




Abstract
The rational design of chemical entities with desired properties for a specific target is a long-standing challenge in drug design. Generative neural networks have emerged as a powerful approach to sample novel molecules with specific properties, termed as inverse drug design. However, generating molecules with biological activity against certain targets and predefined drug properties still remains challenging. Here, we propose a conditional molecular generation net (CMGN), the backbone of which is a bidirectional and autoregressive transformer. CMGN applies large-scale pretraining for molecular understanding and navigates the chemical space for specified targets by fine-tuning with corresponding datasets. Additionally, fragments and properties were trained to recover molecules to learn the structure–properties relationships. Our model crisscrosses the chemical space for specific targets and properties that control fragment-growth processes. Case studies demonstrated the advantages and utility of our model in fragment-to-lead processes and multi-objective lead optimization. The results presented in this paper illustrate that CMGN has the potential to accelerate the drug discovery process.
INTRODUCTION
Identifying high-quality drug candidates with desired properties for specific targets is an ongoing goal in drug discovery, as it is hard for medicinal chemists to consider biological activities, drug-likeness and chemical properties simultaneously [1–3]. A traditional approach is to explore the virtual chemical space by some screening methods, such as machine learning prediction models (Figure 1, left) [4–6]. However, the total number of drug candidates may be as high as 1060 and screening a practically infinite chemical space is not possible [7, 8]. Thus, rapid and efficient methods are required to navigate the chemical space. Recently, generative models have become popular as they generate molecules from scratch, learn probability distributions over a set of molecules and sample molecules from the corresponding chemical space. Such approaches circumvent the need to explore the vast chemical space [9–13]. Generative models inspire hope for a new era in drug discovery, where models help medicinal chemists design molecules with required properties more quickly.
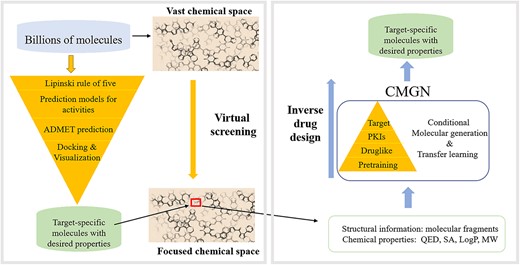
Left: traditional methods to acquire target-specific molecules with desired properties. Right: proposed methods to acquire target-specific molecules with desired properties.
Development of deep generative models has spawned a mass of promising methods to address the structure generation issue in drug design, such as recurrent neural networks (RNNs) [14], variational autoencoders (VAEs) [15] and generative adversarial networks (GANs) [12, 16]. These models have shown their ability to generate valid and novel structures. Further, to tackle the actual problem of generating molecules that exhibit a specific set of properties, different algorithms like Bayesian optimization and reinforcement learning (RL) are applied to make the generator models explore regions of the chemical space where molecules satisfy predetermined constraints. For example, Blaschke et al. [17] combined VAE and GAN to develop a molecule generator and used Bayesian optimization to ensure that molecules with specific properties were created.
Recently, with the advent of transformer models as the state of the art in Natural Language Processing (NLP) tasks, molecular generation based on the representation of molecules in the Simplified Molecular Input Line Entry System (SMILES) has demonstrated the ability to generate molecules with desired properties. Wang J et al. [18] combined conditional transformer, RNN and RL to generate molecules that can satisfy multiple constraints. Additionally, Bagal et al. [7] trained a transformer decoder using masked self-attention for the generation of druglike molecules, and they demonstrated that their model can be trained conditionally to control multiple properties of the generated molecules. Though transformer models have shown potential to solve the inverse problem of drug design, generating molecules with biological activity against specific target and predefined druglike properties still remains challenging.
To address the challenge, we proposed a conditional molecular generation net (CMGN), whose backbone was a bidirectional and autoregressive transformer (BART). To quickly narrow down to certain regions in the chemical space of bioactive compounds, CMGN applied large-scale pretraining for molecular understanding, and fine-tuned on different datasets to navigate the chemical space (Figure 1, right). To solve the inverse problem of properties-guided molecular generation, fragments of molecules and molecular properties, such as molecular weight (MW), quantitative estimates of drug-likeness (QED), LogP and synthetic accessibility (SA), were trained to recover primary molecules and learn the structure–properties relationships (SPR). The effectiveness of CMGN to navigate the chemical space for specific target and generate molecules with desired properties was evaluated using data analysis and case studies. Finally, CMGN was used for multi-objective lead optimization for inhibitors of Bruton’s tyrosine kinase (BTK). CMGN, which combines transfer learning (TL) strategy and conditional molecular generation methods to generate target-specific molecules with desired properties, showed advantages and utility in fragment-to-lead case and multi-objective lead optimization in real drug discovery.
