




Abstract
Liquid–liquid phase separation (LLPS) of proteins and nucleic acids underlies the formation of biomolecular condensates in cell. Dysregulation of protein LLPS is closely implicated in a range of intractable diseases. A variety of tools for predicting phase-separating proteins (PSPs) have been developed with the increasing experimental data accumulated and several related databases released. Comparing their performance directly can be challenging due to they were built on different algorithms and datasets. In this study, we evaluate eleven available PSPs predictors using negative testing datasets, including folded proteins, the human proteome, and non-PSPs under near physiological conditions, based on our recently updated LLPSDB v2.0 database. Our results show that the new generation predictors FuzDrop, DeePhase and PSPredictor perform better on folded proteins as a negative test set, while LLPhyScore outperforms other tools on the human proteome. However, none of the predictors could accurately identify experimentally verified non-PSPs. Furthermore, the correlation between predicted scores and experimentally measured saturation concentrations of protein A1-LCD and its mutants suggests that, these predictors could not consistently predict the protein LLPS propensity rationally. Further investigation with more diverse sequences for training, as well as considering features such as refined sequence pattern characterization that comprehensively reflects molecular physiochemical interactions, may improve the performance of PSPs prediction.
INTRODUCTION
In recent years, liquid–liquid phase separation (LLPS) of biomolecules has garnered considerable interest, as it underlies the formation of subcellular membraneless compartments, also known as biomolecular condensates [1]. Biomolecular LLPS is closely implicated in various cellular events, such as chromatin rearrangement, RNA metabolism and signal transduction and abnormal phase behavior of proteins is associated with neurodegenerative diseases, cancers and other diseases [2–10]. Multivalent weak interactions between/within proteins or between proteins and nucleic acids have been verified to drive the condensates formation through LLPS [11]. Intrinsically disordered proteins (IDPs) or regions (IDRs), particularly low complexity regions (LCRs) such as prion-like domains (PLDs), RNA binding regions, play a critical role in driving LLPS process through multivalent interactions, especially for those forming condensates through self-assembly, named as phase-separating proteins (PSPs) [12, 13]. Although the phase behavior of proteins is regulated by microenvironment, the features encoded in sequences determine their potential LLPS capacity under physiological condition.
With increasing number of PSPs identified by experiments, a number of sequence-based prediction tools have been developed. Early predictors were generally based on specific protein features, and some were not even intended to predict the phase behavior of proteins, as summarized by Vernon et al. [14] and Shen et al. [15]. For example, PLAAC was constructed based on amino acid composition of PLDs [16]; catGranule was based on the features responsible for granule formation, such as RNA-binding propensity, structural disorder content, etc. [17]; R + Y was developed according to the content of arginine and tyrosine in protein sequences, by analyzing the FET family proteins [18]; and Pscore was built on the statistically expected planar π interaction frequency [19]. With the emergence of several databases specific for proteins related LLPS, such as LLPSDB [20], PhaSePro [21], PhaSepDB [22], DrLLPS [23] and RNAGrabule [24], a new generation of sequence-based predictors for PSPs has been developed, such as DeePhase [25], PSAP [26], PSPredictor [27], Droppler [28], FuzDrop [29] and LLPhyScore [30]. Most of these predictors adopt machine learning techniques, and use different sequence features and training datasets. How these current available predictors perform compared with each other, and whether they can predict LLPS propensity of proteins accurately, have not yet been evaluated.
The recently updated database LLPSDB v2.0 composes of entries more than double of previous version LLPSDB, and the newly curated data provide the opportunity to evaluate the performance of current available prediction tools. In this manuscript, we compare eleven available sequence-based PSP predictors, including the first-generation PLAAC, R + Y, catGranule and PScore mentioned in Vernon RM et al.’s review [14], as well as the new generation PSPer, Droppler, PSAP, PSPredictor, DeePhase, FuzDrop and LLPhyScore. We evaluate their performance based on the new data in LLPSDB v2.0, using the Human proteome, folded proteins, and non-PSPs as negative test sets. Furthermore, we discuss the advances and limitations of these current predictors.
METHODS AND MATERIALS
Phase separation predictors
In this work, eleven predictors based on sequence are compared, including four first-generation tools and seven newly developed tools designed specifically for PSPs. Most of these models are built on engineered features, physiochemical properties of PSPs, or/and use natural language models and a variety of machine learning algorithms such as hidden Markov model (HMM), random forest, etc. A brief description of each predictor is shown in Table 1 as well as the following.
