




Abstract
Recent advances in long read technologies not only enable large consortia to aim to sequence all eukaryotes on Earth, but they also allow individual laboratories to sequence their species of interest with relatively low investment. Long read technologies embody the promise of overcoming scaffolding problems associated with repeats and low complexity sequences, but the number of contigs often far exceeds the number of chromosomes and they may contain many insertion and deletion errors around homopolymer tracts. To overcome these issues, we have implemented the ILRA pipeline to correct long read-based assemblies. Contigs are first reordered, renamed, merged, circularized, or filtered if erroneous or contaminated. Illumina short reads are used subsequently to correct homopolymer errors. We successfully tested our approach by improving the genome sequences of Homo sapiens, Trypanosoma brucei, and Leptosphaeria spp., and by generating four novel Plasmodium falciparum assemblies from field samples. We found that correcting homopolymer tracts reduced the number of genes incorrectly annotated as pseudogenes, but an iterative approach seems to be required to correct more sequencing errors. In summary, we describe and benchmark the performance of our new tool, which improved the quality of novel long read assemblies up to 1 Gbp. The pipeline is available at GitHub: https://github.com/ThomasDOtto/ILRA.
INTRODUCTION
The process of assembling a genome can be challenging due to multiple factors, such as variable sample quality and sequencing depth, or large numbers of repeats and/or low complexity regions. These may produce larger numbers of contigs or lower consensus quality. In response, next-generation sequencing techniques have undergone impressive development over the last few years, achieving unparalleled resolution and performance [1]. If enough high molecular weight DNA and funding is available, long read technologies provided by Pacific Bioscience (PacBio) [2] and Oxford Nanopore Technologies (ONT) [3], combined with scaffolding methods such as HiC or Bionano, produce continuous sequences, mainly because complex repeats can be spanned. The availability of these technologies, together with the accompanying drop in price per base, has motivated the formation of large consortia, such as the Earth BioGenome Project that aims to sequence all eukaryotes [4]. These consortia aim to use a range of sequencing technologies and scaffolding methods to generate telomere-to-telomere assemblies with fewer than 1 error per 10 000 bases (i.e. gold standard reference genomes [5, 6]). Individual research groups, however, tend to apply whole genome sequencing to either produce de novo assemblies of a few species of interest or to study genome variation within a species. As resources are typically limited, the resulting assemblies are likely to be more fragmented and additional finishing is required.
Short sequencing reads would be required to address the limitations of ONT and PacBio technologies regarding homopolymer tracts and short tandem repeats (STRs). Although these errors affect the accuracy of the predicted gene models organism-wide [7–9], this issue is more pronounced for organisms with highly skewed base composition, such as the malaria parasite Plasmodium falciparum (GC content ~19%). Notably, up-to-date improvements of sequencing technologies (e.g. PacBio Hi-Fi, Ultima Genomics, ultralong reads by ONT or R10.4.1 flow cells by ONT) may improve but do not completely overcome this issue [10–14]. Similarly, there have been dramatic improvements in assembler software of long reads. For example, HGAP [15], Canu [16] or MaSuRCA [17] are tools that correct reads prior assembly, unlike faster options such as Wtdbg2 [18]. Another option is to combine multiple sequencing technologies (i.e. hybrid approaches [19, 20]), such as MaSuRCA using both short and long reads [21], or Verkko integrating both PacBio and ONT reads [22]. However, none of the above generates telomere-to-telomere assemblies and the consensus sequences contain errors, mainly due to genome complexity or the aforementioned limitations of the input (i.e. erroneous or low quality sequencing reads). Therefore, post-assembly finishing tools are still required.
Historically, finishing steps (e.g. closing sequencing gaps, decontamination or following naming conventions) have taken as much time and manual effort as sequencing and assembly, but since the advent of Illumina sequencing, several steps can now be automated with tools like PAGIT [23]. Other tools such as iCORN2 [24] and Pilon [25] have been developed to leverage the accuracy of Illumina short reads and correct small errors and frameshifts [26, 27]. However, in contrast to the variety of assembler software available, there are very few pipelines to automatically assemble long reads and polish the resulting sequences. Assemblosis [28], ARAMIS [29] and MpGAP [30] are examples, but they may include technology- or sequence-specific software (e.g. particular assembler or error correction tools). These may not be easy to install and run locally and could also be improved by incorporating processing such as decontamination.
In this study, we have overcome the above limitations and further streamlined the automatic finishing of genome sequences by developing Improvement of Long Read Assemblies (ILRA), a pipeline that is easy to use and combines novel and existing tools to improve de novo genome assemblies. We have tested ILRA on human data to investigate any limitation of genome size, and then applied it to several genomes with varying sequencing depth, median read length and sequencing approaches. These include four Plasmodium falciparum genomes, a Trypanosoma brucei assembly by PacBio [31], and two fungi assemblies (Leptosphaeria spp.) by ONT. We conclude that ILRA is generally applicable to long read assemblies across species and technologies, and outperforms existing alternatives.
METHODS
Assembly software and annotation of sequences
To investigate whether different assembler tools provide consistent results and to assess the need for further improvements, we compared HGAP [15], Canu [16] and Wtdbg2 [18], which all use PacBio long reads, and the hybrid MaSuRCA [17] that uses both PacBio long reads and Illumina short reads. As case examples of challenging sequences with very different origins and characteristics, we used four novel sets of reads from three P. falciparum isolates (PfCO01, PfKE07 and Pf2004; Table 1 and Supplementary Table 1).
Overview of P. falciparum assemblies by MaSuRCA and information of the datasets used in this study
| P. falciparum isolates | Pf3D7 reference | PfCO01 | PfCO01 | PfKE07 | Pf2004 |
|---|---|---|---|---|---|
| Library preparation | – | Standard | Standard | WGA | Standard |
| Sequencer (PacBio) | – | RSII | Sequel | RSII | RSII |
| Number of contigs | 16 | 30 | 41 | 259 | 21 |
| Size (Mbp) | 23.33 | 23.36 | 23.43 | 21.76 | 23.30 |
| Number of genes | 5720 | 6145 | 6210 | 5067 | 5701 |
| Number of pseudogenes | 158 | 154 | 163 | 449 | 156 |
| P. falciparum isolates | Pf3D7 reference | PfCO01 | PfCO01 | PfKE07 | Pf2004 |
|---|---|---|---|---|---|
| Library preparation | – | Standard | Standard | WGA | Standard |
| Sequencer (PacBio) | – | RSII | Sequel | RSII | RSII |
| Number of contigs | 16 | 30 | 41 | 259 | 21 |
| Size (Mbp) | 23.33 | 23.36 | 23.43 | 21.76 | 23.30 |
| Number of genes | 5720 | 6145 | 6210 | 5067 | 5701 |
| Number of pseudogenes | 158 | 154 | 163 | 449 | 156 |
Standard—normal DNA preparation. Supplementary Table 1 contains detailed information.