MATERIALS AND METHODS
Dataset preparation
Fifty million molecules in the SMILES format were collected from ZINC for pretraining [19]. Druglike molecules were a subset of the ChEMBL dataset, which were filtered by QED value with a threshold of 0.5 [20]. Protein kinase inhibitors (PKIs), BTK and PAK1 inhibitors were also collected from ChEMBL. BTK and PAK1 inhibitors were not included in the dataset of PKIs. SMILES data were standardized with chirality using RDKit (version 2021.03.1) [21], and duplicate molecules were removed. RECAP and BRICS methods were used to cut the original molecules into fragments, and the original molecules were then removed. Finally, 49 929 577 molecules were retained for pretraining: 973 983 druglike molecules, 37 933 PKIs and 1806 BTK inhibitors. Additionally, 219 PAK1 inhibitors were retained for fine-tuning. Datasets for fine-tuning were randomly split into training and test sets with a 9:1 ratio. Molecular properties of the molecules in the training and test sets are calculated by RDKit.
Data representation
Chemical structures and fragments were represented as SMILES strings. These strings were tokenized through NLP strings, such as ‘C,’ ‘N’ and ‘(’). Further, every SMILES string was expanded with the leading token <SMILES> and lagging token </SMILES>. Similarly, the leading and lagging tokens for fragment SMILES string expansion were < fragment> and </fragment>, respectively [22]. Property ranges are numerical lists of property values. MW ranged within 0–1000 without decimal digits, QED within 0–1 with two decimal digits, LogP within −4 to 7 with one decimal digit and SA within 0–10 with one decimal digit.
Model architecture
The BART backbone in our work was composed of six encoder layers with 12 attention heads, six decoder layers with 12 attention heads and 768-dimensional hidden units. Models were trained using an AdamW optimizer with a batch size of 32 (Figure 2). The model was trained on a server equipped with two Intel Xeon Gold 5320 CPU 32-core 2.20 GHz processors using four NVIDIA A100 GPUs. We pretrained CMGN for two epochs and trained CMGN-DL for 20 epochs. CMGN-PKI, CMGN-BTK and CMGN-PAK are trained for 200 epochs.
Evaluation settings
Conditional metrics were applied to evaluate the ability of CMGN to generate molecules containing input fragments and recover original molecules. Molecular Sets (MOSES) metrics are the main benchmarks for de novo molecular generation.
Conditional metrics:
Same fragment fraction (SFF): percentage of molecules that contain the input fragment.
Recovery: percentage of original molecules generated among test set compounds.
Recovery (Tc ≥ 0.6): generated molecules with Tanimoto similarity coefficient ≥ 0.6 compared with original molecules are regarded as original molecules.
MOSES metrics:
Validity: fraction of valid generated molecules. RDKit was used for validity checks.
Uniqueness: proportion of unique structures generated.
Novelty: proportion of generated molecules not in the training set.
Internal diversity (IntDiv): measure of diversity of the generated molecules calculated as the average Tc in the set of generated molecules.
RESULTS AND DISCUSSION
CMGN approach
BART was employed to pretrain the model to understand the information in a large amount of unlabeled data and capture grammar rules for molecular generation [23]. Conditional training involved the input of fragments and calculated properties of molecules with the goal of recovering the original molecules. Further, a TL strategy was applied to fine-tune on druglike molecules [24]. We then trained the model to generate kinase inhibitors by focusing on PKIs and inhibitors of specific proteins. Eventually, three models were trained: CMGN-DL (based on druglike molecules), CMGN-PKI (based on PKIs) and CMGN-BTK (based on BTK inhibitors).
Quality and properties of molecules generated by CMGN models
Molecules were generated based on single fragments that were expected to exist in the generated structures. SFF was used to assess the capacity of the models to control fragments when generating molecules. In other words, it is the probability that the generated molecule contains a given molecular fragment. The results were listed in Table 1. First, the results showed that the models except CMGN-BTK achieved >90% SFF, demonstrating the ability of CMGN to control for the existence of fragments. The lower SFF for CMGN-BTK might be due to the BTK inhibitor dataset, which was much more focused and unfamiliar with the model compared with the other two sets. Further, recovery rates were low, indicating that the models struggled to recover molecules based on single fragments. Further, the MOSES metrics were evaluated. All models achieved >95% validity, showing that the TL strategy would not affect the model to generate qualified molecules. The models were also tested on extra test sets and the results are presented in Table S1, which was consistent with Table 1. Overall, the quality and properties of generated molecules could be maintained using the TL strategy for downstream tasks.