A brief description of the eleven predictors in this study
| Name | Algorithm description | Datasets | Web server (or codes link) | Reference |
|---|---|---|---|---|
| PLAAC | HMM based on amino acid composition of PLDs | Known prion proteins, yeast and human proteomes | plaac.wi.mit.edu | [15] |
| R + Y | Saturation concentrations of protein LLPS are inversely proportional to the product of the numbers of arginine and tyrosine residues | FET family proteins (FUS, EWSR1 and TAF15) | - | [17] |
| catGranule | Features consist of RNA-binding propensity, structural disorder content, the frequencies of arginine, glycine and phenylalanine and sequence length | RNA binding proteins, etc. | s.tartaglialab.com/update_submission/478780/1b2dd6c97b | [16] |
| Pscore | Statistical planer π-π interaction frequency | PSPs from literatures, folded proteins in PDB | pound.med.utoronto.ca/~JFKlab/#PScore | [18] |
| PSPer | HMM-like model containing 16 logical states representing instances of four conceptual regions (RRM, PLD, Spacer and Other) | Proteins from Uniprot as background | bio2byte.be/b2btools/psp/ | [34] |
| Droppler | Multi-head neural attention neural network model, considered protein sequence and experimental conditions (temperature, salt, protein concentration, pH and presence of crowding agent) | PSPs and non-PSPs in LLPSDB | bitbucket.org/grogdrinker/droppler | [27] |
| PSAP | Random forest model based on 55 features including protein length, hydrophobicity, IDR fraction, etc. | PSPs from literatures, human proteomes from Uniprot | github.com/Guido497/phase-separation | [25] |
| PSPredictor | language model (LM)-word2vec, machine learning algorithm- GBDT | PSPs from LLPSDB, folded proteins in PDB | pkumdl.cn:8000/PSPredictor/ | [26] |
| FuzDrop | Binary logistic regression model, considered probability for disorder in free state and disordered binding, as well as hydrophobic term | PSPs from PhaSepDB, PhaSePro and LLPSDB, proteins from human proteome and a mixture of organisms | fuzdrop.bio.unipd.it/predictor | [28] |
| DeePhase | Random forest classifier, LM-word2vec, Engineered feature (sequence length, hydrophobicity, Shannon entropy, LCRs, IDRs, etc.) | PSPs from LLPSDB, folded proteins in PDB | deephase.ch.cam.ac.uk/ | [24] |
| LLPhyscore | Genetic algorithm to optimize features, three-layer ‘neural network’-like model, 8 physical feature (long-range hydrogen bond, long-range disorder, short-range electrostatic interactions, etc.) | PSPs from literatures, folded proteins in PDB, human proteomes | github.com/julie-forman-kay-lab/LLPhyScore | [29] |
| Name | Algorithm description | Datasets | Web server (or codes link) | Reference |
|---|---|---|---|---|
| PLAAC | HMM based on amino acid composition of PLDs | Known prion proteins, yeast and human proteomes | plaac.wi.mit.edu | [15] |
| R + Y | Saturation concentrations of protein LLPS are inversely proportional to the product of the numbers of arginine and tyrosine residues | FET family proteins (FUS, EWSR1 and TAF15) | - | [17] |
| catGranule | Features consist of RNA-binding propensity, structural disorder content, the frequencies of arginine, glycine and phenylalanine and sequence length | RNA binding proteins, etc. | s.tartaglialab.com/update_submission/478780/1b2dd6c97b | [16] |
| Pscore | Statistical planer π-π interaction frequency | PSPs from literatures, folded proteins in PDB | pound.med.utoronto.ca/~JFKlab/#PScore | [18] |
| PSPer | HMM-like model containing 16 logical states representing instances of four conceptual regions (RRM, PLD, Spacer and Other) | Proteins from Uniprot as background | bio2byte.be/b2btools/psp/ | [34] |
| Droppler | Multi-head neural attention neural network model, considered protein sequence and experimental conditions (temperature, salt, protein concentration, pH and presence of crowding agent) | PSPs and non-PSPs in LLPSDB | bitbucket.org/grogdrinker/droppler | [27] |
| PSAP | Random forest model based on 55 features including protein length, hydrophobicity, IDR fraction, etc. | PSPs from literatures, human proteomes from Uniprot | github.com/Guido497/phase-separation | [25] |
| PSPredictor | language model (LM)-word2vec, machine learning algorithm- GBDT | PSPs from LLPSDB, folded proteins in PDB | pkumdl.cn:8000/PSPredictor/ | [26] |
| FuzDrop | Binary logistic regression model, considered probability for disorder in free state and disordered binding, as well as hydrophobic term | PSPs from PhaSepDB, PhaSePro and LLPSDB, proteins from human proteome and a mixture of organisms | fuzdrop.bio.unipd.it/predictor | [28] |
| DeePhase | Random forest classifier, LM-word2vec, Engineered feature (sequence length, hydrophobicity, Shannon entropy, LCRs, IDRs, etc.) | PSPs from LLPSDB, folded proteins in PDB | deephase.ch.cam.ac.uk/ | [24] |
| LLPhyscore | Genetic algorithm to optimize features, three-layer ‘neural network’-like model, 8 physical feature (long-range hydrogen bond, long-range disorder, short-range electrostatic interactions, etc.) | PSPs from literatures, folded proteins in PDB, human proteomes | github.com/julie-forman-kay-lab/LLPhyScore | [29] |
Abbreviation: PSPs—phase-separating proteins; PLD—prion-like Domain; HMM—Hidden Markov model; LM—language model; GBDT—gradient boosting decision tree; PDB—protein data bank; RRM—RNA recognition motif; LCR—low complex region; IDR—intrinsically disordered region
A brief description of the eleven predictors in this study
| Name | Algorithm description | Datasets | Web server (or codes link) | Reference |
|---|---|---|---|---|
| PLAAC | HMM based on amino acid composition of PLDs | Known prion proteins, yeast and human proteomes | plaac.wi.mit.edu | [15] |
| R + Y | Saturation concentrations of protein LLPS are inversely proportional to the product of the numbers of arginine and tyrosine residues | FET family proteins (FUS, EWSR1 and TAF15) | - | [17] |
| catGranule | Features consist of RNA-binding propensity, structural disorder content, the frequencies of arginine, glycine and phenylalanine and sequence length | RNA binding proteins, etc. | s.tartaglialab.com/update_submission/478780/1b2dd6c97b | [16] |
| Pscore | Statistical planer π-π interaction frequency | PSPs from literatures, folded proteins in PDB | pound.med.utoronto.ca/~JFKlab/#PScore | [18] |
