




Abstract
Kinase inhibitors are crucial in cancer treatment, but drug resistance and side effects hinder the development of effective drugs. To address these challenges, it is essential to analyze the polypharmacology of kinase inhibitor and identify compound with high selectivity profile. This study presents KinomeMETA, a framework for profiling the activity of small molecule kinase inhibitors across a panel of 661 kinases. By training a meta-learner based on a graph neural network and fine-tuning it to create kinase-specific learners, KinomeMETA outperforms benchmark multi-task models and other kinase profiling models. It provides higher accuracy for understudied kinases with limited known data and broader coverage of kinase types, including important mutant kinases. Case studies on the discovery of new scaffold inhibitors for membrane-associated tyrosine- and threonine-specific cdc2-inhibitory kinase and selective inhibitors for fibroblast growth factor receptors demonstrate the role of KinomeMETA in virtual screening and kinome-wide activity profiling. Overall, KinomeMETA has the potential to accelerate kinase drug discovery by more effectively exploring the kinase polypharmacology landscape.
INTRODUCTION
Protein kinase-mediated cellular signaling pathways are responsible for a range of physiological and pathological processes. Kinase-targeted inhibitors have emerged as a promising therapeutic approach and have been applied clinically for a wide range of diseases, including cancer and inflammation [1, 2]. However, due to the structural similarity of kinases, many kinase inhibitors modulate multiple target proteins, resulting in either therapeutic effects or unwanted side effects [3]. Multi-target kinase inhibitors capable of modulating several signaling pathways with minimal polypharmacy-related risks hold paramount importance for the development of highly effective targeted therapies. This is particularly significant for addressing severe diseases that require modulation of multiple targets or overcoming therapeutic resistance [4, 5].
To design and optimize kinase-targeted drugs rationally, it is essential to gain insights into the selectivity or promiscuity of inhibitors. Kinome-wide activity profiling can provide multidimensional structure–activity relationships simultaneously against hundreds of kinases, but experimental screening for a broad spectrum of kinases remains time-consuming, technically challenging and costly [5, 6]. Moreover, this type of profiling is usually carried out during the later stages of drug discovery, making it challenging to offer guidance for molecular design. Recent advances in deep learning technology have made in-silico screening assays more reliable, allowing investigators to annotate compounds with the kinase spectrum more rapidly and cost-effectively in the early stages of drug discovery. Various models have been proposed to predict the polypharmacological effects of small molecule kinase inhibitors, such as naive Bayes (NB) [7], Random forest (RF) [8], Support vector machine (SVM) [7, 9–11] and Deep neural network (DNN) [12, 13]. In our previous research, we developed a multi-task deep neural network (MT-DNN) model employing molecular ECFPs to predict the inhibitory effects of small molecules against 391 kinases. This MT-DNN model exhibited remarkable performance superiority over single-task machine learning (ML) models, particularly for kinases with limited activity data. This was attributed to the inherent generalization capability of multi-task transfer learning [14]. Recently, Bao et al. [15] applied graph neural networks (GNNs) to MT learning to predict the inhibition profiles for small molecules against 204 kinases. GNNs can produce task-specific representation for molecules, surpassing models based on pre-defined descriptors. In addition to ligand-based strategies, multidimensional relationships can also be constructed from the perspective of heterogeneous networks that describe the compound-protein interactions, such as graph convolution networks (GCNs)-based IDDkin [16]. However, network-based methods have limitations in the case of large graphs, making it hard to diffuse information for large-scaled datasets of compound-kinase pairs.
Developing virtual profiling methodologies for kinases presents a challenge due to insufficient data for understudied kinases. This limitation is a significant bottleneck for accurately predicting a broader spectrum of kinases, particularly ‘dark’ or mutant kinases, where the number of known active compounds is insufficient for building accurate models. Additionally, existing models for predicting kinases are often not extensible, making it difficult to generalize to new kinases that have not been included in training (unseen tasks). Even if additional data are obtained from new literature or wet-lab experiments, traditional models struggle to incorporate this new data and extend the predictable kinase spectrum.
Meta-learning is a promising algorithm that can address the challenge of low-data, which leverages previous knowledge acquired from data to solve novel tasks quickly and efficiently [17]. Meta-learning has been adopted in some fields of drug discovery, such as molecule optimization [18], drug response prediction [19], chemical–protein interactions prediction [20], T-cell receptor-antigen binding recognition [21], etc. Therefore, meta-learning has the potential to become a new paradigm for the future drug discovery, which demands a closed-loop automated procedure that synergies between the components of the conventional discovery procedures and extensible AI methods [22].
This study presents KinomeMETA, a general framework for profiling the activity of small molecule kinase inhibitors across a panel of 661 kinases. One of the key challenges in virtual profiling methodologies for kinases is the limited availability of data, particularly for some mutant forms and understudied kinases. To address this challenge, KinomeMETA utilizes a modified meta-learning strategy integrated with a GNN. This strategy enables KinomeMETA to effectively learn from limited data and enhance its predictive capabilities, thus expanding the coverage of kinome-profiling. Additionally, the framework incorporates fast fine-tuning that enables it to generalize to unseen kinases with high accuracy, overcoming the limitations of previous machine learning models that were restricted to specific kinases within their modeling domains (Figure 1). We assess the performance of KinomeMETA for three different application scenarios, including kinome-wide activity profiling, mutant kinase selectivity prediction and rapid adaptation to previously unseen kinases. Moreover, we apply KinomeMETA in these practical scenarios, such as the discovery of new scaffold inhibitors for the membrane-associated tyrosine- and threonine-specific cdc2-inhibitory kinase (PKMYT1) and retrospective analysis of selective inhibitors for drug-resistant mutants of fibroblast growth factor receptors (FGFRs). By integrating it into the iterative predict-experiment cycles, we show that KinomeMETA can aid in the rational design of kinase inhibitors with a more favorable selectivity profile. This paves the way for the development of more effective targeted therapies.

The overall architecture of KinomeMETA.