Performance comparison of models with different training stages
| Metrics | Pretraining | CMGN-TL | |||
|---|---|---|---|---|---|
| CMGN-DL | CMGN-PKI | CMGN-BTK | |||
| Conditional metrics | SFF (%) | 90.659 | 96.592 | 92.676 | 78.493 |
| Recovery (%) | 2.587 | 9.647 | 5.257 | 1.845 | |
| Recovery (%) (Tc ≥ 0.6) | 12.400 | 23.170 | 15.28 | 9.833 | |
| MOSES metrics | Validity (%) | 98.039 | 99.829 | 99.384 | 98.924 |
| Uniqueness (%) | 40.573 | 63.923 | 48.891 | 40.910 | |
| Novelty (%) | – | – | 58.62 | 58.53 | |
| IntDiv | 0.589 | 0.603 | 0.661 | 0.682 | |
| Metrics | Pretraining | CMGN-TL | |||
|---|---|---|---|---|---|
| CMGN-DL | CMGN-PKI | CMGN-BTK | |||
| Conditional metrics | SFF (%) | 90.659 | 96.592 | 92.676 | 78.493 |
| Recovery (%) | 2.587 | 9.647 | 5.257 | 1.845 | |
| Recovery (%) (Tc ≥ 0.6) | 12.400 | 23.170 | 15.28 | 9.833 | |
| MOSES metrics | Validity (%) | 98.039 | 99.829 | 99.384 | 98.924 |
| Uniqueness (%) | 40.573 | 63.923 | 48.891 | 40.910 | |
| Novelty (%) | – | – | 58.62 | 58.53 | |
| IntDiv | 0.589 | 0.603 | 0.661 | 0.682 | |
Performance comparison of models with different training stages
| Metrics | Pretraining | CMGN-TL | |||
|---|---|---|---|---|---|
| CMGN-DL | CMGN-PKI | CMGN-BTK | |||
| Conditional metrics | SFF (%) | 90.659 | 96.592 | 92.676 | 78.493 |
| Recovery (%) | 2.587 | 9.647 | 5.257 | 1.845 | |
| Recovery (%) (Tc ≥ 0.6) | 12.400 | 23.170 | 15.28 | 9.833 | |
| MOSES metrics | Validity (%) | 98.039 | 99.829 | 99.384 | 98.924 |
| Uniqueness (%) | 40.573 | 63.923 | 48.891 | 40.910 | |
| Novelty (%) | – | – | 58.62 | 58.53 | |
| IntDiv | 0.589 | 0.603 | 0.661 | 0.682 | |
| Metrics | Pretraining | CMGN-TL | |||
|---|---|---|---|---|---|
| CMGN-DL | CMGN-PKI | CMGN-BTK | |||
| Conditional metrics | SFF (%) | 90.659 | 96.592 | 92.676 | 78.493 |
| Recovery (%) | 2.587 | 9.647 | 5.257 | 1.845 | |
| Recovery (%) (Tc ≥ 0.6) | 12.400 | 23.170 | 15.28 | 9.833 | |
| MOSES metrics | Validity (%) | 98.039 | 99.829 | 99.384 | 98.924 |
| Uniqueness (%) | 40.573 | 63.923 | 48.891 | 40.910 | |
| Novelty (%) | – | – | 58.62 | 58.53 | |
| IntDiv | 0.589 | 0.603 | 0.661 | 0.682 | |
Evaluation of the TL strategy for generating molecules for specific targets
The Fréchet ChemNet distance (FCD) was used to assess the similarity of generated molecules to the corresponding training set [25]. We initially focused on the impact of training epochs of the fine-tuning stage on the FCD value. As shown in (Figure 3), with the increase of training rounds, the FCD value decreased continuously and remained unchanged after 100 epochs. For the fine-tuning stage of BTK, the trend was consistent with that of PKIs, and the FCD reached a minimum value at ~200 epochs. Our models gradually learned distributions of training sets and ultimately generated similar molecules to the training set. More intuitively, we sampled molecules to compare the chemical spaces between CMGN-DL, CMGN-PKI and CMGN-BTK. An MW range of 450–550 was used to sample 10 000 molecules. Molecules generated by CMGN-DL were distributed in a completely different space compared with BTK inhibitors. Instead, the chemical space of CMGN-PKI-generated molecules covered the space of BTK inhibitors with an FCD value of 15.09 (Figure 4). The chemical space of molecules generated by CMGN-BTK was covered by the space of BTK inhibitors and the FCD value was 3.85, which was relatively lower than that of CMGN-PKI, indicating that fine-tuning on the BTK dataset made the generated molecules more like BTK inhibitors. Simultaneously, the novelty value of CMGN-BTK reached 55.2%. Thus, TL strategy enabled the generation of molecules similar to the training set while maintaining a certain novelty.