| PSPer | HMM-like model containing 16 logical states representing instances of four conceptual regions (RRM, PLD, Spacer and Other) | Proteins from Uniprot as background | bio2byte.be/b2btools/psp/ | [34] |
| Droppler | Multi-head neural attention neural network model, considered protein sequence and experimental conditions (temperature, salt, protein concentration, pH and presence of crowding agent) | PSPs and non-PSPs in LLPSDB | bitbucket.org/grogdrinker/droppler | [27] |
| PSAP | Random forest model based on 55 features including protein length, hydrophobicity, IDR fraction, etc. | PSPs from literatures, human proteomes from Uniprot | github.com/Guido497/phase-separation | [25] |
| PSPredictor | language model (LM)-word2vec, machine learning algorithm- GBDT | PSPs from LLPSDB, folded proteins in PDB | pkumdl.cn:8000/PSPredictor/ | [26] |
| FuzDrop | Binary logistic regression model, considered probability for disorder in free state and disordered binding, as well as hydrophobic term | PSPs from PhaSepDB, PhaSePro and LLPSDB, proteins from human proteome and a mixture of organisms | fuzdrop.bio.unipd.it/predictor | [28] |
| DeePhase | Random forest classifier, LM-word2vec, Engineered feature (sequence length, hydrophobicity, Shannon entropy, LCRs, IDRs, etc.) | PSPs from LLPSDB, folded proteins in PDB | deephase.ch.cam.ac.uk/ | [24] |
| LLPhyscore | Genetic algorithm to optimize features, three-layer ‘neural network’-like model, 8 physical feature (long-range hydrogen bond, long-range disorder, short-range electrostatic interactions, etc.) | PSPs from literatures, folded proteins in PDB, human proteomes | github.com/julie-forman-kay-lab/LLPhyScore | [29] |
| Name | Algorithm description | Datasets | Web server (or codes link) | Reference |
|---|---|---|---|---|
| PLAAC | HMM based on amino acid composition of PLDs | Known prion proteins, yeast and human proteomes | plaac.wi.mit.edu | [15] |
| R + Y | Saturation concentrations of protein LLPS are inversely proportional to the product of the numbers of arginine and tyrosine residues | FET family proteins (FUS, EWSR1 and TAF15) | - | [17] |
| catGranule | Features consist of RNA-binding propensity, structural disorder content, the frequencies of arginine, glycine and phenylalanine and sequence length | RNA binding proteins, etc. | s.tartaglialab.com/update_submission/478780/1b2dd6c97b | [16] |
| Pscore | Statistical planer π-π interaction frequency | PSPs from literatures, folded proteins in PDB | pound.med.utoronto.ca/~JFKlab/#PScore | [18] |
| PSPer | HMM-like model containing 16 logical states representing instances of four conceptual regions (RRM, PLD, Spacer and Other) | Proteins from Uniprot as background | bio2byte.be/b2btools/psp/ | [34] |
| Droppler | Multi-head neural attention neural network model, considered protein sequence and experimental conditions (temperature, salt, protein concentration, pH and presence of crowding agent) | PSPs and non-PSPs in LLPSDB | bitbucket.org/grogdrinker/droppler | [27] |
| PSAP | Random forest model based on 55 features including protein length, hydrophobicity, IDR fraction, etc. | PSPs from literatures, human proteomes from Uniprot | github.com/Guido497/phase-separation | [25] |
| PSPredictor | language model (LM)-word2vec, machine learning algorithm- GBDT | PSPs from LLPSDB, folded proteins in PDB | pkumdl.cn:8000/PSPredictor/ | [26] |
| FuzDrop | Binary logistic regression model, considered probability for disorder in free state and disordered binding, as well as hydrophobic term | PSPs from PhaSepDB, PhaSePro and LLPSDB, proteins from human proteome and a mixture of organisms | fuzdrop.bio.unipd.it/predictor | [28] |
| DeePhase | Random forest classifier, LM-word2vec, Engineered feature (sequence length, hydrophobicity, Shannon entropy, LCRs, IDRs, etc.) | PSPs from LLPSDB, folded proteins in PDB | deephase.ch.cam.ac.uk/ | [24] |
| LLPhyscore | Genetic algorithm to optimize features, three-layer ‘neural network’-like model, 8 physical feature (long-range hydrogen bond, long-range disorder, short-range electrostatic interactions, etc.) | PSPs from literatures, folded proteins in PDB, human proteomes | github.com/julie-forman-kay-lab/LLPhyScore | [29] |
Abbreviation: PSPs—phase-separating proteins; PLD—prion-like Domain; HMM—Hidden Markov model; LM—language model; GBDT—gradient boosting decision tree; PDB—protein data bank; RRM—RNA recognition motif; LCR—low complex region; IDR—intrinsically disordered region
PLAAC: Originally developed to identify prion-like domains (PLDs), this predictor uses a hidden Markov model trained on a set of 28 known prion proteins, on the basis of amino acid composition [16]. The predictor was trained against yeast proteome initially [31] and later extended to human proteome [32]. Although it was designed to screen PLDs associated with amyloid aggregates, a number of human RNA-binding proteins that can potentially form condensates through LLPS are also identified using this method [33].
R + Y: According to the extensive mutagenesis experimental results of the FET family proteins (including FUS, EWSR1 and TAF15), Wang et al. found that the phase separation of these proteins is governed by the interactions between tyrosine residues from PLDs and arginine residues from RNA-binding domains [34]. They further developed the model R + Y to predict phase separation property of proteins beyond FET family, assuming that the saturation concentration of protein phase separation is inversely proportional to the product of the numbers of arginine and tyrosine residues.
catGranule: This algorithm is designed to predict proteins that take part in foci-formation through LLPS. It is built on the discovery that proteins in yeast becoming toxic with the increase of concentration share physiochemical properties with those participating in LLPS under physiological condition [17]. The predictor is trained on the yeast proteome, taking into account the combination of structural disorder content, RNA-binding propensity, sequence length and the frequencies of arginine, glycine and phenylalanine, and it performs well when applied to mammalian proteomes.
