary Tables S8, S9 and S10 across eight cell lines, illustrate significant insights. Enhancer-MDLF with transfer_1 demonstrates notable improvement in prediction performance, albeit falling short compared with predicting enhancers in the same cell line. However, this outcome underscores the effectiveness of transfer learning. In contrast, Enhancer-MDLF with transfer_2, leveraging additional enhancer information from multiple cell lines during the model pre-training, exhibits exceptional performance. Compared with the model directly trained with random initial weights (Supplementary Table S11, Supplementary Figure S2), the results indicate that pre-training the model with enhancers from diverse cell lines yields more effective predictions in new cell lines. This is attributed to the more representative initial weights acquired from the pre-trained model, highlighting its potential for accurately labeling other unannotated data, especially under the constraint of limited annotated data size. Particularly noteworthy is the observation that Enhancer-MDLF with transfer_3 exhibits optimal performance across the eight cell lines.
These findings present a practical scenario for addressing enhancer-specific prediction challenges. If Enhancer-MDLF is pre-trained using as many (or even all) cell line enhancers following our transfer learning strategies, a comprehensive pre-trained model may be obtained, serving as an effective initial model for enhancer prediction tasks in various cell lines. Even with potential cost constraints in obtaining enhancers from numerous cell lines, Enhancer-MDLF with transfer_2 demonstrates robust performance with only a few cell lines’ enhancers. Overall, the utilization of transfer learning provides an effective and promising solution to overcome prediction challenges arising from enhancer specificity in practical applications. We have provided the pre-trained model, utilizing enhancers from these eight cell lines, available on GitHub for further exploration and utilization.
Performance evaluation on other datasets
To demonstrate the superiority of our approach comprehensively, we subject Enhancer-MDLF to evaluation using the generic dataset created by Liu et al. [21], a widely used benchmark in enhancer identification tasks. To maintain comparability with prior studies, we utilize the same training set, test set and evaluation metrics. The outcomes reveal that Enhancer-MDLF achieves optimal performance on the independent test set, with MCC, SN and ACC of 0.6067, 0.84 and 0.8025, respectively (Figure 5D, Supplementary Table S12). These results highlight the robustness and generality of Enhancer-MDLF, demonstrating its efficacy and versatility as a powerful tool for predicting enhancers in human cell lines.
Besides, existing studies have demonstrated that enhancers and promoters exhibit similar sequence structures [49, 50]. Leveraging datasets derived from iPro-WAEL [33], we conduct an investigation to assess whether Enhancer-MDLF effectively distinguishes between enhancers and promoters. Given the limitations of some enhancer prediction methods for this dataset, as detailed in the Supplementary Information, we restrict our comparison to Enhancer-MDLF, ienhancer-2L and enhancer-IF. The results unequivocally showcase Enhancer-MDLF’s robust capacity to discriminate between promoters and enhancers, effectively capturing the distinct information associated with these two regulatory elements despite their similar sequence structures (Figure 5E–H and Supplementary Table S14).
Exploration of TFBS motifs in enhancer regions
Investigating TFBS motifs within enhancer regions is crucial for understanding the regulatory mechanisms of TFs. TFs typically exert their transcriptional influence by recognizing specific TFBS motifs and binding to the regulatory regions of genes. Enhancers, along with the associated TFs, play a significant role in human diseases and biological processes [35, 51, 52]. To identify key motifs associated with enhancers, we utilize the Shapley Additive exPlanations (SHAP) framework [53, 54] to interpret the input features of the motif module in Enhancer-MDLF. The details of the SHAP framework are provided in the Supplementary Information.
Figure 6 displays the top 50 motifs with the most significant impact on the model’s output across eight cell lines. The x-axis represents the SHAP value, where a positive value indicates a positive effect on the model’s output, while a negative value indicates a negative effect. Taking SP2 in Figure 6A as an example, higher feature values predominantly cluster in the region where SHAP values >0, indicating that SP2 has a positive effect on predicting the samples as enhancers. It can be seen from Figure 6 that the contributions of different motifs to the model’s output vary across different cell lines, providing further insight into the cell specificity of enhancers at the motif level.