Overview of P. falciparum assemblies by MaSuRCA and information of the datasets used in this study
| P. falciparum isolates | Pf3D7 reference | PfCO01 | PfCO01 | PfKE07 | Pf2004 |
|---|---|---|---|---|---|
| Library preparation | – | Standard | Standard | WGA | Standard |
| Sequencer (PacBio) | – | RSII | Sequel | RSII | RSII |
| Number of contigs | 16 | 30 | 41 | 259 | 21 |
| Size (Mbp) | 23.33 | 23.36 | 23.43 | 21.76 | 23.30 |
| Number of genes | 5720 | 6145 | 6210 | 5067 | 5701 |
| Number of pseudogenes | 158 | 154 | 163 | 449 | 156 |
| P. falciparum isolates | Pf3D7 reference | PfCO01 | PfCO01 | PfKE07 | Pf2004 |
|---|---|---|---|---|---|
| Library preparation | – | Standard | Standard | WGA | Standard |
| Sequencer (PacBio) | – | RSII | Sequel | RSII | RSII |
| Number of contigs | 16 | 30 | 41 | 259 | 21 |
| Size (Mbp) | 23.33 | 23.36 | 23.43 | 21.76 | 23.30 |
| Number of genes | 5720 | 6145 | 6210 | 5067 | 5701 |
| Number of pseudogenes | 158 | 154 | 163 | 449 | 156 |
Standard—normal DNA preparation. Supplementary Table 1 contains detailed information.
Annotation was performed with Companion [32]. Full information for all the assemblies and software used are in Supplementary Table 1 and Supplementary Methods.
Comparison of iCORN2 and Pilon correction
The software iCORN2 v1.0 and Pilon v1.24 were evaluated for their performance and accuracy when correcting small indels and single base pair errors in long read assemblies of P. falciparum and T. brucei. We first used an uncorrected long read assembly of P. falciparum 3D7 as a test case. We corrected it with Illumina short reads of different lengths (75–300 bp) and then compared the results with the Pf3D7 reference version v3.1 [33] (i.e. the ground truth). MEGAblast v2.2.26 [34] was used to evaluate the correction steps (Table 2 and Supplementary Table 2). For the same experiment with T. brucei, we used an uncorrected long read assembly [31], two concatenated sets of Illumina short reads and the T. brucei Lister strain 427 2018 as reference. The results are provided in Supplementary Table 3. The step-by-step details for these analyses are provided in Supplementary Methods and Table 3 shows the final results.
Summary of the correction of a P. falciparum sequence with Pilon and iCORN2
| Polisher | No. of remaining indels versus reference | No. of remaining SNPs versus reference | No. of annotated pseudogenes | |
|---|---|---|---|---|
| (None, uncorrected sequence) | 32 702 | 2314 | 520 | |
| (None, reference sequence) | – | – | 158 | |
| Correction with Illumina reads of: | ||||
| 75 bp | Pilon | 26 037 | 1973 | 531 |
| iCORN2 | 9768 | 1961 | 164 | |
| 100 bp | Pilon | 17 905 | 1879 | 255 |
| iCORN2 | 6402 | 2235 | 143 | |
| 300 bp | Pilon | 19 473 | 1749 | 197 |
| iCORN2 | 4485 | 3919 | 141 | |
| Polisher | No. of remaining indels versus reference | No. of remaining SNPs versus reference | No. of annotated pseudogenes | |
|---|---|---|---|---|
| (None, uncorrected sequence) | 32 702 | 2314 | 520 | |
| (None, reference sequence) | – | – | 158 | |
| Correction with Illumina reads of: | ||||
| 75 bp | Pilon | 26 037 | 1973 | 531 |
| iCORN2 | 9768 | 1961 | 164 | |
| 100 bp | Pilon | 17 905 | 1879 | 255 |
| iCORN2 | 6402 | 2235 | 143 | |
| 300 bp | Pilon | 19 473 | 1749 | 197 |
| iCORN2 | 4485 | 3919 | 141 | |
Illumina short reads of different lengths are used. A published Pf3D7 PacBio assembly by HGAP was alternatively corrected with Pilon and iCORN2 (mapping of Illumina short reads with Bowtie2). The results were compared against the P. falciparum 3D7 reference using MegaBLAST to obtain the number of differences between the reference and corrected sequence. SNPs and indels correspond to potential remaining errors, so lower numbers as output of the MegaBLAST imply better correction (i.e. reference and better-corrected sequences would be more similar). The number of annotated pseudogenes is included in all cases. Supplementary Table 2 provides more details.
Summary of the correction of a P. falciparum sequence with Pilon and iCORN2
| Polisher | No. of remaining indels versus reference | No. of remaining SNPs versus reference | No. of annotated pseudogenes | |
|---|---|---|---|---|
| (None, uncorrected sequence) | 32 702 | 2314 | 520 | |
| (None, reference sequence) | – | – | 158 | |
| Correction with Illumina reads of: | ||||
| 75 bp | Pilon | 26 037 | 1973 | 531 |
| iCORN2 | 9768 | 1961 | 164 | |
| 100 bp | Pilon | 17 905 | 1879 | 255 |
| iCORN2 | 6402 | 2235 | 143 | |
| 300 bp | Pilon | 19 473 | 1749 | 197 |
| iCORN2 | 4485 | 3919 | 141 | |
| Polisher | No. of remaining indels versus reference | No. of remaining SNPs versus reference | No. of annotated pseudogenes | |
|---|---|---|---|---|
| (None, uncorrected sequence) | 32 702 | 2314 | 520 | |
| (None, reference sequence) | – | – | 158 | |
| Correction with Illumina reads of: | ||||
| 75 bp | Pilon | 26 037 | 1973 | 531 |
| iCORN2 | 9768 | 1961 | 164 | |
| 100 bp | Pilon | 17 905 | 1879 | 255 |
| iCORN2 | 6402 | 2235 | 143 | |
| 300 bp | Pilon | 19 473 | 1749 | 197 |
| iCORN2 | 4485 | 3919 | 141 | |
Illumina short reads of different lengths are used. A published Pf3D7 PacBio assembly by HGAP was alternatively corrected with Pilon and iCORN2 (mapping of Illumina short reads with Bowtie2). The results were compared against the P. falciparum 3D7 reference using MegaBLAST to obtain the number of differences between the reference and corrected sequence. SNPs and indels correspond to potential remaining errors, so lower numbers as output of the MegaBLAST imply better correction (i.e. reference and better-corrected sequences would be more similar). The number of annotated pseudogenes is included in all cases. Supplementary Table 2 provides more details.
Final results for the comparison of the polishing of uncorrected P. falciparum and T. brucei sequences by Pilon and iCORN2
| Polishing tool (5 iterations) | No. of annotated pseudogenes | Runtime (hours) | |
|---|---|---|---|
| P. falciparum | (None, uncorrected) | 520 | – |
| (None, reference) | 158 | – | |
| Pilon | 210 | 1.91 | |
| iCORN2 | 143 | 9.04 | |
| T. brucei | (None, uncorrected) | 5346 | – |
| (None, reference) | 4925 | – | |
| Pilon | 5037 | 22.51 | |
| iCORN2 | 5179 | 19.35 |
| Polishing tool (5 iterations) | No. of annotated pseudogenes | Runtime (hours) | |
|---|---|---|---|
| P. falciparum | (None, uncorrected) | 520 | – |
| (None, reference) | 158 | – | |
| Pilon | 210 | 1.91 | |
| iCORN2 | 143 | 9.04 | |
| T. brucei | (None, uncorrected) | 5346 | – |
| (None, reference) | 4925 | – | |
| Pilon | 5037 | 22.51 | |
| iCORN2 | 5179 | 19.35 |
The Bowtie2 aligner to map Illumina short reads (100 bp in the case of P. falciparum) and 5 iterations in both iCORN2 and Pilon were used. The number of annotated pseudogenes by Companion in the pre-polished sequences is included as reference. We compared the number of annotated pseudogenes for each correction and organism. The number of pseudogenes in T. brucei may be due to the presence of large numbers of incomplete copies of variant surface genes. Supplementary Tables 2 and 3 provide detailed information.