Pscore: This predictor is developed to distinguish PSPs based on planar π contact frequency [19]. The algorithm is built on the observation that long-rang planer π-π interactions are more prevalent for intrinsically disordered PSPs. It outputs scores for sequences with no less than 140 residues. A recent report indicates that PScore’s predicted results have a large overlap with those from catGranule [14].
PSPer: It is a screening tool for prion-like RNA-binding PSPs [35], built on a rule-based and unsupervised probabilistic HMM-like model, and the empirical rules for the identification of FUS-like PSPs include PLDs, RNA-recognition motifs (RRMs), disordered and arginine-rich regions.
Droppler: This predictor is designed to predict the likelihood of proteins to undergo LLPS given a specific experimental condition. It is built on a neural network model and uses a novel multi-head neural attention mechanism [28]. Using the data in LLPSDB, the model contains 8891 trainable parameters and is trained on protein sequences as well as corresponding experimental conditions including temperature, salt, protein concentration, buffer pH and presence of crowding agent. According to the finding that no clear correlation exists between experimental conditions and LLPS capacity of proteins [28], in this work, the experimental conditions are not considered when compared with other predictors.
PSAP: Phase separation analysis and prediction (PSAP) is a prediction tool to determine the LLPS likelihood for proteins, constructed based on 90 known human PSPs curated from literatures [26]. It used a random forest model trained on a total of 55 features from the sequence of PSPs, such as protein length, hydrophobicity and IDR fraction.
PSPredictor: It is designed to identify PSPs using data in LLPSDB and single-domain folded proteins in protein data bank (PDB). The model is not built on any specific empirical feature of PSP, instead uses a natural language processing technique word2vec to encode protein sequences. Gradient boosting decision tree (GBDT) technique outperforms a number of machine learning algorithms and is adopted in the final model for prediction [27].
FuzDrop: It is a method developed to identify both ‘droplet driving’ proteins, which undergo LLPS spontaneously, and ‘droplet clients’ proteins, which is dependent on specific droplet-promoting regions in protein sequence to generate interactions with partners [29]. The method is based on the protein conformational entropy of free state and its disordered binding modes in droplets, which includes the probability of disorder in free state, probability of disordered binding, as well as hydrophobic term. The method uses a binary logistic regression model trained on data from PhaSepDB, PhaSePro, LLPSDB, as well as proteins from human proteome and mixture organisms. In this work, we use the part of predicting ‘droplet driving’ proteins for evaluation.
DeePhase: It is a tool to predict protein phase behavior. It combines a neural network-based language model and a machine learning classifier based on knowledge-based features [25]. The authors extracted engineered features (EF) such as sequence length, hydrophobicity, Shannon entropy, LCRs and IDRs, trained on PSPs from LLPSDB, folded proteins from PDB and SwissProt database through a random forest classifier. It indicates a comparative prediction performance when only using natural language model word2vec. The final model combines EF and word2vec to give an average prediction score.
LLPhyScore: It is a predictor of IDR-driven PSPs [30], constructed on a set of optimized features involving LLPS physical interactions or structure pattern, including long-range hydrogen bond, long-range disorder, short-range electrostatic interactions, etc. Through a ‘neural network’-like model, the model is trained on 565 known PSPs from literatures as positive set, and different class of proteins as negative set, including folded proteins in PDB, proteins from human proteomes or both. In this work, we pick the model trained on both folded proteins in PDB and proteins from human proteomes.
Dataset construction
Positive dataset
P_All: To evaluate the performance of the eleven predictors for PSPs, we choose protein sequences in LLPSDB v2.0, which can undergo LLPS in one protein system as positive test sets. We remove proteins that form hetero-oligomer or contain non-natural amino acid. The sequences recorded in LLPSDB are excluded since a couple of predictors (such as Droppler, PSPredictor, FuzDrop and DeePhase) were trained on them. The proteins used to extract features or as positive training data for PLAAC, R + Y, Pscore, FuzDrop, PSAP and LLPhyScore are also excluded. Finally, we obtain a total of 337 protein sequences with length ranging from 140 to 3000 as positive test set P_All. Within P_All, 84 sequences are folded (labeled as Pall_Fold), 73 sequences are entirely disordered (labeled as Pall_IDP), and the left ones include both folded and disordered regions. All other positive datasets described in the following are based on P_All.
P_Self: We select proteins undergoing LLPS alone without the effect of nucleic acids from the dataset P_All, and construct dataset P_Self (including 293 sequences).
P_Physiol: Although it is generally conceived that most of proteins might be able to phase separate to form liquid condensates when the experimental conditions are suitable, those undergoing LLPS under physiological conditions are more important for their biological function and related diseases. Here, we set physiological conditions as — Temperature 0–40 °C, salt concentration 50–200 mM and pH 5–8, and screen proteins from P_All and obtain the test dataset P_Physiol (including 200 sequences).
P_Rep: To reduce the sequence similarity in P_All, we apply the CD-hit algorithm [36] with a cutoff value of 0.4 and collect the representative sequence in each cluster to construct the positive test dataset P_Rep (including 94 sequences).