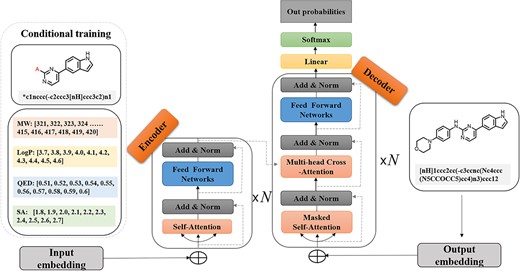
Architecture of the CMGN.
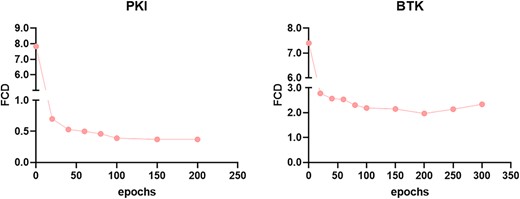
Impact of training epochs of CMGN-PKI and CMGN-BTK on the FCD values compared with those of BTK inhibitors.
So far, we had analyzed how CMGN was able to generate molecules for specific targets. In the following text, we evaluated the ability of CMGN to generate molecules that exhibit specific properties. Thus, we trained CMGN-DL models under different conditions for evaluation. Additionally, since GuacaMol had a wide range in property values, the extra data set, a subset of GuacaMol, was employed to test the models’ ability to control molecular properties trained on it.
Quality and properties of the generated molecules of CMGN-DL models with different input conditions
The performances of models with a single fragment and a single property as input were evaluated. The results showed that the models all achieved ~96% SFF (Table 2), indicating that addition of properties would not affect model’s ability to retain input fragments. Importantly, the addition of a single property improved recovery. Moreover, the addition of four properties increased the recovery to 29.46%. The results indicated that our model had learned the SPR and the properties had helped the model to recover the original molecules. In addition, since our methods of fragmentation cut molecules into 6 to 10 fragments, we tried to add more fragments to the model (Figure S1). Not surprisingly, two fragments increased the recovery by 26%, and three fragments improved it to 63.25%. Finally, CMGN-DL with three fragments and four properties recovered 85.74% of original molecules in the extra test set, indicating that our model could accept both structural and chemical information of structures in generation process. For MOSES metrics, it was observed that when more structure information was added to the model, the validity uniqueness and IntDiv values all decreased slightly. The models were also tested on extra test sets and the results were presented in Table S2, which was consistent with Table 2.