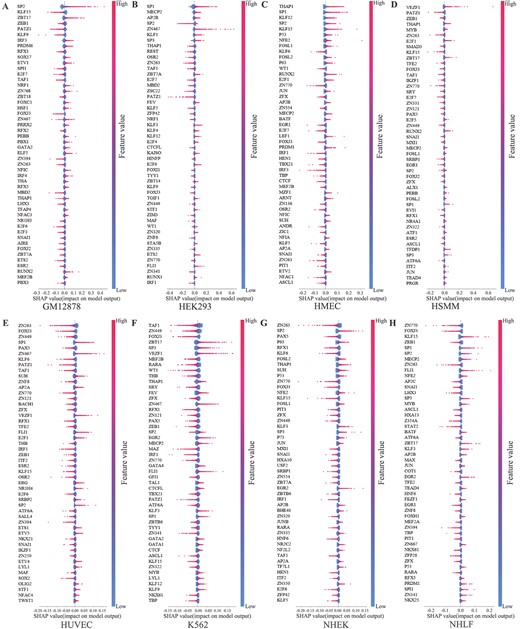
Analysis of top 50 important motif features across eight cell lines. (A–H) This study analyzes the top 50 motifs on eight cell lines that impact the model’s output utilizing the SHAP framework. The x-axis represents the SHAP values that impact the model’s output. Each point on the graph represents a sample, and the color of each sample point ranges from blue to red, representing the corresponding feature values of each sample. The color transition reflects the variation in feature values from low (blue) to high (red).
Besides, we find that certain important motifs identified in our study align well with findings from previous studies. For instance, the BACH1 motif emerges exclusively among the top 50 important features in the HUVEC cell line. Previous studies have shown that the heme-binding factor BACH1 can bind to multiple Maf recognition elements in the heme oxygenase 1 (HO-1) enhancer, thereby inhibiting its activity and participating in gene regulation [55]. Similarly, the GATA1 motif is uniquely present among the top 50 important features solely in the K562 cell line. Previous studies have revealed that the GATA1 protein binds to the GATA-A site in the intronic WT1 enhancer in vitro in K562 cells, transactivating the enhancer and markedly increasing the CAT reporter activity 10–15-fold [56].
To visualize the impact of these features on the model’s output, force plots of SHAP are employed, and specific details are available in Supplementary Figure S1. Additionally, to enhance the user’s interactive experience, we have provided HTML files on GitHub (https://github.com/HaoWuLab-Bioinformatics/Enhancer-MDLF). These files showcase the contributions of each feature to the model’s output on the test sets of eight cell lines. Users can interactively select various features and samples on the graph, facilitating a dynamic exploration of their influence on the model’s output.
Furthermore, the SP1 motif significantly contributes to enhancer predictions across numerous cell lines, as it has been shown to regulate chromatin loops between enhancers and distal promoters, thereby influencing transcriptional activity [57]. Additionally, our analysis reveals certain key motifs, such as RUNX and MAX, which have not been extensively studied but are highlighted in TargetFinder [58], These results underscore that enhancers can exert their effects through the involvement of certain proteins in specific cells, emphasizing the complexity of gene regulation involving enhancers.
DISCUSSION AND CONCLUSION
The identification of cell-specific enhancers is of significant importance for understanding cell-specific gene regulation and deciphering tissue development. In this study, we develop Enhancer-MDLF, a deep learning framework with multi-inputs, designed for accurate identification of cell-specific enhancers by integrating the dna2vec module and motif module. Experimental analyses demonstrate that Enhancer-MDLF outperforms existing methods across multiple cell-specific enhancer datasets, and significantly outperforms previous studies on general datasets and enhancer–promoter datasets, highlighting the robustness and versatility of Enhancer-MDLF. While our evaluations are constrained by the scale of the annotated dataset to specific cell lines, various experimental results instill confidence in the generalization ability of our model to extend to broader datasets. In summary, our proposed Enhancer-MDLF emerges as a superior and efficient tool for identifying enhancers.