P_Ident: Experimentally, several typical proteins have been investigated systematically in different research labs at different time, through mutation and truncation of specific regions (such as FUS, TDP-43 [37, 38]) or comparing with different species (such as hnRNPA1 [18, 39]. Although small distinction in sequence such as a few amino acids mutation or the truncation of N-/C-terminal may result in different phase behavior, similar sequences have a large probability to exhibit similar phase state. Therefore, we further examine the sequence similarity between the training proteins used in the eleven predictors and those in P_All, with sequence identity defined as,
where A and B denote sequence from the dataset P_All and positive training dataset in predictors (the training data in catGranule and PSPer are not available and therefore are not considered), respectively. L indicates sequence length, and Nm means the number of aligned amino acids between sequence A and B, which is calculated with Biopython [40]. The largest Identity value between each sequence in P_All and the positive training proteins in predictors is calculated (as the distribution shown in Figure S1). Sequences with a max ‘Identity’ less than 0.5 are used as the positive set P_Ident (including 112 sequences).
Negative dataset
Three negative test datasets N_Human, N_Fold, N_No are constructed in this work, with the sequence length of proteins ranging from 140 to 3000 amino acids.
N_Fold: According to the negative dataset in PSPredictor, in which 5258 single-domain proteins with full-length solved three-dimensional structures from PDB are collected, we construct dataset N_Fold through randomly sampling 1685 sequences (five times of sequence number in P_All) from it. PSPredictor uses 353 sequences that were randomly selected out of the 5258 sequences set in the final model, occupying less than 7%; therefore, the comparison result in this work will not be influenced much by taking N_Fold as test set. The evaluation is performed five times for any positive test set described above, and 337 sequences in N_Fold are used in each time.
N_Human: We screen human proteome (GRCh38 fasta file from NCBI [GCA_000001405.15]) with those sequences in P_All excluded, and finally 75,271 non-redundant sequences are kept, from which 1685 sequences are sampled randomly to construct dataset N_Human. Additionally, two subsets N_HumanIDP1(524 sequences) and N_HumanIDP2 (81 sequences) are screened from human proteome (UP000005640), which exclusively contain entirely disordered sequences with consecutive amino acid number of folded region less than 30 (identified by IUPred3 [41] with iupred_type set as long and other parameters as default and by MobiDB [42], respectively). Similar to N_Fold, 337 sequences from N_Human are used with any positive test set in each of the five evaluations. For the evaluation using N_HumanIDP1 and N_HumanIDP2, all the sequences within them are used once.
N_No: We collect proteins that are recorded as not undergoing LLPS in LLPSDB v2.0 (also labeled as non-PSPs) to construct dataset N_No, remove those used as negative training data in DeePhase and finally keep 153 sequences. Usually, these proteins were investigated comparably with those PSPs under similar experimental conditions (123/153 under physiological conditions as defined above in P_Physiol dataset), and their sequences are mutant or truncation of those comparable PSPs, as the distribution of the largest Identity value of each sequence in N_No comparing with those in P_All shown in Figure S2. Therefore, N_No provides an effective test set for evaluating whether these predicting algorithms could identify the subtle change in sequences.
Evaluation parameters
The receiver operating characteristic (ROC) curve and the area under the curve (AUC) are calculated for evaluation. In addition, to assess the prediction performance of the predictors (including PSPredictor, FuzDrop, DeePhase, as their scoring thresholds are available in corresponding literatures), the accuracy of prediction (ACC), Matthew’s correlation coefficient (MCC), sensitivity, specificity and F1 score are calculated.
In the above equations, TP, TN, FP and FN stand for true positive, true negative, false positive and false negative, respectively.
RESULTS AND DISCUSSIONS
Before evaluating the performance of the studied predictors, we first analyzed their ability to differentiate between folded PSPs and disordered PSPs using KS-test on datasets Pall_Fold and Pall_IDP, based on prediction scores. The results shown in Table S1 reveal that all the predictors show a clear distinction between Pall_Fold and Pall_IDP, with P-values <0.05. Generally, the scores of sequences in Pall_IDP are much larger than those in Pall_Fold, suggesting that these predictors exhibit a bias towards disordered proteins even in the positive test set.
Prediction performance with negative test set N_Fold
Considering single-domain folded proteins may be generally difficult to form liquid condensates through self-assembling, we firstly compared the performances of the eleven predictors with the negative test set N_Fold. Figure 1 and Table S2 show the AUC value of ROC (AUROC) for each predictor with different positive test sets (as described in section ‘Dataset construction’). Other evaluation parameters for PSPredictor, FuzDrop, DeePhase are presented in Table S3. Figure 1 indicates that most of the predictors exhibit larger AUROCs on P_All, P_Self and P_Physiol than on P_Rep and P_Ident, except PLAAC and R + Y, which display no apparent difference for the five positive test sets. This is expected because P_Rep is a low-redundant set and more diverse; meanwhile, P_Ident is composed of sequences with low similarity to those in the training set. The prediction performance is not influenced much by nucleic acid (P_Self) and physiological condition confinement (P_Physiol) in the comparison.
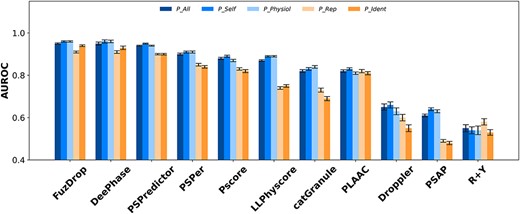
The AUROCs for the eleven predictors with different positive datasets and N_Fold as negative test set.