Performance comparison of CMGN-DL with different input conditions
| Metrics | Single fragment + | Two fragments | Three fragments | Three fragments + All | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| – | MW | LogP | QED | SA | All | |||||
| Conditional metrics | SFF (%) | 96.59 | 96.31 | 96.51 | 96.68 | 96.71 | 95.71 | 91.76 | 83.36 | 77.64 |
| Recovery (%) | 9.65 | 20.38 | 14.69 | 12.00 | 12.46 | 29.46 | 35.87 | 63.25 | 85.74 | |
| Recovery (%) (Tc > 0.6) | 23.17 | 26.87 | 24.97 | 24.26 | 25.03 | 36.92 | 61.11 | 84.44 | 91.59 | |
| MOSES metrics | Validity (%) | 99.83 | 99.29 | 99.80 | 99.82 | 99.80 | 97.49 | 99.47 | 99.01 | 86.48 |
| Uniqueness (%) | 63.92 | 56.71 | 57.34 | 58.92 | 56.61 | 55.33 | 33.67 | 26.93 | 33.41 | |
| IntDiv | 0.60 | 0.64 | 0.60 | 0.59 | 0.58 | 0.64 | 0.47 | 0.41 | 0.46 | |
| Metrics | Single fragment + | Two fragments | Three fragments | Three fragments + All | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| – | MW | LogP | QED | SA | All | |||||
| Conditional metrics | SFF (%) | 96.59 | 96.31 | 96.51 | 96.68 | 96.71 | 95.71 | 91.76 | 83.36 | 77.64 |
| Recovery (%) | 9.65 | 20.38 | 14.69 | 12.00 | 12.46 | 29.46 | 35.87 | 63.25 | 85.74 | |
| Recovery (%) (Tc > 0.6) | 23.17 | 26.87 | 24.97 | 24.26 | 25.03 | 36.92 | 61.11 | 84.44 | 91.59 | |
| MOSES metrics | Validity (%) | 99.83 | 99.29 | 99.80 | 99.82 | 99.80 | 97.49 | 99.47 | 99.01 | 86.48 |
| Uniqueness (%) | 63.92 | 56.71 | 57.34 | 58.92 | 56.61 | 55.33 | 33.67 | 26.93 | 33.41 | |
| IntDiv | 0.60 | 0.64 | 0.60 | 0.59 | 0.58 | 0.64 | 0.47 | 0.41 | 0.46 | |
Performance comparison of CMGN-DL with different input conditions
| Metrics | Single fragment + | Two fragments | Three fragments | Three fragments + All | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| – | MW | LogP | QED | SA | All | |||||
| Conditional metrics | SFF (%) | 96.59 | 96.31 | 96.51 | 96.68 | 96.71 | 95.71 | 91.76 | 83.36 | 77.64 |
| Recovery (%) | 9.65 | 20.38 | 14.69 | 12.00 | 12.46 | 29.46 | 35.87 | 63.25 | 85.74 | |
| Recovery (%) (Tc > 0.6) | 23.17 | 26.87 | 24.97 | 24.26 | 25.03 | 36.92 | 61.11 | 84.44 | 91.59 | |
| MOSES metrics | Validity (%) | 99.83 | 99.29 | 99.80 | 99.82 | 99.80 | 97.49 | 99.47 | 99.01 | 86.48 |
| Uniqueness (%) | 63.92 | 56.71 | 57.34 | 58.92 | 56.61 | 55.33 | 33.67 | 26.93 | 33.41 | |
| IntDiv | 0.60 | 0.64 | 0.60 | 0.59 | 0.58 | 0.64 | 0.47 | 0.41 | 0.46 | |
| Metrics | Single fragment + | Two fragments | Three fragments | Three fragments + All | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| – | MW | LogP | QED | SA | All | |||||
| Conditional metrics | SFF (%) | 96.59 | 96.31 | 96.51 | 96.68 | 96.71 | 95.71 | 91.76 | 83.36 | 77.64 |
| Recovery (%) | 9.65 | 20.38 | 14.69 | 12.00 | 12.46 | 29.46 | 35.87 | 63.25 | 85.74 | |
| Recovery (%) (Tc > 0.6) | 23.17 | 26.87 | 24.97 | 24.26 | 25.03 | 36.92 | 61.11 | 84.44 | 91.59 | |
| MOSES metrics | Validity (%) | 99.83 | 99.29 | 99.80 | 99.82 | 99.80 | 97.49 | 99.47 | 99.01 | 86.48 |
| Uniqueness (%) | 63.92 | 56.71 | 57.34 | 58.92 | 56.61 | 55.33 | 33.67 | 26.93 | 33.41 | |
| IntDiv | 0.60 | 0.64 | 0.60 | 0.59 | 0.58 | 0.64 | 0.47 | 0.41 | 0.46 | |
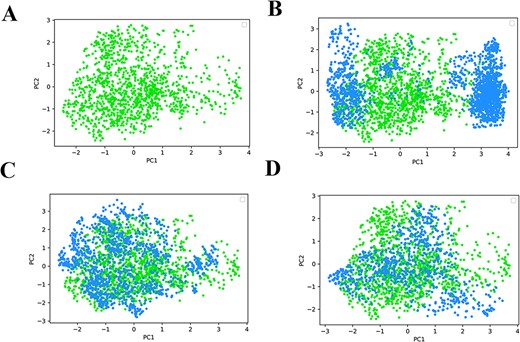
(A) Distributions of molecules of BTK inhibitors; Molecules generated by (B) CMGN-DL, (C) CMGN-PKI and (D) CMGN-BTK.