METHOD
Processing kinase inhibitor data
The structure and activity data of kinase-compound pairs were collected and integrated from open-source databases, ChEMBL [23], PubChem [24] and DKKB [25] (Dark Kinase Knowledgebase). Briefly, those kinase-compound pairs passing the following filters were retained: (i) the assay type was binding, (ii) the bioactivity type was Ki, Kd, IC50, EC50 or %Inh and (iii) the target is single protein kinase whose confidence score is 9 in ChEMBL. For building classification model, bioactivity data were converted to two classes: positive (pKi/pKd/pIC50/pEC50 ≥ 6 or %Inh ≥ 80%) and negative (pKi/pKd/pIC50/pEC50 < 6 or %Inh < 80%). The distribution of compound concentrations for the %Inh data is illustrated in Figure S1 available online at http://bib.oxfordjournals.org/. Only kinases with positive compounds were kept. Specifically, the final datasets encompass over 612 000 manually curated bioactivity datapoints, spanning over more than 160 000 distinct compounds and 661 kinases, including 118 mutants. The specific data statistics are shown in Figure 2. As meta-learning reduces the demand for training data, a large number of kinase domains with data records less than 50 were retained to build a broader task panel, including kinases that have not been fully studied and some rare mutation types (Figure 2A and B). There are 543 wild-type kinases that mainly belong to Homo sapiens, Mus musculus and Rattus norvegicus (Figure 2C), along with 118 mutations involving 44 different kinases, mostly human tyrosine kinases (TK) and tyrosine kinase-like (TKL) kinases (Figure 2D).
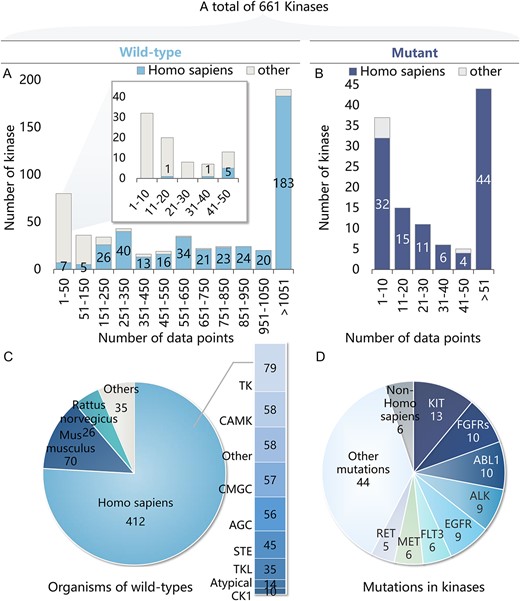
Statistics of the dataset. (A) Statistics of wild-type kinase data: Histogram illustrates the distribution of tasks across different ranges of data sizes. Each task represents a class of kinase and data points correspond to the inhibitors of the kinase. (B) Statistics of mutant kinase data. (C) Distribution of organisms and subfamilies of wild-type kinases. (D) Distribution of subfamilies of mutant kinases.
Negative sampling
In addition, we have built a credible set of negative samples to reduce false positive predictions caused by the imbalanced distribution of samples. In real-world virtual screening scenarios, negative results often greatly outnumber positive results, but the experimentally validated negative samples are often insufficient. Here, three property-matched decoys [26] have been sampled for each positive data from BindingDB database, resulting in a negative–positive ratio of approximately 1:5 in the final datasets. The distribution of molecular weight and logP of decoys is relatively consistent with that of the original samples (see ‘Negative sampling’ in Supplementary materials and Figure S2 available online at http://bib.oxfordjournals.org/). This approach encourages the model to learn an information-rich representation of molecules rather than biases in their properties, thus improving its prediction accuracy.
Metrics
For model quality assessment, the auROC (area under the ROC curve), Recall, Precision, F1-score, Matthews correlation coefficient (MCC) and balanced accuracy (BACC) were evaluated (Table 1). The MCC is the metric we mainly discussed in this work, as is a more reliable statistical rate which produces a high score only if the prediction obtained good results in all of the four confusion matrix categories. It can produce a more informative and truthful score in evaluating label-imbalanced binary classifications than auROC, accuracy and F1-score [27].
Description of the evaluation metrics
| Evaluation metric | Equation |
|---|---|
| Recall | TP/ (TP + FN) |
| Precision | TP/ (TP + FP) |
| F1-score | |$2\times\displaystyle\frac{Precision \times Recall}{Precision + Recall} $| |
| MCC | |$\displaystyle\frac{ TP\times TN-FP\times FN}{\sqrt{(TP+FP)(TP+FN)(TN+FP)(TN+FN)} } $| |
| BACC | (|$\displaystyle\frac{TP}{TP+FN}+\frac{TN}{FP+TN} $|)/2 |
| Evaluation metric | Equation |
|---|---|
| Recall | TP/ (TP + FN) |
| Precision | TP/ (TP + FP) |
| F1-score | |$2\times\displaystyle\frac{Precision \times Recall}{Precision + Recall} $| |
| MCC | |$\displaystyle\frac{ TP\times TN-FP\times FN}{\sqrt{(TP+FP)(TP+FN)(TN+FP)(TN+FN)} } $| |
| BACC | (|$\displaystyle\frac{TP}{TP+FN}+\frac{TN}{FP+TN} $|)/2 |
aTP is the number of correctly predicted actives (true positives). TN is the number of correctly predicted inactive (true negatives). FP is the number of incorrectly identified actives (false positives), and FN is the number of incorrectly identified inactive (false negatives).
Description of the evaluation metrics
| Evaluation metric | Equation |
|---|---|
| Recall | TP/ (TP + FN) |
| Precision | TP/ (TP + FP) |
| F1-score | |$2\times\displaystyle\frac{Precision \times Recall}{Precision + Recall} $| |
| MCC | |$\displaystyle\frac{ TP\times TN-FP\times FN}{\sqrt{(TP+FP)(TP+FN)(TN+FP)(TN+FN)} } $| |
| BACC | (|$\displaystyle\frac{TP}{TP+FN}+\frac{TN}{FP+TN} $|)/2 |
| Evaluation metric | Equation |
|---|---|
| Recall | TP/ (TP + FN) |
| Precision | TP/ (TP + FP) |
| F1-score | |$2\times\displaystyle\frac{Precision \times Recall}{Precision + Recall} $| |
| MCC | |$\displaystyle\frac{ TP\times TN-FP\times FN}{\sqrt{(TP+FP)(TP+FN)(TN+FP)(TN+FN)} } $| |
| BACC | (|$\displaystyle\frac{TP}{TP+FN}+\frac{TN}{FP+TN} $|)/2 |
aTP is the number of correctly predicted actives (true positives). TN is the number of correctly predicted inactive (true negatives). FP is the number of incorrectly identified actives (false positives), and FN is the number of incorrectly identified inactive (false negatives).
The implementation of KinomeMETA
KinomeMETA is a general framework designed to assess the probabilities of a molecule inhibiting a panel of kinases. The framework integrates a model-agnostic meta-learning algorithm, Reptile [28], with Attentive-FP [29], a graph attention neural network-based molecular representation model (see ‘The details of KinomeMETA’ in Supplementary materials available online at http://bib.oxfordjournals.org/). Through this algorithm, a well-generalized meta-learner is generated by training it on diverse kinase tasks intrinsically related. Consequently, the meta-learner can transfer new tasks with just a few training samples, making it suitable for few-shot learning for kinases not thoroughly studied.