It is evident that for all the five positive test sets, FuzDrop, DeePhase and PSPredictor outperform other predictors, as shown Figure 1 and Table S1, with AUROCs ranging from 0.94 to 0.96 on P_All, P_Self and P_Physiol, from 0.90 to 0.94 on P_Rep and P_Ident. Other parameters such as accuracy and MCC (Table S3) of the three tools are also similar to each other. PSPer, PScore and LLPhyscore display moderate AUROCs (0.87–0.91 on P_All, P_Self and P_Physiol; 0.74 ~ 0.85 on P_Rep and P_Ident). PLAAC displays a somehow abnormal behavior, with larger AUROCs on P_Rep and P_Ident but smaller ones on P_All, P_Self and P_Physiol than LLPhyscore. It is noteworthy that both DeePhase and PSPredictor use folded proteins in PDB as negative training set to build the predictors; therefore, the same type of test set N_Fold may account for their better prediction performance. Furthermore, DeePhase and PSPredictor both adopt natural language processing technique word2vec, in which the protein sequence is encoded according to its contexts, without requiring artificial feature design or expert knowledge. The comparison results here suggest that this technique may have an advantage to identify PSPs although it does not explicitly consider physiochemical property of the sequence in the model. FuzDrop uses the probabilities of protein disorder in both free and binding states, as well as sequence hydrophobic property, to build the model, which implies that the conformational entropy and hydrophobic property may crucially contribute to identify PSPs.
To find which of the studied predictors could identify folded PSPs from general folded proteins better, we compared the AUROCs on Pall_Fold as positive set and N_Fold as negative set. The results shown in Table S1 indicate that FuzDrop, DeePhase and PSPredictor still perform better than other predictors.
Prediction performance with negative test set N_Human
Recently, using sequences of folded proteins as the negative training set to identify proteins undergoing LLPS has raised controversy, as it may result in the predictor bias to distinguish between disordered and folded proteins rather than identify PSPs [30]. We then randomly select sequences in human proteome as negative test set (N_Human) to evaluate the performance of the eleven predictors. Figure 2 and Table S4 display the AUROCs of these predictors. Table S5 shows other evaluation parameters for PSPredictor, FuzDrop and DeePhase. Comparing the results with those taking N_Fold as negative test set (Figure 1 and Table S2), it is clear that the corresponding AUROCs decrease for all predictors except LLPhyscore. The decrease of AUROCs may be due to more ‘potential false negatives’ since N_Human definitely includes proteins that can undergo LLPS but have not been experimentally verified, and the predictors determine them as ‘false positives’. The slightly higher AUROCs for LLPhyscore on N_Human in comparison with N_Fold may be attributed to the model being trained on a negative dataset comprising folded proteins in PDB and proteins in human proteomes. Furthermore, the training dataset excludes sequences with high values in CRAPome that are considered prone to phase separation, which probably reduces the number of ‘potential false negatives’. Meantime, similar to Figure 1, the positive test sets P_All, P_Self and P_Physiol show larger AUROCs than P_Rep and P_Ident in all predictors except PLAAC and R + Y.
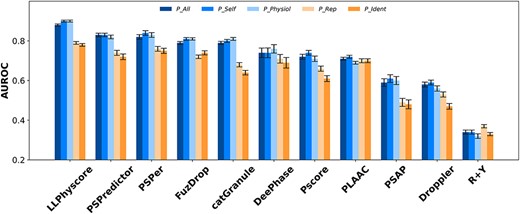
The AUROCs for different predictors with different positive datasets and N_Human as negative test set.
In addition, LLPhyscore displays a better performance than other predictors as the AUROCs are larger on all five positive sets (0.88–0.9 on P_All, P_Self and P_Physiol; 0.78–0.79 on P_Rep and P_Ident), as shown in Figure 2 and Table S4. PSPredictor, PSPer and FuzDrop show AUROCs a bit smaller than LLPhyscore but larger than other else on all positive sets, except FuzDrop has similar AUROCs with catGranule on P_All, P_Self and P_Physiol. DeePhase and PLAAC show moderate AUROCs (around 0.7) on P_Rep and P_Ident, slightly smaller than the four predictors mentioned above. LLPhyscore was trained on negatives including folded proteins from PDB and proteins from the Human proteome. Eight LLPS optimized features presenting weak interactions and structural patterns, such as long-range pi-pi interaction, long-range disorder, Kinked beta-strands similarity, etc. were refined to build the predictor. The evaluation results suggest these features may be more crucial than others in recognizing PSPs from general proteins. In addition, PSPredictor exhibits a bit surprise, since it is trained on folded proteins as negative set, but the performance is comparable with PSPer and FuzDrop (their negative training set is proteome(s)) and better than others except LLPhyscore.
Although predictor such as PSPredictor claims that it is not an IDPs prediction model [27], it is widely acknowledged that IDRs play a crucial role in LLPS, and most predictors use IDR as a feature for training either explicitly or implicitly. Whether these predictors can distinguish PSPs from general IDPs has not been discussed. To address this, we conduct a test using Pall_IDP as positive set and N_HumanIDP1 (obtained through IUPred3) and N_HumanIDP2 (obtained through MobiDB) as negative sets. Although the AUROCs of the studied predictors in Table S1 differ slightly between N_HumanIDP1 and N_HumanIDP2, LLPhyScore, catGranule and Pscore exhibit better identification performance than other predictors on both negative sets (with AUROCs greater than 0.9). These results suggest that the method and features utilized in these predictors may be more competent in identifying LLPS sequences from general IDPs.
Comparing the AUROCs on Pall_fold/N_fold with those on Pall_IDP/N_humanIDP (1 and 2) shown in Table S1, we observe that FuzDrop and DeePhase outperform other predictors in recognizing folded PSPs from folded proteins but do not perform as well in identifying disordered PSPs from general IDPs; LLPhyscore, catGranule and Pscore exhibit opposite performance; PSPredictor performs reasonably well in both cases. These results suggest that natural language processing may be a potent technique for identifying PSPs from general proteins.