Final results for the comparison of the polishing of uncorrected P. falciparum and T. brucei sequences by Pilon and iCORN2
| Polishing tool (5 iterations) | No. of annotated pseudogenes | Runtime (hours) | |
|---|---|---|---|
| P. falciparum | (None, uncorrected) | 520 | – |
| (None, reference) | 158 | – | |
| Pilon | 210 | 1.91 | |
| iCORN2 | 143 | 9.04 | |
| T. brucei | (None, uncorrected) | 5346 | – |
| (None, reference) | 4925 | – | |
| Pilon | 5037 | 22.51 | |
| iCORN2 | 5179 | 19.35 |
| Polishing tool (5 iterations) | No. of annotated pseudogenes | Runtime (hours) | |
|---|---|---|---|
| P. falciparum | (None, uncorrected) | 520 | – |
| (None, reference) | 158 | – | |
| Pilon | 210 | 1.91 | |
| iCORN2 | 143 | 9.04 | |
| T. brucei | (None, uncorrected) | 5346 | – |
| (None, reference) | 4925 | – | |
| Pilon | 5037 | 22.51 | |
| iCORN2 | 5179 | 19.35 |
The Bowtie2 aligner to map Illumina short reads (100 bp in the case of P. falciparum) and 5 iterations in both iCORN2 and Pilon were used. The number of annotated pseudogenes by Companion in the pre-polished sequences is included as reference. We compared the number of annotated pseudogenes for each correction and organism. The number of pseudogenes in T. brucei may be due to the presence of large numbers of incomplete copies of variant surface genes. Supplementary Tables 2 and 3 provide detailed information.
ILRA methods
We implemented ILRA as a bash script to automatically improve long read assemblies. Supplementary Figure 1 shows the overview of the pipeline. Contig cleaning, merging overlapping contigs, ordering contigs against a reference, correcting homopolymer errors, circularizing plasmids, decontamination and quality control are automatically performed by ILRA. Full technical details on the pipeline steps are included in Supplementary Methods and the ILRA GitHub page.
Sequence datasets
To develop and test our pipeline, we used five species and different sets of sequences (i.e. sequencing reads to generate assemblies or existing assemblies):
(i) As a proof of concept, we tested the limits of ILRA by manually taking subsets (from 100 Mbp to 1 Gbp) of the human genome HG00733/GCA_003634875 [35].
(ii) To test ILRA with multiple organisms and sequencing technologies, we used a T. brucei uncorrected PacBio assembly [31], and Leptosphaeria maculans Nz-T4 and Leptosphaeria biglobosa G12–14 ONT assemblies [36].
(iii) To test ILRA with challenging sequences we have expertise with [37, 38], we generated several P. falciparum assemblies from sequencing datasets with different library generation approaches, including Whole Genome Amplification (WGA), and different read characteristics (e.g. PacBio Sequel and PacBio RSII).
Details on all datasets above are in Supplementary Methods and Supplementary Table 1.
Existing finishing pipelines
Assemblosis, ARAMIS and MpGAP were run on two of our novel P. falciparum datasets (Pf2004 and PfCO01 from RSII reads), together with ARAMIS on the human, fungi and T. brucei sequences. The output was compared to the results post-ILRA correction. The details and parameters for each software are reported in Supplementary Methods. We summarized the features and limitations of all pipelines, including ILRA, in Supplementary Table 4.
RESULTS
In this study, we developed a new pipeline to assist in the finishing of draft long read assemblies. We first show that independently of the choice of assembler software, corrections are always necessary. We next compared tools to correct sequencing errors with the aim of integrating them into our pipeline. Next, we applied the ILRA pipeline to multiple assemblies of PacBio and ONT reads from five species (human, T. brucei, L. maculans, L. biglobosa and P. falciparum). Finally, we also compared our results with other assembly and polishing pipelines and explored limitations, such as genome size or fragmentation.
Comparison of assembler software
First, we evaluated the impact of using different assemblers on the number of contigs and the presence of potential frameshifts due to homopolymer tracts and STR. We used a set of particularly challenging sequences from P. falciparum, a typically difficult-to-sequence organism due to extreme AT content. We included PacBio reads from RSII and Sequel (PfCO01 RSII/Sequel, Pf2004 RSII), which were also of different overall qualities, and one dataset (PfKE07) whose RSII reads were generated from a library subjected to WGA (Supplementary Table 1). The four sets of P. falciparum reads were assembled with HGAP, Canu, MaSuRCA and Wtdbg2, to determine the optimum algorithm. As a measure of assembly quality, we used the genome size, number of contigs, and number of annotated genes and pseudogenes (see Methods). For comparison, the P. falciparum reference (Pf3D7 v3.1) contains 23.33 Mpb in 16 contigs, with 5720 and 158 genes and pseudogenes, respectively. The differences between the top three assemblers (HGAP, Canu and MaSuRCA) were minimal. MaSuRCA generally generated the best results, except for the Pf2004 assembly. However, there was only 1 contig difference between the HGAP (20 contigs) and the MaSuRCA assemblies (21 contigs) and the number of annotated genes and pseudogenes was closer to the reference in the case of MaSuRCA (5701 genes and 156 genes), as opposed to HGAP (5735 and 205 pseudogenes). Table 1 summarizes the information for all assemblies. Supplementary Table 1 shows full details, including runtimes and standardized scores by BUSCO [39] and QUAST [40]. Similar to above, the assemblies with the highest scores were also from MaSuRCA. The BUSCO score was highest for the MaSuRCA assembly, followed by HGAP, in all cases except for the more fragmented PfKE07, with slightly higher score in the HGAP assembly. Despite choosing the best assembler based on multiple evidence, our results with test case sequences of P. falciparum still show that the number of contigs (up to 259) was higher than expected based on the curated Pf3D7 reference (14 chromosomes, 1 mitochondrion and 1 apicoplast). A wide range of 5067–6210 genes and 154–449 pseudogenes were also annotated, in contrast to the reference (5720 genes and 158 pseudogenes). Our results suggest that further polishing is required.
Comparison of methods for the correction of homopolymer tracts and STR
Homopolymer tracts in long read sequencing technologies are known to insert artificial frameshifts, which cause the truncation of gene models and their annotation as pseudogenes. Therefore, the excessive numbers of annotated genes and pseudogenes in the de novo P. falciparum assemblies emphasize the importance of polishing and show that P. falciparum is a good test case (Figure 1).