To implement KinomeMETA, the dataset comprising 661 kinase tasks was divided into training tasks |${T}^{train}$|, validation tasks |${T}^{valid}$| and test tasks |${T}^{test}$| at the task level. |${T}^{train}$| was used to train a meta-learner with the optimal initialization parameters, while both |${T}^{valid}$| and |${T}^{test}$| were used to build kinase-specific base-learners for evaluating the generalization of the meta-learner in fine-tuning scenarios. Specifically, |${T}^{valid}$| was used for optimizing the meta-learner, whereas |${T}^{test}$| was used for the comparisons with other methods. At the instance level for each kinase task, 20% of the compounds within a task were segregated as ‘Test’ for testing the performance of the kinase-specific base-learners. In the training tasks (|${T}^{train}$|), the remaining 80% compounds, referred to as ‘Train’ were further randomly divided into a support set and query set at a ratio of 4:1, similar to a conventional meta-learning process (Figure 3A).
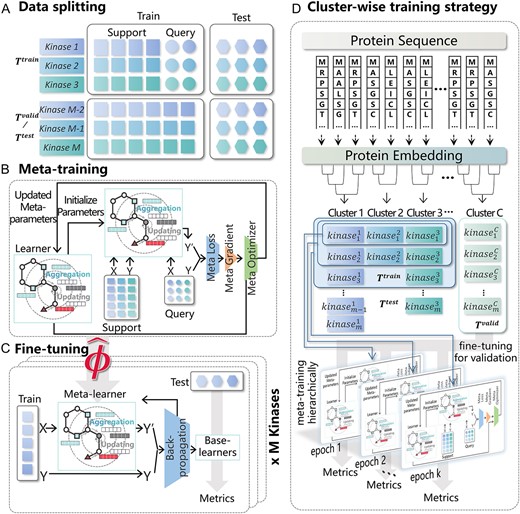
Data splitting strategy and training process. (A) Data Splitting Strategy. (B) The meta-training phase. (C) The fine-tuning phase. (D) Cluster-wise training strategy.
During the meta-training phase, the meta-learner performs multiple gradient descents within the task by utilizing the support set. This process generates a model that updates its parameters based on the prediction errors made on the query set, as shown in Figure 3B. In the fine-tuning phase, the meta-learner undergoes additional gradient steps on the support set of M test tasks, creating M task-specific base-learners that can effectively classify examples in the query set for each of the M test tasks, as shown in Figure 3C. To build KinomeMETA for the specific case of N-way K-shot few-shot classification problems, we selected the strategy of 2-way 3-shot classification. This means that for each of the two classes of compounds (N = 2), namely positive and negative, we sampled three compounds (K = 3, determined through hyperparameter searching as shown in Figure S3 and Table S1 available online at http://bib.oxfordjournals.org/) for the selected kinase tasks.
Cluster-wise training strategy
Task heterogeneity is a critical challenge in meta-learning, which is limited to settings where the current task is closely related to previous ones [30]. However, distribution heterogeneity on sequences and structures of the protein kinases [31], means that in practical transfer learning scenarios the new kinase task might not be tightly linked to previous kinases learned in the meta-learner. In addition, given the imbalanced kinase groups, overfitting to a dominant group such as TK will impair global generalization. Thus, we do not need to train the meta-model with a fixed partition ratio of tasks to establish a generalizable meta-learner, but only with a few representative kinase tasks. We have modified the meta-training strategy by training the meta-learner hierarchically in a task cluster-wise manner, based on the hierarchical cluster of the task representations for 661 kinases. We sampled kinases from every cluster as equally as possible and progressively added them into the meta-learner as training tasks (Figure 3D for details, as well as ‘Building meta-learner in a cluster-wise manner’ and Figure S4 in Supplementary materials available online at http://bib.oxfordjournals.org/).
RESULTS
KinomeMETA demonstrates high performance for kinase profiling
During the meta-training phase, the meta-learner was established in a task cluster-wise manner, involving 113 kinases. The meta-learner shows an average MCC of 0.73 on validation task, outperforming the baselines by adding random tasks iteratively or by adding all tasks in the first iterations (see ‘Building meta-learner in a cluster-wise manner’ and Figure S4c in Supplementary materials available online at http://bib.oxfordjournals.org/). By progressively training the meta-learner in a cluster-wise manner, we can balance the knowledge learned across heterogeneous kinase tasks, preventing the meta-model from being dominated by closed tasks in the majority cluster and improving its generalization.
In the subsequently fine-tuning phase, the meta-learner was optimized for unseen tasks originating from a diverse attribute space. To enable comprehensive polypharmacology profiling across the kinome, a panel of fine-tuned models encompassing 661 kinases including training, validation and test tasks needed to be constructed and assessed. We assess the performance of KinomeMETA from three different perspectives, corresponding to different application scenarios including kinome-wide activity profiling, mutant kinase selectivity prediction and rapid adaptation to previously unseen kinases. We implemented a baseline method that uses a multi-task transfer learning approach based on GNN. The multi-task GNN model (MTGNN) is based on our previous work [14], which was initially pre-trained on |${T}^{train}$| and subsequently fine-tuned on each task in |${T}^{test}$|, following the similar procedure as KinomeMETA. Further implementation details can be found in the Supplementary materials and Figure S5 available online at http://bib.oxfordjournals.org/.
Overall performance on kinome-wide tasks
KinomeMETA outperforms MTGNN on both meta-training tasks |${T}^{train}$| and unseen tasks |${T}^{test}$|. It adapts well to tasks within the training field (Figure 4A) while maintaining a high level of generalization to unseen kinases. Figure 4B showed that most kinase-specific learners built from KinomeMETA have MCCs above 0.6 (452 out of 517 test tasks). This suggests that KinomeMETA can provide accurate predictions for kinome-wide activity profiles. Specifically, for 412 predictable wild-type human kinases, MCCs of 189 kinases are higher than 0.8, while only 12 of them have MCCs less than 0.4 (Figure 4C and Table S5 provides details on each task). These results confirmed that KinomeMETA has overcome the limitations of previous models that could only predict a small range of targets, achieving the goal of large-scale kinase prediction and selectivity analysis.