Prediction performance with negative test set N_No
It is difficult to confirm with certainty that a protein cannot undergo LLPS under any condition. Consequently, a definitive and completely negative dataset of PSPs does not yet exist. Only LLPSDB and LLPSDB v2.0 contain records of proteins that do not form liquid condensates themselves under specific condition (refer to non-PSPs). These proteins are generally the mutant, truncation or modification of corresponding PSPs recorded in the database. Although these proteins may form condensed droplet themselves under other favorable conditions, their experimental conditions are nearly identical to those of the corresponding PSPs in LLPSDB/LLPSDB v2.0. We use these non-PSPs as a negative test set (dataset N_No) to examine the capability of the predictors in identifying minor sequence variations that result in significantly different phase behavior under the same condition. The performance of the eleven predictors is shown in Figure 3, Table S6 and Table S7. It is apparent that all the AUROCs are much smaller than those on N_Fold and N_Human, with the largest value of 0.65 from LLPhyscore on P_Self as positive test set. For P_Rep and P_Ident, the largest AUROC of 0.62 and 0.58 is obtained from R + Y, respectively, indicating that it performs slightly better than others in this challenging test. Therefore, all the predictors in this study cannot identify non-PSPs rationally. More nuanced and diverse positive and negative training data, as well as optimized features with a deeper understanding of physiochemical mechanism in protein LLPS may improve the performance of prediction tools in the future.
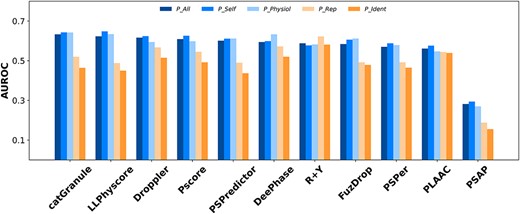
The AUROCs for different predictors with different positive datasets and N_No as negative test set.
Correlation of LLPS prediction between predictors
Each predictor assigns a score to a protein sequence, indicating its propensity to undergo LLPS. To investigate whether these predictors generate similar predicted LLPS propensities, Spearman coefficients are calculated for the predicted scores of sequences in P_Rep across all pairs of predictors. The resulting correlation matrix is depicted in Figure 4 (bottom-left blue triangle matrix). The largest Spearman coefficient is 0.79 observed between FuzDrop and PSAP, followed by the value 0.78 obtained between FuzDrop and PSPredictor, despite they adopt quite different algorithms. In addition, the Spearman coefficient between PSAP and PSPredictor (0.76), and that between LLPhyscore and PSAP (0.75), are relatively high compared to other pairs left. The top-right triangle green matrix of Figure 4 displays the sequence number of overlapped top 20 ranked predictions between any two models on P_Rep. The pair Pscore/catGranule shows the largest overlapped number of 17, followed by 16 from catGranule/LLPhyscore, and 14 from catGranule/PSPer as well as PSPer/LLPhyscore. Overall, PSPer and LLPhyscore and catGranule exhibit more overlapped sequences with other predictors (most of the overlapped number being no less than 10 for them). On the other hand, both the blue and the green grid regions indicate that R + Y has less common prediction tendency with other predictors. The results for other positive test sets (shown in Figure S3) indicate that the Spearman correlation coefficients generally exhibit a similar trend, but the overlapped numbers of top 20 sequences of predictor pair display larger differences for different positive sets.
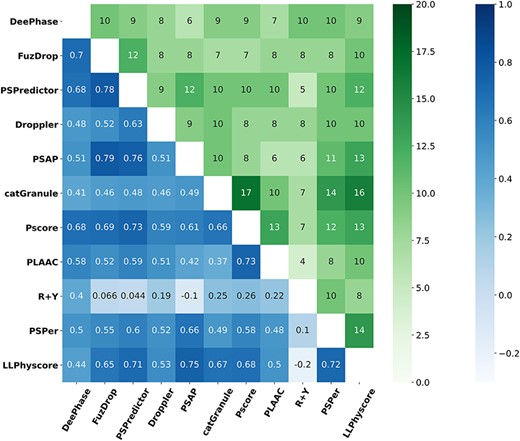
Spearman correlation coefficients of prediction score (bottom–left triangle blue matrix) and overlapped sequence number of ranked top 20 predictions (top–right triangle green matrix) between any two predictors on sequences in P_Rep.
Prediction performance of LLPS propensity
Recently, using the prion-like low-complexity domain (PLCD) from isoform A of human hnRNPA1(referred to A1-LCD hereafter) as model protein, Bremer et al. investigated systematically the role of different types of amino acid in LLPS through extensive mutation experiments [43]. The measured saturation concentrations of various mutated proteins, which form condensates through LLPS at the same pH, salt concentration, temperature, etc., provide valuable dataset for evaluating the performance of predictors in LLPS propensity prediction. We select two group of the mutant sequences from Bremer et al.’s experiments based on the experimental temperature, one including 17 sequences with mutations primarily focused on aromatic or charged residues and the saturation concentrations measured at 4 °C (Table S8), the other including 13 sequences focusing on the role of residue Gly and Ser with the saturation concentrations measured at 20 °C (Table S9).