Example of a frameshift error in one of the gene models in our long read genome assemblies of P. falciparum due to the presence of a homopolymer tract. Artemis visualization of a PacBio genome assembly (bottom panel) and the aligned Illumina short reads (top panel, horizontal blue bars). Reads mapping to the forward strand are on top, and to the reverse below. Sequencing errors in the Illumina short reads are marked with vertical light red lines. A homopolymer tract of 17 A’s is highlighted in yellow. The quality of the reads drops after the homopolymer, and accordingly it can be seen that reads on the forward strand have just few sequencing errors, but after the homopolymer tract the error rate is high. This tract is not sequenced correctly, it generates a frameshift and therefore causes a gene model to be wrongly annotated as a pseudogene. In the bottom panel, the two light blue boxes represent exons that due to the indel are split into two. Ab initio gene finders could try to build an intron here (losing exon sequence) or to generate a pseudogene. In the zoom-in visualization (right), the dark red vertical lines in the aligned Illumina short reads point to bases that are missing from the short repetition in the assembly, resulting in the homopolymer tract causing the frameshift.
For the task of correcting errors around homopolymers, we compared iCORN2 and Pilon. We used these to correct a PacBio assembly of P. falciparum 3D7 by others, using the Pf3D7 reference as the ground truth set and Illumina short reads of different length (Table 2 and Supplementary Table 2). In the comparison we include the number of SNPs, indels and pseudogenes (see Methods for details). We argue that comparing the number of annotated pseudogenes in corrected assemblies is a robust method to assess the outcome of tools since overcorrections are unlikely and functional genes can be confirmed with other approaches, such as protein evidence.
We observed that the iterative approach of iCORN2 generally recognizes and corrects more indel errors than Pilon (Table 2 and Supplementary Table 2). For instance, when correcting with 75 bp Illumina short reads, out of the 32 702 indels present in the uncorrected sequence compared with the reference, ~26 000 (531 pseudogenes) were still uncorrected after Pilon and ~9750 after iCORN2 (164 pseudogenes, closer to the reference 158 pseudogenes). Annotated pseudogenes were lower due to the better correction of frameshifts in gene models. Figure 2 shows examples of this differential correction. Overall, iCORN2 performs better and corrects around 2–4-fold more indels than Pilon (Table 2 and Supplementary Table 2).

Differential frameshifts correction by Pilon and iCORN2. ACT visualization of a section of the Pf3D7 reference genome sequence, the corresponding section of an uncorrected P. falciparum 3D7 PacBio genome assembly, and the Pilon-corrected and iCORN2-corrected sequences. Syntenic regions (BLAST) are indicated in gray bars between the reference and the uncorrected assembly. Annotated genes in the reference are colored. Black vertical lines mark the absence of open reading frames (ORFs). Red squares mark the frameshifts within ORFs in the uncorrected genome sequences. These are differentially processed by Pilon and iCORN2, with multiple iterations of iCORN2 correcting more frameshifts than a single Pilon run. Green squares mark the correct and successively corrected ORFs, which based on the reference could be annotated as correct gene models instead of an excessive and incorrect annotation of pseudogenes.
We expected the number of both types of corrections (SNPs and indels) to increase with the read length independently of the tool since longer reads align more accurately and span larger indels. Indeed, the correction improved when increasing the read length, but we still observed indels, leaving the question open if those are genuine differences of the ground truth versus the new DNA stock, or errors in the ground truth. We cannot address these questions, as the genome of P. falciparum is of extremely low complexity. However, the number of pseudogenes dropped with longer read length, which shows that in less complex regions there is little difference between the ground truth and our best simulations. Another observation is that the number of SNPs (i.e. one-base pair substitutions) increased with the read length. Manual inspection showed that reads of 300 bp covered several homopolymer tracts and that the quality of the reads was lower (Supplementary Figure 2). In particular, it dropped after each homopolymer tract with C/G bases replaced by A/T. Therefore, we would not recommend long reads for polishing.
To explore whether the better performance of iCORN2 is limited to an AT-extreme organism, we also applied the approach above to a T. brucei uncorrected assembly (GC content ~43%). We confirmed that iCORN2 outperforms Pilon. When compared with the T. brucei Lister strain 427 2018 reference, around 20% fewer indels were found in the iCORN2-corrected sequences than in the Pilon-corrected ones and fewer pseudogenes were annotated (Supplementary Table 3). These differences in performance could be due to the iterative nature of iCORN2. Therefore, we also tested the performance of Pilon with the same number of iterations as iCORN2 on the uncorrected P. falciparum and T. brucei genome sequences (i.e. 5 iterations and with the better-quality 100 bp Illumina reads in the case of P. falciparum). We observed that despite being ~5 times slower with default parameters, the annotation of the iCORN2-corrected P. falciparum yielded one-third fewer pseudogenes than the Pilon-corrected version: 143 versus 210 pseudogenes (Table 3). Thanks to the iterative approach, Pilon also corrected ~20% more pseudogenes: 210 versus 255 pseudogenes (Supplementary Table 2). In contrast, Pilon-corrected T. brucei sequences displayed fewer pseudogenes than iCORN2-corrected, but the difference was very low (~3% of the pseudogenes) and the runtime was increased. The improvement in using Pilon iteratively was also noticeable, with 5037 annotated pseudogenes after 5 iterations versus 7902 after a single iteration (Supplementary Table 3). Overall, the better performance of iCORN2 led us to incorporate it as the default in our pipeline, together with the alternative choice of Pilon.
Automatic finishing of de novo genome assemblies by the ILRA pipeline
Next, we applied the ILRA pipeline (more details in Methods and Supplementary Figure 1) to several datasets. Table 4 and Supplementary Tables 1 and 5 summarize all results from the ILRA pipeline in this section. First, to explore limitations, we simulated larger sequences by taking subsets of the human genome of various sizes: ~100 Mbp, ~150 Mbp, ~300 Mbp, 500 Mbp and 1 Gbp (see Methods). We report that ILRA scaled well and successfully processed larger sequences, requiring larger runtimes and slightly more memory usage (Supplementary Table 5). Next, we ran ILRA with the T. brucei uncorrected PacBio assembly used to test polishing above. As expected, the ILRA pipeline successfully corrected the sequences, improving contiguity from 1232 to 616 contigs (reference genome = 317) and genome size from 65.5 to 57.8 Mbp (reference genome = 50.1 Mbp). The number of annotated pseudogenes also decreased by ~900 (5346 versus 4420). Afterwards, we addressed de novo genome assemblies from ONT reads. We ran ILRA on two recently published fungal genome sequences and reported the correction of thousands of SNPs and indels, together with improvements in contiguity and annotation of pseudogenes. While no good reference genomes are available for Leptosphaeria spp, a L. maculans strain Nz-T4 assembly was improved from 288 to 101 contigs and the annotated pseudogenes decreased from 540 to 433, which is closer to the number reported in previous assemblies. Similar improvements were also observed for a L. biglobosa G12–14 strain assembly, with 33.3% fewer contigs and 15.7% fewer annotated pseudogenes.