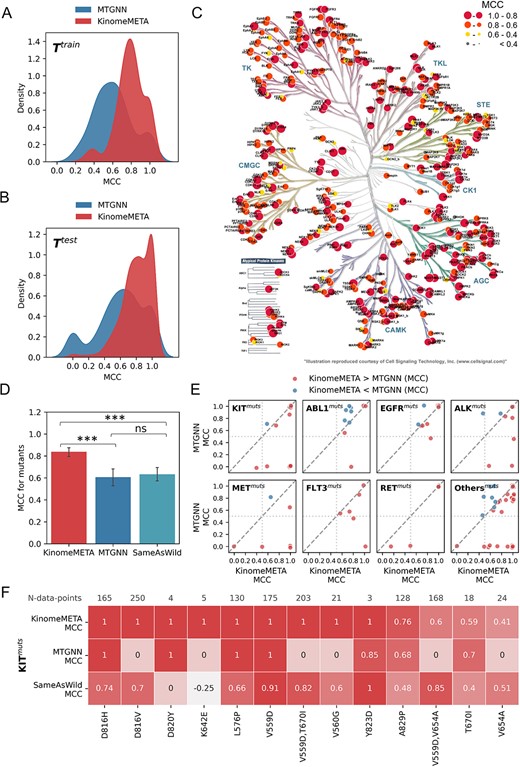
The performance of KinomeMETA on overall tasks and mutations tasks. (A) The MCC-based performance comparison between KinomeMETA and MTGNN on the training tasks. (B) The MCC-based performance comparison between KinomeMETA and MTGNN on test tasks. (C) The phylogenetic tree of 412 predictable wild-type human kinases, with each point representing the MCC of a specific kinase. (D) The MCC-based performance comparison between KinomeMETA, MTGNN and ‘SameAsWild’ on human kinase mutation tasks. The statistical analyses were performed by one-tailed Wilcoxon Signed-Rank test. (*) 0.01 < p ≤ 0.05; (**) 0.001 < p ≤ 0.01; (***) p ≤ 0.001. (E) The performance of KinomeMETA and MTGNN for kinases with over four mutation types (KITmuts, ABL1muts, EGFRmuts, ALKmuts, METmuts, FLT3muts and RETmuts), as well as for kinases with less than four mutation types (Othersmuts). The scatter plot visualizes the relationship between KinomeMETA and MTGNN MCC values for different mutants of corresponding wild-type kinases. Each point on the plot represents a mutant form. (F) The performance of KinomeMETA for KIT mutants, comparing it to MTGNN and the ‘SameAsWild’ prediction, all measured by MCC.
Performance on mutations tasks
Generalizing to mutants of kinases is a challenging task. This challenge stems not only from insufficient data but also from inherent conflicts resulting from data distribution differences among tasks. In other words, molecules for wild-type kinases and corresponding mutant tasks may have some opposing labels due to drug resistance caused by mutations. In transfer learning, these distribution differences can lead to negative transfer, especially when model parameters are extensively shared across all tasks, such as in MTGNN [32]. To examine whether KinomeMETA can adapt to the different distribution between wild-type kinases and their mutant forms, we compared it with MTGNN and another baseline model called ‘SameAsWild’. This baseline represents an overfitted model that can only learn the label distribution from wild-type kinases and transfer this ‘naïve knowledge’ to their mutant forms.
When compared to MTGNN, KinomeMETA demonstrates significantly superior performance on mutation tasks (Figure 4D). Further evaluation for each kinase with at least four mutation types reveals robust performance of KinomeMETA for the majority of mutations. As shown in Figure 4E, KinomeMETA consistently maintains a higher predictive performance than MTGNN across the majority of mutations, as evidenced by the significantly greater number of red points, which are predominantly concentrated in the first and fourth quadrants. In contrast, MTGNN shows MCCs of 0 for many tasks. Comparing with ‘SameAsWild’, KinomeMETA also exhibits substantial superiority (Figure 4D). This is true even when modeling data is scarce, as exemplified by KIT-D820Y, KIT-K642E and KIT-Y823D shown in Figure 4F (refer to Figure S6 for labels of samples in KIT and its mutant forms and Figure S7 displays the heatmaps for other kinases’ mutant forms available online at http://bib.oxfordjournals.org/). In addition, MTGNN demonstrates moderate performance similar to ‘SameAsWild’ (Figure 4D) and even performs worse than ‘SameAsWild’ in certain mutant tasks due to negative transfer. These results demonstrate that KinomeMETA can identify the drug resistance of wild-type kinase’s active compounds to their mutant forms, rather than simply overfitting to the training data. This ability can be attributed to the advantage of the meta-learning approach employed by KinomeMETA, which mitigates overfitting through two different time scales of learning: slow learning for common features of molecular interactions with kinases during meta-training phase, and fast learning for task-specific aspects of kinase inhibition in the fine-tuning phase. This combination allows meta-learning to develop an understanding of a wide range of kinase tasks from more stable parts, while also enabling faster adaptation for changes with less data [33]. This ability is crucial for the successful screening of inhibitors targeting drug-resistant kinase mutants.
Performance on few-shot learning tasks
We evaluated KinomeMETA’s performance on few-shot kinase tasks that were not incorporated during the model’s training phase. Few-shot tasks corresponded to kinases with limited characterization, making them particularly worth exploring. We conducted a statistical analysis of model performance across kinases with varying data sizes and found that KinomeMETA is effective for kinases with less than 50 active samples (as shown in Figure 5A). In fact, KinomeMETA significantly outperformed MTGNN for all ranges of data sizes (Figure 5B), showcasing its remarkable ability to generalize and rapidly learn from a limited set of examples to tackle novel tasks quickly.
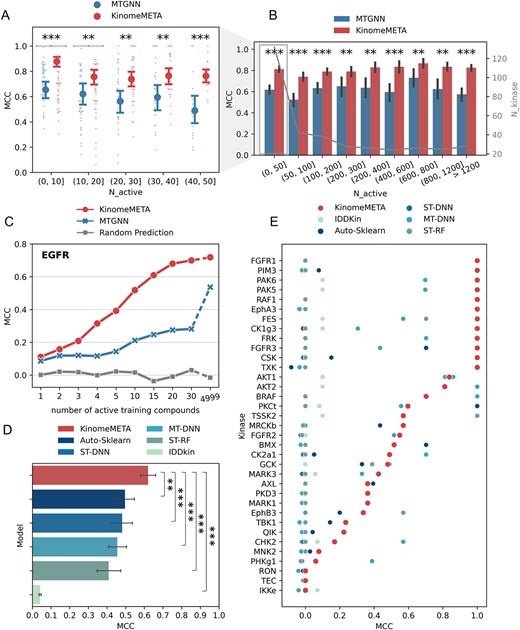
KinomeMETA’s performance on few-shot learning tasks and comparison with previous models. (A) Performance comparison between KinomeMETA and MTGNN on few-shot learning setting, where the range of training active compounds is from 1 to 50. For each range, small scatters represent the Matthews correlation coefficient (MCC) of each task in the corresponding range, while the large scatters with error bars represent the average and standard deviation of them. (B) Overall performances comparison between KinomeMETA and MTGNN using a bar plot. Each bar indicates the average MCC of tasks within the underlying range, and its error bar indicates standard deviation of them. (C) Performance comparison between KinomeMETA and MTGNN in the data-adding experiment, in terms of MCC. (D) Comparing KinomeMETA’s performance with RF, MT-DNN, IDDkin, ST-DNN and Auto-Sklearn based on the dataset of IDDkin. Bar plots of model performance on MCC. A bar indicates the average MCC of all modelling kinase tasks, and its error bar indicates standard deviation of them. (E) Comparing KinomeMETA’s performance with RF, MT-DNN, IDDkin, ST-DNN and Auto-Sklearn based on the dataset of IDDkin on few-shot kinase tasks (< 10 active data points) measured by MCC. All Statistical analyses were performed by one-tailed Student’s t-test. (*) 0.01 < p ≤ 0.05; (**) 0.001 < p ≤ 0.01; (***) p ≤ 0.001.