The Spearman coefficient between saturation concentration and prediction score of the two group of sequences are calculated for nine predictors in this work (except PScore due to sequence length limitation, and FuzDrop due to the online server does not work for unknown reason). Figure 5a and Table S8 show the results for the first group, and Figure 5b and Table S9 show the results for the second group. The results in Figure 5a indicate that all the predictors exhibit a low correlation between prediction score and saturation concentration of LLPS, as the best Spearman coefficient is −0.271 from R + Y, which means none of them could predict LLPS propensity properly. Figure 5b shows that for the sequences in the second group, the correlation is better for most of the nine predictors. The Spearman coefficient from catGranule reaches −0.83. This relative high correlation value may arise from that catGranule is built on the Gly, Arg and Phe content, and the mutant sequences in group two just reflect changes on Gly, Ser, Phe and Tyr content. The Spearman coefficient from PSPredictor in Figure 5b is −0.68, while its prediction scores for these sequences are within a quite small range of 0.9929 ~ 0.9992. Meanwhile, PSAP, R + Y and DeePhase exhibit moderate correlation performance, with Spearman coefficient of −0.678, −0.629 and − 0.585, respectively. Overall, although a couple of predictors in this study can predict LLPS propensity for some sequences to a certain extent, their prediction performance for other sequences is relatively poor.
![The prediction score of nine predictors versus LLPS saturation concentration of protein A1-LCD and mutations from ref [43]. The Spearman coefficient ρ is shown on each panel. (a) Saturation concentrations at 4 °C come from Figures 1f, 2b, 3d, 4b, 4d and 4f in ref [43]; (b) saturation concentrations at 20 °C come from Figure 7d in ref [43].](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/bib/24/4/10.1093_bib_bbad213/1/m_bbad213f5.jpeg?Expires=1749126840&Signature=j0XuxxS614X3HJ66XSuYEI1uaWxm81Xa-ZY-NGpj4Qg9Z6sKmEwr3v-G2mn9b2PIZ-ZbKTJ8FRmwzFSPeHEWc8TQUNOYPqJ3~RmAghJrZ9v4HeknjAULet78vbdtiXCJZKFzgu-A3cH9sv04Mrpc7mutYvjV6TTyluXQHiFq98AdwXf89JdxDKPWXf4jDrTA2hL-jECnEZAyPsTYBu4nywJZ8r2FzYlFZ4vNXnrckQwyFaTRtp9aMEBavrlBdBRq1T0N3bVP-x12wEDcXkD1DoMHtsrBNurQAksbI-IaNFVNE1tf74JgpCLL2p5OgcM9QTRNCEu5pEqGiWXrTQBkgg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
The prediction score of nine predictors versus LLPS saturation concentration of protein A1-LCD and mutations from ref [43]. The Spearman coefficient ρ is shown on each panel. (a) Saturation concentrations at 4 °C come from Figures 1f, 2b, 3d, 4b, 4d and 4f in ref [43]; (b) saturation concentrations at 20 °C come from Figure 7d in ref [43].
CONCLUSIONS
The rapid increasing investigations in biomolecular phase separation have accumulated extensive experimentally measured data, which provide valuable sources for the development of PSP prediction tools. In this paper, based on the updated data in LLPSDBv2.0, we evaluate 11 predictors for PSPs, ranging from first-generation tools such as PLAAC, PScore, to recently developed ones such as PSPredictors, FuzDrop and LLPhyScore, on different negative test sets, as well as experimental measured data. The AUROCs indicate FuzDrop, DeePhase and PSPredictor perform better on folded proteins as the negative set, while LLPhyScore outperforms on human proteome as the negative set, compared with other prediction models. However, these new generation predictors do not perform well in more challenging evaluations, because none of the eleven prediction tools could recognize non-PSPs well, which are usually mutants or truncations of PSPs measured under the same conditions. In addition, the correlation analysis between predicted scores and experimentally measured saturation concentrations of protein LLPS indicates that the tools could not consistently predict protein LLPS propensity properly for various mutant sequences.
Although new generation predictors, which employ language models, machine learning techniques, and refined features, demonstrate improved PSP prediction performance on some datasets, including distinguishing folded PSPs from general folded proteins and disordered PSPs from general IDPs, the fact that all eleven models fail to identify non-PSPs and exhibit inadequate protein LLPS propensity prediction highlights the need for further efforts. To enhance prediction tools’ performance, it is necessary to incorporate more diverse training and test data, as well as essential features such as phosphorylation frequency, as recently reported by Chen et al. [44]. Additionally, features reflecting multiple physiochemical interactions and regulatory mechanisms in protein LLPS may also contribute to future improvements.
Systematical evaluation of PSPs prediction performance for eleven predictors on LLPSDB v2.0 and different negative datasets is performed.
FuzDrop, DeePhase and PSPredictor outperform other tools on folded proteins as negative set, and LLPhyScore shows a better performance on human proteome as negative set.
None of the predictors could reliably distinguish non-PSPs and most of them are inadequate in accurately predicting protein LLPS propensity.
ACKNOWLEDGEMENTS
We thank Dr. Youjun Xu for the calculations through PSPredictor, as well as Professor Guido van Mierlo for the calculations through PSAP, as well as Mr. Tinglan Wang for technique assistant.
FUNDING
This work was supported by the National Natural Science Foundation of China [32071250, 31870718, 21633001] and by the Fundamental Research Funds for the Central Universities.
DATA AVAILABILITY
All data relevant to the study are included in the article or uploaded as supplementary information.
Author Biographies
Shaofeng Liao is a master student at College of Life Sciences, University of Chinese Academy of Sciences. His research interests include machine learning and bioinformatics.
Yujun Zhang is a master student at College of Life Sciences, University of Chinese Academy of Sciences. Her research interests include bioinformatics and data integration.
Yifei Qi is currently an associate professor at School of Pharmacy, Fudan University. His research focuses on applying machine learning in computational biology.
Zhuqing Zhang is currently a professor at College of Life Sciences, University of Chinese Academy of Sciences. Her research focuses on bioinformatics investigation in biomolecules phase separation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}