Overview of the ILRA improvements step by step in the datasets used in this study
| H. sapiens 100 Mbp | H. sapiens 1 Gbp | T. brucei | L. maculans Nz-T4 | L. biglobosa G12–14 | PfCO01 RSII | PfCO01 Sequel | PfKE07 RSII | Pf2004 RSII | |
|---|---|---|---|---|---|---|---|---|---|
| Pre-ILRA | |||||||||
| Assembly size (Mbp) | 100.44 | 1036.48 | 65.53 | 43.43 | 34.95 | 23.36 | 23.43 | 21.76 | 23.30 |
| #Contigs | 518 | 535 | 1232 | 288 | 156 | 30 | 41 | 259 | 21 |
| #Pseudogenes | 763 | – | 5346 | 540 | 498 | 154 | 163 | 449 | 156 |
| ILRA correction | |||||||||
| #Excluded contigs <5 kb | 0 | 0 | 94 | 0 | 0 | 3 | 4 | 3 | 0 |
| #Excluded contained contigs | 98 | 108 | 254 | 0 | 0 | 0 | 1 | 2 | 0 |
| #Excluded contigs not covered and merged | 76 | 55 | 33 | 0 | 0 | 0 | 0 | 1 | 0 |
| #Contigs merging and reference ordering | 29 | 20 | 231 | 186 | 51 | 7 | 12 | 180 | 4 |
| #Excluded contigs contamination | 0 | 0 | 6 | 1 | 1 | 2 | 7 | 0 | 0 |
| Post-ILRA | |||||||||
| Assembly size (Mbp) | 64.60 | 574.58 | 57.84 | 43.43 | 34.93 | 22.64 | 22.64 | 21.79 | 23.30 |
| #Contigs | 315 | 352 | 614 | 101 | 104 | 18 | 17 | 73 | 17 |
| #Pseudogenes | 489 | - | 4420 | 433 | 420 | 112 | 109 | 223 | 118 |
| H. sapiens 100 Mbp | H. sapiens 1 Gbp | T. brucei | L. maculans Nz-T4 | L. biglobosa G12–14 | PfCO01 RSII | PfCO01 Sequel | PfKE07 RSII | Pf2004 RSII | |
|---|---|---|---|---|---|---|---|---|---|
| Pre-ILRA | |||||||||
| Assembly size (Mbp) | 100.44 | 1036.48 | 65.53 | 43.43 | 34.95 | 23.36 | 23.43 | 21.76 | 23.30 |
| #Contigs | 518 | 535 | 1232 | 288 | 156 | 30 | 41 | 259 | 21 |
| #Pseudogenes | 763 | – | 5346 | 540 | 498 | 154 | 163 | 449 | 156 |
| ILRA correction | |||||||||
| #Excluded contigs <5 kb | 0 | 0 | 94 | 0 | 0 | 3 | 4 | 3 | 0 |
| #Excluded contained contigs | 98 | 108 | 254 | 0 | 0 | 0 | 1 | 2 | 0 |
| #Excluded contigs not covered and merged | 76 | 55 | 33 | 0 | 0 | 0 | 0 | 1 | 0 |
| #Contigs merging and reference ordering | 29 | 20 | 231 | 186 | 51 | 7 | 12 | 180 | 4 |
| #Excluded contigs contamination | 0 | 0 | 6 | 1 | 1 | 2 | 7 | 0 | 0 |
| Post-ILRA | |||||||||
| Assembly size (Mbp) | 64.60 | 574.58 | 57.84 | 43.43 | 34.93 | 22.64 | 22.64 | 21.79 | 23.30 |
| #Contigs | 315 | 352 | 614 | 101 | 104 | 18 | 17 | 73 | 17 |
| #Pseudogenes | 489 | - | 4420 | 433 | 420 | 112 | 109 | 223 | 118 |
Pre- and post-ILRA are the statistics of the assemblies prior to and after correction. The improvement (i.e. discarded or merged contigs) by each module of ILRA is shown.
Overview of the ILRA improvements step by step in the datasets used in this study
| H. sapiens 100 Mbp | H. sapiens 1 Gbp | T. brucei | L. maculans Nz-T4 | L. biglobosa G12–14 | PfCO01 RSII | PfCO01 Sequel | PfKE07 RSII | Pf2004 RSII | |
|---|---|---|---|---|---|---|---|---|---|
| Pre-ILRA | |||||||||
| Assembly size (Mbp) | 100.44 | 1036.48 | 65.53 | 43.43 | 34.95 | 23.36 | 23.43 | 21.76 | 23.30 |
| #Contigs | 518 | 535 | 1232 | 288 | 156 | 30 | 41 | 259 | 21 |
| #Pseudogenes | 763 | – | 5346 | 540 | 498 | 154 | 163 | 449 | 156 |
| ILRA correction | |||||||||
| #Excluded contigs <5 kb | 0 | 0 | 94 | 0 | 0 | 3 | 4 | 3 | 0 |
| #Excluded contained contigs | 98 | 108 | 254 | 0 | 0 | 0 | 1 | 2 | 0 |
| #Excluded contigs not covered and merged | 76 | 55 | 33 | 0 | 0 | 0 | 0 | 1 | 0 |
| #Contigs merging and reference ordering | 29 | 20 | 231 | 186 | 51 | 7 | 12 | 180 | 4 |
| #Excluded contigs contamination | 0 | 0 | 6 | 1 | 1 | 2 | 7 | 0 | 0 |
| Post-ILRA | |||||||||
| Assembly size (Mbp) | 64.60 | 574.58 | 57.84 | 43.43 | 34.93 | 22.64 | 22.64 | 21.79 | 23.30 |
| #Contigs | 315 | 352 | 614 | 101 | 104 | 18 | 17 | 73 | 17 |
| #Pseudogenes | 489 | - | 4420 | 433 | 420 | 112 | 109 | 223 | 118 |
| H. sapiens 100 Mbp | H. sapiens 1 Gbp | T. brucei | L. maculans Nz-T4 | L. biglobosa G12–14 | PfCO01 RSII | PfCO01 Sequel | PfKE07 RSII | Pf2004 RSII | |
|---|---|---|---|---|---|---|---|---|---|
| Pre-ILRA | |||||||||
| Assembly size (Mbp) | 100.44 | 1036.48 | 65.53 | 43.43 | 34.95 | 23.36 | 23.43 | 21.76 | 23.30 |
| #Contigs | 518 | 535 | 1232 | 288 | 156 | 30 | 41 | 259 | 21 |
| #Pseudogenes | 763 | – | 5346 | 540 | 498 | 154 | 163 | 449 | 156 |
| ILRA correction | |||||||||
| #Excluded contigs <5 kb | 0 | 0 | 94 | 0 | 0 | 3 | 4 | 3 | 0 |
| #Excluded contained contigs | 98 | 108 | 254 | 0 | 0 | 0 | 1 | 2 | 0 |
| #Excluded contigs not covered and merged | 76 | 55 | 33 | 0 | 0 | 0 | 0 | 1 | 0 |
| #Contigs merging and reference ordering | 29 | 20 | 231 | 186 | 51 | 7 | 12 | 180 | 4 |
| #Excluded contigs contamination | 0 | 0 | 6 | 1 | 1 | 2 | 7 | 0 | 0 |
| Post-ILRA | |||||||||
| Assembly size (Mbp) | 64.60 | 574.58 | 57.84 | 43.43 | 34.93 | 22.64 | 22.64 | 21.79 | 23.30 |
| #Contigs | 315 | 352 | 614 | 101 | 104 | 18 | 17 | 73 | 17 |
| #Pseudogenes | 489 | - | 4420 | 433 | 420 | 112 | 109 | 223 | 118 |
Pre- and post-ILRA are the statistics of the assemblies prior to and after correction. The improvement (i.e. discarded or merged contigs) by each module of ILRA is shown.