We designed a data-adding experiment to demonstrate the usage of KinomeMETA in a low-data scenario for drug discovery. Specifically, we selected EGFR, a kinase with a large dataset of 6250 active compounds in |${T}^{test}$| to simulate the low-data scenario. We trained EGFR-specific base-learners with increasing data size to determine when we could achieve satisfactory performance. This process is referred to as a ‘data-adding experiment’. We evaluated the performances of KinomeMETA, MTGNN, and a ‘random prediction’ baseline model (randomly assigned active/inactive labels to test compounds and calculate the metrics with true labels) on 20% of the compounds, which were randomly and consistently split. The results are shown in Figure 5C measured by MCC and Figure S8 measured by auROC. With one-shot learning setting, i.e. training with one active and one inactive compound, KinomeMETA’s EFGR model achieved an MCC of 0.11 and an auROC of 0.62, while the ‘random prediction’ model achieved an MCC of 0 and an auROC of 0.5. These findings demonstrated that KinomeMETA can effectively learn from a very small amount of data. Furthermore, KinomeMETA was able to be optimized more quickly than MTGNN when the data size was increased by adding an active and an inactive data point each time. The performance gap between KinomeMETA and MTGNN increased as well. Within only 20-shot learning, KinomeMETA achieved a strong performance (MCC = 0.68, and auROC = 0.91), which was close to the performance achieved when training with all 4999 active compounds in the training set (MCC = 0.72 and auROC = 0.92). In contrast, MTGNN had slower performance growth and earlier convergence, resulting in significantly lower performance metrics compared to KinomeMETA.
KinomeMETA demonstrated impressive performance on different fine-tuning shot numbers, highlighting its practical value in low-data scenarios for understudied kinases. Additionally, modeling with few-shot kinases offers an added benefit of quickly training new models for previously unseen kinases. With as few as 10 samples, KinomeMETA can be used to construct a model with decent performance for a new task. This means that KinomeMETA can be continuously extended to effectively address the problem of previous machine learning models that could only predict specific kinases within their modeling domain.
KinomeMETA outperforms previous kinase profiling models
We conducted a comparative analysis of KinomeMETA and several machine learning methods for kinase profiling, including RF [8], multi-task DNN [14] (MT-DNN) and network-based IDDkin [16]. Additionally, we evaluated two widely used general target prediction methods: single-task DNN (ST-DNN) and Auto-Sklearn [34]. Since IDDkin is not suitable for large graphs due to the use of the GCN, we used the modelling dataset reported in IDDkin to train the meta-learner of KinomeMETA and fine-tuned it on these kinases. The remaining methods were also trained using the same dataset as IDDkin. The implement of each method can be found in ‘Baseline models’ in Supplementary Materials, and the parameter settings for each model are provided in Table S2 available online at http://bib.oxfordjournals.org/.
The results show that KinomeMETA outperformed all other methods significantly (Figure 5D and Table S3). Figure 5E illustrates the kinase-specific performance of each algorithm on 35 few-shot learning tasks that have less than 10 positive training samples. KinomeMETA exhibited superior performance on 22 tasks, with MCC over 0.4 for 23 tasks, demonstrating its robustness. Single-task algorithms exhibit quite unstable performances on these tasks. Auto-Sklearn achieved MCC values higher than 0.4 on four tasks, while ST-DNN failed to achieve values higher than 0.4 on any task. In contrast, the performance of multi-task algorithms, such as MT-DNN, was relatively stable on few-shot kinases, with 11 kinases’ MCC above 0.4, as previously reported [14]. However, the average MCC of MT-DNN is moderate, possibly due to negative transfer for some tasks caused by the hard parameter sharing mechanism. IDDkin, the network model, performed only slightly better than random prediction when measured by MCC. Although IDDkin has previously reported high auROC values, it had very low F1-scores, which can be attributed to treating all unknown kinase-compound pairs as negative samples during training, resulting in biased prediction. In conclusion, KinomeMETA consistently outperformed other models in most kinases, particularly in those with less known data, regardless of whether it is a single-task, multi-task or network algorithm. The robustness of KinomeMETA further supports its ability to enhance kinome-wide virtual profiling.
KinomeMETA-aided discovery of kinase inhibitors
Discovery new scaffold inhibitors for PKMYT1.
The Dark Kinase Knowledgebase (DKKB; https://darkkinome.org) is specifically focused on developing a better understanding of the approximately 160 understudied kinases whose function in human biology is poorly understood [25]. Among these, the membrane-associated tyrosine- and threonine-specific cdc2-inhibitory kinase (PKMYT1), a compelling target for treating specific types of DNA damage response-related cancers due to its established synthetic lethal connection with CCNE1 amplification [35], is of particular interest and has been selected in the early stages of DKKB project. PKMYT1 is a highly selective kinase that is difficult to inhibit compared to other kinases [36, 37]. According to a comprehensive analysis of kinase inhibitor selectivity by Davis et al. [36] only 4.17% of the tested compounds inhibit PKMYT1, while only 2% of all the tested kinases have such low propensity for compound binding. Platzer et al. [37] tested 800 compounds from kinase inhibitor libraries PKIS I and II and identified only 10 PKMYT1 inhibitors with IC50 values in the nanomolar and micromolar range (e.g. GW559768X in Figure 6C). Currently, PKMYT1 inhibitors are limited to several scaffolds, including pyridopyrimidine, azastilbenes, 4-aminoquinolines, aminopyrimidines and pyrrolo[2,3-b]pyridin-2-amine (Figure 6C) [38]. Apart from RP-6306 [39], the first potent, selective, and orally bioavailable PKMYT1 inhibitor reported in 2022, most of these inhibitors are pan-kinase inhibitor.