Additionally, in the course of cross-cell-line evaluation, we observe a notable challenge wherein models trained on one cell line often exhibit unsatisfactory performance when applied to predict enhancers on other cell lines. This limitation stems from the inherent cell specificity of enhancers, suggesting that a model trained on a specific cell line achieves optimal prediction performance solely within that context, posing a substantial limitation in practical applications. To overcome this limitation, we endeavor to mitigate it through the implementation of transfer learning. The pre-trained model with transfer_2 achieves remarkably precise predictions with only a modest amount of annotated data (e.g. 292 samples, constituting 10% of the training set, in the NHLF cell line). In practical scenarios, the scarcity of annotated data in private datasets may face challenges for the effective application of supervised learning. While unsupervised learning methods, such as clustering, can partially emulate similar functionality, their accuracy often falls significantly below the standards required for practical applications. Consequently, we assert that this approach substantiates an effective strategy for addressing the challenges posed by the cell-specific nature of enhancers. Among the three transfer learning strategies employed, transfer_3, a pre-trained model trained on all cell lines, demonstrates the most promising performance. This outcome suggests that with a sufficiently extensive dataset encompassing a diverse range of cell lines as input to our model, the development of a universal pre-trained model for identifying human enhancers becomes a plausible avenue of exploration—a prospect that holds significant interest and potential in research endeavors.
Moreover, we employ a computational approach to analyze potentially important TFBS motifs associated with enhancers. Our analysis reveals several motifs that have undergone extensive study in previous research, such as BACH1, GATA1, SP1, RUNX and MAX. Additionally, we identify motifs like FOXJ3 and SP2, which significantly contribute to enhancer predictions across numerous cell lines but have received limited attention in prior studies. We infer that these less-explored motifs may play roles in gene regulation through intricate and as-yet-unidentified processes. While our current research conditions constrain further validation and exploration of the relationships between these new motifs and enhancer function, we believe these findings open new avenues for future enhancer-related research in the field of biology. Notably, Whalen et al. [58] extensively explored the critical role of YY1 in enhancer–promoter interactions by interpreting ML models. Later on, it was further confirmed that YY1 regulates enhancer–promoter chromatin loops [59].
We propose a novel deep learning framework, called Enhancer-MDLF, employing multi-module inputs to identify the cell-specific enhancers.
We confirm that Enhancer-MDLF substantially outperforms state-of-the-art predictors without the need for parameter tuning.
We incorporate three transfer learning strategies into our model to address the challenges posed by enhancer specificity for cross-cell lineage prediction.
We analyze the conservation and specificity of enhancers at the motif level, exploring the most important TFBS motifs within enhancer regions utilizing the SHAP framework.
ACKNOWLEDGMENTS
We thank members of the group for their valuable discussions and comments. The scientific calculations in this study have been done on the HPC Cloud Platform of Shandong University.
FUNDING
This work is supported by the National Natural Science Foundation of China (Grant No. 62272278 and 61972322), the National Key Research and Development Program (Grant No. 2021YFF0704103) and the Fundamental Research Funds of Shandong University. The funders did not play any role in the design of the study, the collection, analysis and interpretation of data or the writing of the manuscript.
AUTHOR CONTRIBUTIONS STATEMENT
H.W., Y.Z. and P.Z. conceived the experiments. Y.Z. and P.Z. conducted and analyzed the experiments. Y.Z. and P.Z. wrote the manuscript. H.W. reviewed the manuscript.
Author Biographies
Yao Zhang is currently a graduate student at the School of Software, Shandong University. Her research interests focus on bioinformatics and artificial intelligence.
Pengyu Zhang is currently pursuing a PhD degree at Xi'an Jiaotong University, China, with his research interests focused on computational biology, genomics, bioinformatics and deep learning.
Hao Wu is currently an associate professor at the School of Software, Shandong University. His research interests include artificial intelligence, data mining, and biomedical big data mining.
References
Author notes
Yao Zhang and Pengyu Zhang contributed equally to this work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}