To assess the effect of the different read quality and characteristics, we used the novel P. falciparum assemblies generated above from three isolates with diverse origin: Colombia (PfCO01), Kenya (PfKE07) and Ghana (Pf2004). Table 1 shows that the quality of the assemblies obtained using MaSuRCA varied between isolates. Contig numbers ranged from 21 to 259 and annotated pseudogenes from 154 to 449. These differences can be attributed to contamination, different median read lengths, different sequencing technologies for PfCO01 or library preparation protocols (e.g. PfKE07 was based on WGA). We observed that the sets of PacBio RSII reads were assembled into a range of 17–73 contigs (median = 18). These assemblies clearly benefited from the automatic correction by ILRA, with lower contiguity (previous range of 21–259 contigs, median = 30) and fewer annotated pseudogenes. For example, ILRA improved contiguity and corrected ~40 gene models (wrongly annotated as pseudogenes) in the Pf2004 sample, which finally assembled into 17 contigs with 5728 genes and 118 pseudogenes. Overall, more than 5200 genes were annotated in all cases, and contiguity and number of annotated pseudogenes were always improved. Moreover, we observed several contigs containing terminal telomere-associated repeats, which could indicate fully assembled chromosomes (e.g. 6 out of the 17 contigs in the Pf2004 assembly post-ILRA had both telomeres attached). Despite the general improvements after correction by ILRA, there were also some assemblies of lower quality, such as PfKE07. For these sequences, the numbers of contigs and gaps were still large, and we observed artifacts and mis-assemblies due to library preparation (WGA). These errors may be due to the polymerase switching between strands during the amplification step, which results in inverted chimeras generating mis-assemblies that ILRA cannot address. Figure 3 displays a schematic case example of an error due to WGA and chimeric reads.
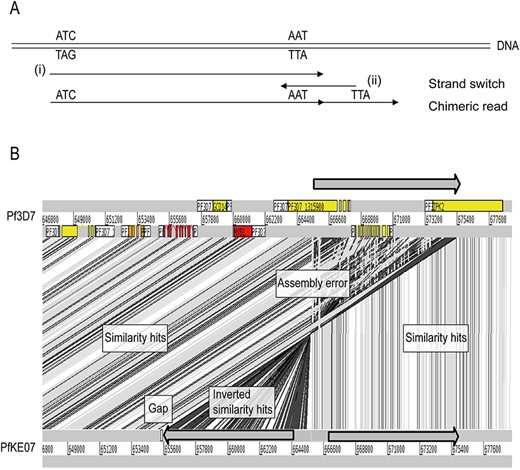
Whole Genome Amplification (WGA) errors in the PfKE07 assembly. (A) Schematic error of WGA. DNA gets amplified (i), but then the polymerase strand switches and generates the reverse strand (ii). This generates a chimeric read that generates mis-assemblies. (B) These chimeric reads generate assembly errors, as seen in an ACT view. The top part of the reference genome (gray arrow) is duplicated in the WGA-amplified genome. The assembly errors generally occur at the contig end, so gaps are generated. Syntenic regions when comparing to the reference genome (BLAST similarity hits) are indicated in gray. Mis-assemblies (inverted similarity hits) are indicated in black.
Furthermore, to compare the ILRA performance on different types of reads, we sequenced the Colombian sample (PfCO01) using the PacBio Sequel chemistry (an early release of chemistry), in addition to RSII. The same library was sequenced with both chemistries, but the mean read length was longer in the RSII (9413 versus 7668), while the read depth was higher in the Sequel run (198 versus 168). The initial assembly with the PacBio RSII reads was of considerably higher quality, with ~25% less contigs compared to Sequel. However, after correction by ILRA, the sequences coming from Sequel reads assembled into 17 contigs and similarly, the assembly from RSII reads was composed of 18 contigs. Another improvement by ILRA in this case was the identification and removal of multiple contigs identified as Mycoplasma arginini contamination [41, 42], which also caused an excessive number of annotated genes and pseudogenes in both PfC01 assemblies pre-ILRA (Table 4).
Finally, we observed that choosing iCORN2 or Pilon within the full ILRA pipeline led to variable but similar numbers of annotated pseudogenes. For PfCO01 (Sequel run) and Pf2004 assemblies, the annotations post-iCORN2 and post-Pilon differed in only 1 pseudogene, while the PfCO01 from the RSII run and the PfKE07 assemblies differed in 8 pseudogenes (6.7% fewer in iCORN2 correction) and in 12 pseudogenes (5.1% fewer in iCORN2 correction), respectively (Supplementary Table 1).
Comparison between the ILRA pipeline and similar tools for finishing genome sequences
To further test the quality of the ILRA pipeline, we compared it with some existing alternative software for the assembly and correction of sequences displaying different features, called ARAMIS, Assemblosis and MpGAP (Table 5, Supplementary Tables 1, 4 and 5). For the already-assembled sequences that we used above (i.e. human, T. brucei and L. biglobosa), we compared ARAMIS and ILRA. We report that ILRA consumed less resources than ARAMIS (in particular for larger assemblies) and provided better results in terms of contiguity and annotation in all cases. The size of the human assemblies dropped with the ILRA correction, but this was expected as the majority of size reduction was due to the collapse of redundant contigs containing alternative alleles. Accordingly, the number of annotated genes and pseudogenes decreased more after ILRA correction in comparison to ARAMIS: for the subset of 100 Mbp, 21 183 genes and 763 pseudogenes annotated after ARAMIS versus 12 715 genes and 489 pseudogenes post-ILRA (Table 5). As to PfCO01, we observed an increase in the number of annotated pseudogenes of 32.1, 30.4 and 13.4%, respectively, for the ARAMIS-, Assemblosis- and MpGAP-processed sequences, when compared with the ILRA-corrected assembly. The contiguity post-ILRA was also better (18 contigs versus 30, 34 and 35 after ARAMIS, Assemblosis and MpGAP). The number of annotated genes was ~5500 for the ILRA-corrected assembly and ~5800 after Assemblosis, while it reached 6200 and 6300 after ARAMIS and MpGAP. In the case of Pf2004, when compared with ILRA, the assemblies generated by ARAMIS, Assemblosis and MpGAP displayed very similar numbers of annotated genes (~5700), but more contigs (17 contigs in ILRA versus 21, 22 and 29 contigs in the alternative software), and they contained 20.3, 40.7 and 9.3% more annotated pseudogenes, respectively. Overall, we showed that ILRA outperforms alternative tools.