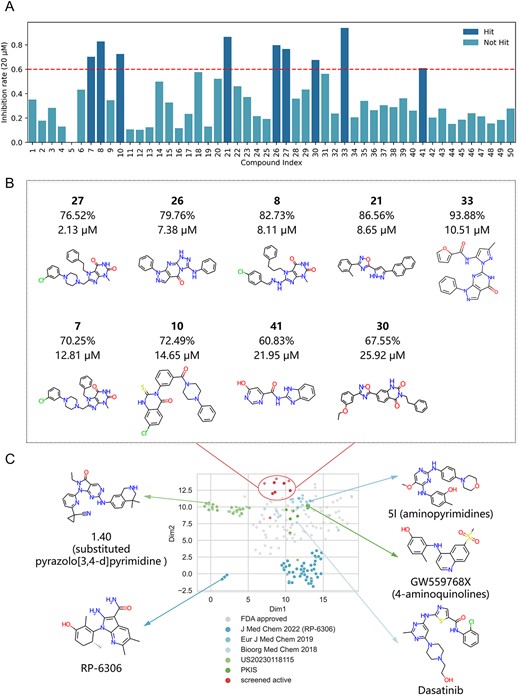
The screening results of PKMYT1. (A) The inhibition rates for 50 screened compounds at an inhibitor concentration of 20 μM. (B) The inhibition rates, IC50 and structures for nine compounds with inhibition rate > 60%. (C) Uniform manifold approximation and projection (UMAP) and representative chemical structures for FDA approved kinase-targeted drugs, new identified PKMYT1 inhibitors, previously reported PKMYT1 inhibitors.
The lack of potent and selective PKMYT1 inhibitor with new scaffolds has hindered further research. To address this challenge, a virtual screening with KinomeMETA combined with molecular docking was performed to discover new PKMYT1 inhibitor scaffolds. From HTS Compound collection of Life Chemical Screening Library, which contains over 525 000 drug-like compound, 50 candidates were selected and purchased for further experimental evaluation (see ‘Virtual screening and experimental evaluation for PKMYT1’ in Supplementary materials; details of these compounds are shown in Table S6 available online at http://bib.oxfordjournals.org/). Nine compounds showed inhibition rate > 60% in the initial screening performed at 20 μM inhibitor concentration (Figure 6A). The IC50 values were determined for these hits using the FP binding assay, and the potent compounds with inhibitory activity in the low-micromolar range were confirmed (Figure 6B and Figure S9 available online at http://bib.oxfordjournals.org/). Despite the difficulty of inhibiting PKMYT1, as evidenced by the hit rate of 4.17% reported by Davis et al. [36] and 1.25% (10/800) reported by Platzer et al. [37] from known kinase inhibitor libraries, KinomeMETA can distinctly enrich PKMYT1 inhibitors from a large-scale compound library with a high hit rate of 18.00% (9/50), demonstrating its effectiveness for virtual screening.
As shown in Figure 6C, the spatial projection of newly identified PKMYT1 inhibitors is far from that of known PKMYT1 inhibitors and FDA approved kinase-targeted drugs. This demonstrated that KinomeMETA can identify new PKMYT1 inhibitors with structural novelty, providing different scaffolds such as xanthine (compound 27, 8 and 7), pyrazolo[3,4-d] [1, 2, 4]triazolo[4,3-a]pyrimidin-5-one (compound 26), 1,2,4-oxadiazole (compound 21). These scaffolds are not present in any known PKMYT1 inhibitors and therefore are not used in the training of PKMYT1’s model. However, KinomeMETA can still identify these scaffolds since some of them have been identified active for other kinases and trained in the meta-learner. For example, xanthine and its derivatives have been revealed as potential apoptotic antitumor agents that inhibit EGFR, KDR and BRAF [40–42]. KinomeMETA has incorporated a methylxanthines derivative in the meta-training process on KDR task. Similarly, 1,2,4-Oxadiazole derivatives have been reported to target EGFR and c-Met degradation in TKI resistant NSCLC [43]. In contrast, to the best of our knowledge, pyrazolo[3,4-d] [1,2,4]triazolo[4,3-a]pyrimidin-5-one has not been included in any previously approved drugs or known kinase inhibitors. This new scaffold can be referred to as ‘settler’ that contributes to important advancements for new therapies [44].
In summary, the identification of PKMYT1 inhibitors with new scaffolds highlights the KinomeMETA’s effectiveness in large-scale virtual screening scenarios. KinomeMETA significantly improves the hit rate in experimental screens for challenging-to-inhibit kinases with limited available inhibitors. Additionally, KinomeMETA’s strong generalization ability, which results from its fitting to diverse chemical spaces during meta-training process, facilitates innovation in small molecule drugs by enabling the discovery of structurally novel compounds. This potential for target-specific therapies holds promise for the development of novel and effective treatments.
Retrospective analysis of the inhibitors targeting understudied FGFR mutants
In the treatment of various types of tumors, although FGFRs inhibitors have been successfully used [45], gain-of-function mutations in FGFRs can lead to drug resistance. Identifying new agents that target the gatekeeper and other high incidence resistant mutant of FGFRs could provide therapeutic promise for a subclass of patients of lung cancers, gastric cancer, breast cancers, etc. [46, 47].
We conducted a retrospective analysis to assess the effectiveness of KinomeMETA in identifying effective and selective inhibitors for FGFR drug-resistant mutants. In our previous study, we obtained a high-potency lead compound, compound 15 [48], a multi-target FGFRs inhibitor that acts as a multi-target inhibitor of FGFRs. We subsequently synthesized and evaluated a series of derivatives of compound 15 [49, 50]. Among these compounds, we identified compound 2e as a highly selective inhibitor for the drug-resistant mutant FGFR2-N549H (Figure 7A) [50]. The experimental profiling from Kinomescan profiling by Eurofins Discovery against a panel of 412 kinases at 1 μM concentration indicates that compound 2e could selectively inhibit FGFR2-N549H mutant with an IC50 values of 16 nM, and has no substantial inhibitory effect against 95.7% (397/415) tested kinases. Here, we utilized KinomeMETA to predict the kinome spectrum of compound 15 and compound 2e, aiming to evaluate its ability to profile the kinome-wide potency and selectivity of these compounds.
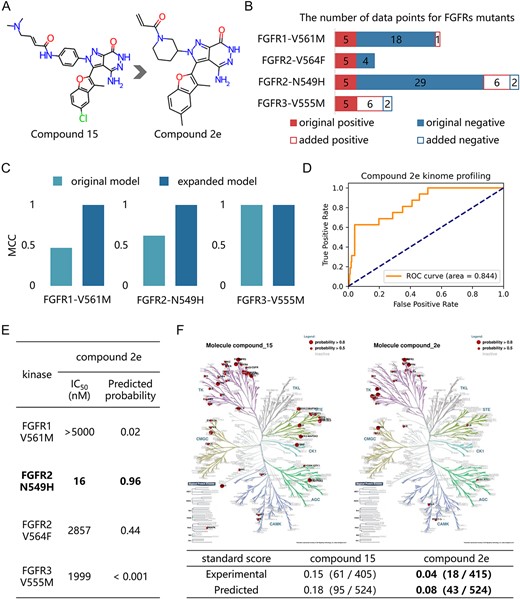
The retrospective analysis of the inhibitors targeting understudied FGFR mutants. (A) Chemical structures of compound 15 and 2e. (B) Distribution of data points in the base-learners for FGFR1-V561M, FGFR1-V564F, FGFR2-N549H and FGFR3-V555M. The solid bars indicate the original number of samples, while the hollow bars indicate the number of samples added. (C) Model performance measured by MCC for the original and expanded base-learners for FGFR1-V561M, FGFR2-N549H and FGFR3-V555M. (D) Model performance measured by ROC curve of compound 2e against 331 predictable kinases screened in the Kinomescan panel. (E) Inhibitory activities and predicted active probability of compound 2e against FGFR mutants. (F) Phylogenetic tree depicting predicted profiles for compound 15 (left) and compound 2e (right). The table presents standard scores obtained from the experimental and predicted profiles for these compounds.
There is limited knowledge about selective inhibitors for some understudied FGFR mutants, such as FGFR1-V561M, FGFR2-V564F, FGFR2-N549H and FGFR3-V555M. These mutants pose a few-shot learning challenge for KinomeMETA. To address this, we fine-tuned the kinase-specific models according to newly added data. The base-learners for FGFR1-V561M, FGFR2-N549H and FGFR3-V555M were expanded through fast adaption to recently reported compounds (Figure 7B) [51–57]. Compared to the moderate performance of the original models of FGFR1-V561M and FGFR2-N549H, the performance of these finetuned models improved, while the performance of FGFR3-V555M remained consistently high (Figure 7C). Further details regarding the data and model performance of the FGFRs are shown in Table S4 available online at http://bib.oxfordjournals.org/.
Using these fine-tuned models, we conducted kinome profiling to determine the inhibitory probabilities of compound 15 and its derivatives against predictable human kinases. These predictions agree well with the experimental results for compound 2e. For the 331 predictable kinases in the Kinomescan panel, the auROC between experimental and predicted profile of KinomeMETA is up to 0.84 (Figure 7D). Finally, KinomeMETA identified compound 2e as a high-selective inhibitor of drug-resistant mutant FGFR2-N549H, while inactive toward FGFR1-V561M, FGFR2-V564F and FGFR3-V555M (Figure 7E). Moreover, we calculated the standard scores [3] (see ‘Standard score’ in Supplementary materials available online at http://bib.oxfordjournals.org/) of both the lead compound 15 and the optimized compound 2e. The experimental and predicted profiles for these compounds showed consistent trends, indicating the reliability and accuracy of KinomeMETA in assessing selectivity (Figure 7F). All experimental and predicted results are shown in Table S7 available online at http://bib.oxfordjournals.org/.
To comprehensively assess of the selectivity of kinase inhibitors, we used KinomeMETA to predict the activity profiles and selectivity scores of 243 kinase inhibitors that are either approved for clinical use or in clinical trials. The obtained results agreed well with the selectivity scores determined by a chemical proteomic approach [58] (Figure S10 available online at http://bib.oxfordjournals.org/), showing a significant rank correlation (Spearman correlation = 0.752) and similar distribution. Notably, when compared with other baseline models trained with the same dataset, KinomeMETA’s selectivity score performance significantly surpassed that of MTGNN, IDDkin, MT-DNN, ST-DNN, ST-RF and Auto-Sklearn (Figure S11 available online at http://bib.oxfordjournals.org/). The distribution also revealed that compound 2e possesses higher selectivity than most known kinase inhibitors, whereas compound 15 exhibits moderate selectivity. This highlights the potential of KinomeMETA to prioritize inhibitors based on their kinome-wide selectivity.
In conclusion, the experiment confirmed the exceptional predictive performance of KinomeMETA. It may serve as a valuable tool for accurately profiling kinase inhibitors and assessing their selectivity. Moreover, it can effectively address low-data scenarios, such as drug-resistant kinase mutations, due to its ability to incorporate newly added activity compounds.
DISCUSSION
Overall, KinomeMETA is a valuable tool for rational design of multi-targeted selective kinase inhibitors. It not only helps us quickly identify novel hit compounds through large-scale virtual screening, but also contributes to the design of more effective and safer kinase inhibitors through kinome-wide activity profiling. Its scalability and rapid adaptation for new kinases make it well-suited for exploring so far understudied kinases, which may offer new possibilities for treating more diseases and addressing unmet clinical needs.
Although KinomeMETA shows good performance and potential in predicting kinase inhibitory activity, there are still some aspects that can be improved. Firstly, the performance of KinomeMETA is still limited by the quality and quantity of available data, especially for new kinases that differ greatly from those well studied. Therefore, collecting more experimental data to cover a wider range of the kinome will be a direction for further improvement. Secondly, the prediction ability of KinomeMETA may be affected by the diversity and feature representation of compounds. In addition to collecting more data, improving the method of molecular representation is a feasible option. Incorporating 3D information in the molecular representation of KinomeMETA is a promising direction, considering the successful application of these strategies in predicting molecular properties [59]. Additionally, characterizing molecules by phenotypic profiles, such as gene-expression profiles and Cell Painting images, may enable models to capture the connections between biological features of compounds, mitigating the constraints imposed by chemical structural similarity [60].
Moving forward, our efforts will persist in refining kinase inhibition prediction through meticulous curation of high-quality data, refining molecular representation techniques, and delving into more advanced few-shot learning approaches. Our vision is that KinomeMETA will evolve into an important component in the future kinase drug discovery and development.
KinomeMETA uses a meta-learning strategy integrated with a GNN, leveraging a large-scale dataset of kinase inhibitor data for 661 kinases. This meta-learning strategy addresses the challenge of the scarcity of data, thereby improving the accuracy and coverage of kinome profiling.
KinomeMETA incorporates fast fine-tuning, allowing it to generalize to unseen kinases with high accuracy, overcoming the limitations of previous machine learning models that were restricted to specific kinases within their modeling domains.
KinomeMETA’s performance excels across three distinct perspectives, corresponding to diverse application scenarios including kinome-wide activity profiling, mutant kinase selectivity prediction, and rapid adaptation to previously unseen kinases.