Overview of the sequences alternatively corrected by different tools and pipelines, namely, ARAMIS and ILRA applied over assemblies, or Assemblosis and MpGAP to assembly and correct sequences (assembly statistics and the annotation by Companion are included)
| Isolates/tools | T. brucei | L. biglobosa | H. sapiens 100 Mbp | PfCO01 RSII | Pf2004 RSII | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MaSuRCA | MaSuRCA | |||||||||||||
| ARAMIS | ILRA | ARAMIS | ILRA | ARAMIS | ILRA | Assemblosis | MpGAP | ARAMIS | ILRA | Assemblosis | MpGAP | ARAMIS | ILRA | |
| Size (Mbp) | 65.53 | 57.84 | 34.95 | 34.93 | 100.44 | 64.60 | 23.05 | 23.57 | 23.36 | 22.64 | 23.43 | 23.32 | 23.30 | 23.30 |
| No. of contigs | 1232 | 616 | 156 | 104 | 518 | 315 | 34 | 35 | 30 | 18 | 22 | 29 | 21 | 17 |
| No. of genes | 14 650 | 13 031 | 9172 | 9246 | 21 183 | 12 715 | 5763 | 6298 | 6159 | 5.521 | 5743 | 5707 | 5718 | 5723 |
| No. of pseudogenes | 5333 | 4420 | 498 | 420 | 763 | 489 | 146 | 127 | 148 | 112 | 166 | 129 | 142 | 119 |
| Runtime (hours, 60 cores) | 0.94 | 7.46 | 0.39 | 5.90 | 0.85 | 5.16 | 10.21 | 23.48 | 0.30 | 4.131 | 9.33 | 11.05 | 0.23 | 3.139 |
| Isolates/tools | T. brucei | L. biglobosa | H. sapiens 100 Mbp | PfCO01 RSII | Pf2004 RSII | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MaSuRCA | MaSuRCA | |||||||||||||
| ARAMIS | ILRA | ARAMIS | ILRA | ARAMIS | ILRA | Assemblosis | MpGAP | ARAMIS | ILRA | Assemblosis | MpGAP | ARAMIS | ILRA | |
| Size (Mbp) | 65.53 | 57.84 | 34.95 | 34.93 | 100.44 | 64.60 | 23.05 | 23.57 | 23.36 | 22.64 | 23.43 | 23.32 | 23.30 | 23.30 |
| No. of contigs | 1232 | 616 | 156 | 104 | 518 | 315 | 34 | 35 | 30 | 18 | 22 | 29 | 21 | 17 |
| No. of genes | 14 650 | 13 031 | 9172 | 9246 | 21 183 | 12 715 | 5763 | 6298 | 6159 | 5.521 | 5743 | 5707 | 5718 | 5723 |
| No. of pseudogenes | 5333 | 4420 | 498 | 420 | 763 | 489 | 146 | 127 | 148 | 112 | 166 | 129 | 142 | 119 |
| Runtime (hours, 60 cores) | 0.94 | 7.46 | 0.39 | 5.90 | 0.85 | 5.16 | 10.21 | 23.48 | 0.30 | 4.131 | 9.33 | 11.05 | 0.23 | 3.139 |
Overview of the sequences alternatively corrected by different tools and pipelines, namely, ARAMIS and ILRA applied over assemblies, or Assemblosis and MpGAP to assembly and correct sequences (assembly statistics and the annotation by Companion are included)
| Isolates/tools | T. brucei | L. biglobosa | H. sapiens 100 Mbp | PfCO01 RSII | Pf2004 RSII | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MaSuRCA | MaSuRCA | |||||||||||||
| ARAMIS | ILRA | ARAMIS | ILRA | ARAMIS | ILRA | Assemblosis | MpGAP | ARAMIS | ILRA | Assemblosis | MpGAP | ARAMIS | ILRA | |
| Size (Mbp) | 65.53 | 57.84 | 34.95 | 34.93 | 100.44 | 64.60 | 23.05 | 23.57 | 23.36 | 22.64 | 23.43 | 23.32 | 23.30 | 23.30 |
| No. of contigs | 1232 | 616 | 156 | 104 | 518 | 315 | 34 | 35 | 30 | 18 | 22 | 29 | 21 | 17 |
| No. of genes | 14 650 | 13 031 | 9172 | 9246 | 21 183 | 12 715 | 5763 | 6298 | 6159 | 5.521 | 5743 | 5707 | 5718 | 5723 |
| No. of pseudogenes | 5333 | 4420 | 498 | 420 | 763 | 489 | 146 | 127 | 148 | 112 | 166 | 129 | 142 | 119 |
| Runtime (hours, 60 cores) | 0.94 | 7.46 | 0.39 | 5.90 | 0.85 | 5.16 | 10.21 | 23.48 | 0.30 | 4.131 | 9.33 | 11.05 | 0.23 | 3.139 |
| Isolates/tools | T. brucei | L. biglobosa | H. sapiens 100 Mbp | PfCO01 RSII | Pf2004 RSII | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MaSuRCA | MaSuRCA | |||||||||||||
| ARAMIS | ILRA | ARAMIS | ILRA | ARAMIS | ILRA | Assemblosis | MpGAP | ARAMIS | ILRA | Assemblosis | MpGAP | ARAMIS | ILRA | |
| Size (Mbp) | 65.53 | 57.84 | 34.95 | 34.93 | 100.44 | 64.60 | 23.05 | 23.57 | 23.36 | 22.64 | 23.43 | 23.32 | 23.30 | 23.30 |
| No. of contigs | 1232 | 616 | 156 | 104 | 518 | 315 | 34 | 35 | 30 | 18 | 22 | 29 | 21 | 17 |
| No. of genes | 14 650 | 13 031 | 9172 | 9246 | 21 183 | 12 715 | 5763 | 6298 | 6159 | 5.521 | 5743 | 5707 | 5718 | 5723 |
| No. of pseudogenes | 5333 | 4420 | 498 | 420 | 763 | 489 | 146 | 127 | 148 | 112 | 166 | 129 | 142 | 119 |
| Runtime (hours, 60 cores) | 0.94 | 7.46 | 0.39 | 5.90 | 0.85 | 5.16 | 10.21 | 23.48 | 0.30 | 4.131 | 9.33 | 11.05 | 0.23 | 3.139 |
DISCUSSION
With the advent of long read technologies, the rapid fall in cost and the development of associated algorithms for the analysis, genome sequencing has become more popular and accessible for many laboratories worldwide [43]. However, researchers performing de novo assemblies typically have limited bioinformatics knowledge. Here, we demonstrate that automatic finishing of assemblies is essential to improve the quality of the final genome sequences. We present the ILRA pipeline, which in all cases improved the sequencing outcome, despite lower quality or variable technologies and sequencing reads. We also explored the impact of different assembler and polisher software, and showed that ILRA scales up to sequences of 1 Gbp.
First, we investigated the impact of the use of different assembler software on the finishing process. Although the results were similar for some, MaSuRCA (i.e. a hybrid assembler including PacBio and Illumina reads) outperforms the others. It is important to outline that assembly and post-assembly is a rather sequential process: first the longest reads are corrected with shorter reads, they are then assembled, resulting contigs may be further scaffolded and at last post-assembly tools like ILRA would process the sequences. Notably, despite up-to-date development like reduced error rates in ONT [12, 14, 27], there is an everlasting need of correcting both the sequencing reads and ensuing assemblies. Hybrid assemblers may also use input from different sequencing technologies to compensate the errors that are intrinsic to each one independently, achieving better results that are however still far from perfect [20, 44]. Overall, we advocate that the best assembly would be the one most continuous and containing most of the sequence. In our tests, the assemblies from MaSuRCA ranked first.
To test the how different assembler software may impact the ILRA pipeline, we chose P. falciparum as a case example of extremely low GC content and we generated assemblies with highly variable reads from different library preparations and chemistries. For example, DNA was insufficient for the sample PfKE07 and a WGA approach was performed to ensure enough DNA for sequencing, which resulted in a fragmented assembly with artifacts. PacBio recently generated new methods to obtain long reads from as little as 100 ng of DNA [45], but these methods may need to be first popularized and implemented by more sequencing facilities. Furthermore, even if the final results are far from perfect, we demonstrated that ILRA is capable of largely improving WGA-based assemblies. PfCO01 assemblies are another example illustrating how the initial quality of the reads may impact the results. These genome assemblies were obtained from reads from the same biological samples, alternatively sequenced by PacBio Sequel or PacBio RSII technologies, and displayed differences (~36.7% more contigs in PfCO01 Sequel). We report that the ILRA pipeline was able to correct them to the same extent (difference of one contig). Overall, high quality reads and primary assemblies are crucial for obtaining gold standard genomes. However, these are not always available, and when large consortia like the Earth BioGenome aim to obtain genomes from more challenging samples (e.g. due to environments within other species, or common difficulties to obtain adequate samples when DNA is limited or contaminated) automatic finishing tools like ILRA must be incorporated.