KinomeMETA has proven its efficacy in the identification of novel scaffold inhibitors for PKMYT1 and the retrospective analysis of selective inhibitors for drug-resistant mutants of FGFRs. These instances underscore KinomeMETA’s potential in efficiently navigating the kinase polypharmacology landscape through comprehensive virtual screening and kinome-wide activity profiling.
AUTHOR CONTRIBUTIONS
M.Y.Z. and X.T.L. conceived the project and were responsible for the decision to submit the manuscript. Q.R. and N.Q. implemented the KinomeMETA model and conducted computational analysis. J.J.S, J.L., L.N., X.C.T., Z.M.Z, X.T.K., Y.M.W and Y.T.W. accessed, verified and analyzed the data. S.L.Z. provided the equipment, materials, and guidance for the biochemical experiments. J.Y.Z conducted the biochemical experiments. Q.R., X.T.L, N.Q. and M.Y.Z. wrote the paper. All authors discussed the results and commented on the manuscript.
FUNDING
National Key Research and Development Program of China (2022YFC3400504 to M.Y.Z.), National Natural Science Foundation of China (T2225002 and 82273855 to M.Y.Z., 82204278 to X.T.L.), Lingang Laboratory (LG202102–01-02 to M.Y.Z.), SIMM-SHUTCM Traditional Chinese Medicine Innovation Joint Research Program (E2G805H to M.Y.Z.), China Postdoctoral Science Foundation (2022 M720153 to X.T.L.), and Shanghai Municipal Science and Technology Major Project.
DATA AVAILABILITY
The source codes and related data of KinomeMETA are available at: https://github.com/myzheng-SIMM/KinomeMETA. The raw data is accessible at: https://doi.org/10.5281/zenodo.10082625.
ABBREVIATIONS
AI: Artificial Intelligence; PKMYT1: Membrane-associated tyrosine- and threonine-specific cdc2-inhibitory kinase; FGFR: Fibroblast growth factor receptor; KIT: KIT Proto-Oncogene, Receptor Tyrosine Kinase; ABL1: ABL proto-oncogene 1, non-receptor tyrosine kinase; EGFR: Epidermal growth factor receptor; ALK: Anaplastic lymphoma kinase; MET: its encoding protein called c-Met, cellular-mesenchymal to epithelial transition factor; FLT3: Fms-like Receptor Tyrosine Kinase 3; RET: Rearranged during transfection receptor tyrosine kinase; CDK1: cyclin-dependent kinase 1; CCNE1: cyclin E1; Wee1: WEE1 G2 checkpoint kinase; KDR: Kinase insert domain receptor; BRAF:B-Raf Proto-Oncogene, Serine/Threonine Kinase; NB: Naive Bayes; RF: Random Forest; SVM: Support Vector Machine; DNN: Deep Neural Network; ST: Single-Task; MT: Multi-Task; ECFP: extended connectivity fingerprint; DMSO: Dimethyl Sulfoxide; ML: Machine Learning; DL: Deep Learning; GNN: Graph Neural Network; GCN: Graph Convolution Network; MAML: Model-Agnostic Meta-Learning; SGD: Stochastic gradient descent; UMAP: Uniform manifold approximation and projection; DKKB: Dark Kinase Knowledgebase; PKIS: protein kinase inhibitor set; FDA: Food and Drug Administration; PDB: Protein Data Bank; IC50: half maximal inhibitory concentration; Ki: inhibitor constant; EC50: half maximal effective concentration; Kd: dissociation constant; %Inh: percentage inhibition rate; pKi: −log10Ki; pKd: −log10Kd; pIC50: −log10IC50; pEC50: −log10EC50; TK: Tyrosine kinase; TKL: Tyrosine Kinase-Like kinase; TKI: Tyrosine Kinase Inhibitor; NSCLC: Non-Small Cell Lung Cancer; HTS: High Throughput Screening; PAINS: pan assay interference compounds; logP: log10 (Partition Coefficient); μM: Micromolar; nM: Nanomolar; FP binding assay: Fluorescence Polarization binding assay; auROC: area under the ROC curve; MCC: Matthews correlation coefficient; BACC: Balanced accuracy.
Author Biographies
Qun Ren obtained her Master Degree at Nanjing University of Chinese Medicine and Shanghai Institute of Materia Medica. Her research interest is artificial intelligence-assisted multi-target kinase drug design.
Ning Qu is a Ph.D. student at Shanghai Institute of Materia Medica. His research interests are artificial intelligence-assisted pharmacology and pharmacodynamics prediction.
Jingjing Sun is a Master student at Shanghai Institute of Materia Medica. Her research interest is artificial intelligence-assisted multi-target kinase drug design.
Jingyi Zhou is a Ph.D. student at ShanghaiTech University. Her research interest is molecular biology, biochemistry and cell biology.
Jin Liu is a Ph.D. student at College of Pharmaceutical Sciences, Zhejiang University. Her research interest is artificial intelligence-assisted drug safety assessment.
Lin Ni obtained her Master Degree at Nanjing University of Chinese Medicine and Shanghai Institute of Materia Medica. Her research interest is artificial intelligence-assisted multi-target kinase drug design.
Xiaochu Tong is a Ph.D. student at Shanghai Institute of Materia Medica. Her research interests are artificial intelligence-assisted pharmacology and pharmacodynamics prediction.
Zimei Zhang is a Master student at Shanghai Institute of Materia Medica. Her research interest is artificial intelligence-assisted molecular representation.
Xiangtai Kong is a Master student at Shanghai Institute of Materia Medica. His research interest is artificial intelligence-assisted drug-target interaction prediction.
Yiming Wen is a Master student at Pharmaceutical Science and Technology, Hangzhou Institute for Advanced Study. Her research interest is artificial intelligence-assisted drug design.
Yitian Wang is a Ph.D. student at Shanghai Institute of Materia Medica. Her research interests are artificial intelligence-assisted pharmacology and ADMET prediction.
Dingyan Wang is a researcher at Lingang Laboratory. His research interest is artificial intelligence-assisted drug design.
Xiaomin Luo is a professor at Shanghai Institute of Materia Medica. His research interests are artificial intelligence-assisted drug design, computational chemistry and computational biology.
Sulin Zhang is an associate professor at Shanghai Institute of Materia Medica. His research interest is molecular biology, biochemistry and cell biology.
Mingyue Zheng is a professor at Shanghai Institute of Materia Medica. His research interest is the development of accurate drug design techniques based on artificial intelligence, computational chemistry and computational biology.
Xutong Li is an associate professor at Shanghai Institute of Materia Medica. Her primary research interest is the development of accurate drug design techniques based on artificial intelligence, including multi-target selective drug design, drug-target interactions and explainable deep learning for druggability prediction.
References
Author notes
Qun Ren and Ning Qu contributed equally to this work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}