Long reads by PacBio and ONT are prone to errors around homopolymer tracts and STR, so consensus sequences may also contain errors in the assembled contigs [9, 33]. To address this, we compared two polishing algorithms (Pilon and iCORN2) and concluded that, for our use case with extremely high AT content (P. falciparum), an iterative approach is necessary to correct errors that greatly hamper downstream functional analyses (i.e. differences in annotated genes and pseudogenes). We propose here the number of annotated pseudogenes after correction as a metric for quality control, together with additional evidence that the sequences are correct (i.e. protein evidence or comparison from references or close organisms). We show that in P. falciparum and T. brucei, iCORN2 matched or superseded Pilon. Importantly, even at the cost of larger runtimes, an iterative approach should be implemented in all cases to fully benefit from the short reads when improving long read assemblies. Unlike iCORN2, Pilon does not have a native iterative implementation but when tested iteratively, as done by others [16, 19, 46], Pilon performed better, even if it was still outperformed by iCORN2. Nevertheless, ILRA users can choose either of the tools. Ultimately, the fact that there is not really a ground truth for a ‘perfect’ genome sequence highlights the outstanding dilemma that for some reference genomes it is difficult to determine the final consensus sequence.
Despite the availability of other pipelines to automatically finish genome sequences (e.g. Assemblosis, ARAMIS or MpGAP), none of the tools performs the wealth of post-assembly processing of ILRA, which outperforms them. This is mainly because some of the tools lack some processing steps or include the assembly step by predetermined software with a single round of polishing, which has downsides as shown before. In terms of post-assembly improvement, ARAMIS and MpGAP only perform polishing by Pilon (ARAMIS requires an indel detected by Pilon to be present in a determined fraction of the aligned reads, and MpGAP performs iterative polishing). In terms of contiguity, ARAMIS was the most similar to ILRA because they operate on the same primary assembly, while regarding polishing, the number of annotated pseudogenes by MpGAP was closest to ILRA. The low number of pseudogenes is explained because MpGAP applies an iterative implementation of Pilon via Unicycler until minimal changes are made, but in some cases, this required large runtimes to reach ~20 and ~40 iterations. In general, genomes with a ploidy >1 are difficult to sequence and assemble, so collapsed consensus sequences are more frequent [47, 48]. Of note, the size of the ILRA-corrected human assemblies are nearly halved. This is because the assemblies used included both alleles. Although there are more assemblers that split the haplotypes [49–51], ILRA is designed to collapse the sequences to one allele, so it facilitates the use of the resulting assemblies for NGS analysis.
Overall, ILRA is more efficient, outperforms other tools and provides a reliable output for non-specialist users. Beyond polishing, it has several unique features that are not present in any other tool and enable extensive improvements. These include reordering of contigs based on a reference, removal of sequences not covered by Illumina short reads, analysis of telomere sequences, automatic formatting and filtering following NCBI’s Foreign Contamination Screen, or decontamination step at the assembly level (also in Assemblosis). ILRA also provides comprehensive reports uncovering the contigs that may include contamination, which are discarded, and the errors present in the sequencing reads, which are polished. Accordingly, with this information users may pre-process and reassemble the reads, so ILRA can be applied again over genome assemblies from reads corrected beforehand, so results are further improved.
Finally, the four de novo P. falciparum assemblies from field isolates, generated by MaSuRCA and automatically corrected by ILRA in this study, are now publicly available. In particular, the isolate from Colombia is the first reference genome recently cultured from South America.
CONCLUSIONS
Long read technologies enable generation of almost perfect de novo genome assemblies from any organism, but consensus sequences still need polishing. Furthermore, in many cases it is not possible to assemble reads at the chromosome level due to challenges such as limited amount of DNA, low DNA quality or contamination. In all these cases, the ILRA pipeline is an easy-to-use and accessible tool for any laboratory without deep bioinformatics knowledge. It automatically performs several polishing steps and successfully improves assemblies, making them more continuous and decreasing the number of wrongly assigned pseudogenes due to homopolymer-related errors.
The genome sequences resulting from different library preparation approaches, sequencing techniques and assembler tools are of variable quality, and still require polishing and extensive finishing.
ILRA is an easy-to-use pipeline combining novel and existing tools that automatically performs post-assembly steps, successfully improves sequences and outperforms alternatives.
We corrected genome assemblies from multiple organisms and origins, and we generated novel Plasmodium falciparum genome assemblies of high quality, as a proof of concept addressing a particularly challenging organism.
ACKNOWLEDGEMENTS
We would like to thank the patients who contributed samples and the health workers who assisted with the sample collections. We would also like to thank staff from the Illumina Bespoke Sequencing Team at the Wellcome Sanger Institute for their contribution. This paper is published with permission from the Director of Kenya Medical Research Institute (KEMRI).
FUNDING
This work was supported by the Wellcome Trust [098051, 104111/Z/14/ZR]. E.G.-D. is funded by the Spanish Ministry of Science and Innovation grant no. PID2019-111109RB-I00 and by La Caixa Foundation—Health Research Program (grant no. HR20-00635). J.L.R is funded by a Severo Ochoa Fellowship (BES-2016-076276). D.F.E and J.D.E-P are funded by Colciencias, call 656–2014 ‘EsTiempo de Volver’ award FP44842-503-2014 and ‘Programa Jovenes Investigadores’ special cooperation 552–2015, respectively. M. Marti and N. M. B. Brancucci are funded by WT Investigator Award 110166.
DATA AVAILABILITY
ILRA can be downloaded from GitHub: https://github.com/ThomasDOtto/ILRA. The code and the de novo P. falciparum assemblies corrected in this study are also available in Zenodo—10.5281/zenodo.7516750. Accession numbers of the published data are, T. brucei: ERR1795268/SRR5466319, L. maculans NzT4: PRJEB24469, L. maculans G12–14: PRJEB24467 and P. falciparum reads: ERS037841, ERS001369 and ERS557779, for the 75 bp, 100 bp and 300 bp, respectively. Novel data can also be found on online databases (accession numbers for long reads, short reads and the de novo assemblies in this study, respectively); Colombian (PfCO01, RSII long reads): ERS2460039, ERS1746432, GCA_019802425.1, Colombian (PfCO01, Sequel long reads): ERS2460039, ERS1746432, GCA_019802405.1, Kenyan (PfKE07): ERS2026796, ERS166385, GCA_019802515.1 and Ghanaian (Pf2004): ERS1412916, ERS1306150, GCA_019802415.1. The corrected assemblies and annotation can also be found on the ILRA GitHub page.
Biographical note: ILRA is a pipeline to assist in the post-assembly process and finishing of genome sequences, by filtering contigs, reordering, decontaminating, correcting sequencing errors, circularizing organellar DNA or performing quality control.

{kind=link}
{kind=link}
{kind=link}