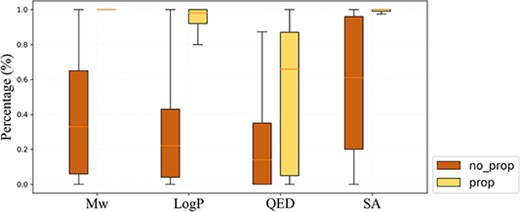
Distribution of the proportion of molecules whose properties are within the given property range among 100 generated molecules for 10 000 target molecules.
Capacity of CMGN-DL to control molecular properties in the process of molecular generation
As shown above, the increase of recovery rate of CMGN benefited from the input properties, demonstrating the potency of CMGN to control the properties of generated molecules. Here, we conducted experiments to check CMGN’s capacity to control molecular properties. The CMGN-DL model was tested with and without a single property as input, and the proportion of molecules whose properties were within the given property range of the target molecule among the 100 generated molecules was calculated. As depicted in Figure 5, molecule generation with property input improved the percentage of molecules satisfying input conditions compared with that of the generated molecules without a property. In addition, the results showed that MW and SA had the best controlling effect in that the average percentages of generated molecules satisfying the input MW and SA all exceeded 95%. The average percentage value for LogP was 92.9%. Meanwhile, QED exhibited a weaker impact, with an average value of 53.11%. QED is dependent on multiple molecular properties simultaneously, making it difficult to control. We believe that the complexity of QED is the reason why it has poor control over structure generation. In addition, it was worth mentioning that when the generation was based on single fragment, the SSF all exceeded 95% as shown above. Overall, CMGN exhibited good control of properties in a fragment-based generation test.
Evaluation of the capacity of CMGN-DL to control user-defined molecular properties
Molecules are often required to have specific properties for drug design. For example, making designed molecules easier to synthesize often requires low values of SA. Additionally, lipophilicity is a primary factor for drugs designed to cross the blood–brain barrier, and LogP values are usually high [26]. Thus, the ability to artificially set properties in the molecular generation process is critical. We set different target property intervals for molecular generation based on the same molecular fragments in the same dataset to evaluate CMGN-DL capacity to control user-defined molecular properties.
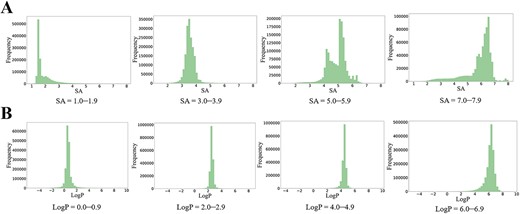
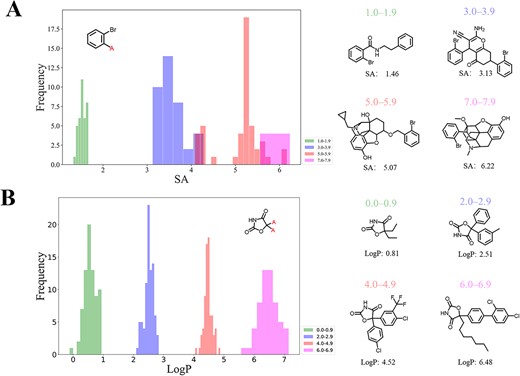
Generated distributions for single SA and single LogP control were shown in Figure 6. When SA ranged within 1–1.9 and 3.0–3.9, the molecular properties were well controlled. Conversely, for the SA range of 5–5.9, only a fraction of generated molecules was within input properties. At 7–7.9, most properties of generated molecules lay between 6 and 7, with a trend toward generating molecules with higher SA (Figure 6A). The SA value of training set molecules lies between 1 and 6 (Figure S2). This finding provides an explanation of the difficulty of CMGN to generate SA values >7. As shown in the Figure 7A, based on the starting fragment, the defined SA values controlled the generation process of CMGN. In detail, the higher the values of SA, the more complex the molecules generated. The performance of the controlling effect of LogP values was also evaluated. As shown in Figure 6B, LogP showed better control over molecular generation than SA values. The example of generated molecules conditioned on LogP value also demonstrated their excellent controlling power (Figure 7B). To be specific, a higher LogP value goal would likely encourage CMGN to assign lipophilic groups to the starting fragment, which to some extent indicated the intelligence of our conditional generation model.
Distribution of properties of generated molecules conditioned on different ranges of (A) SA and (B) LogP.
Generation based on multiple user-defined molecular properties
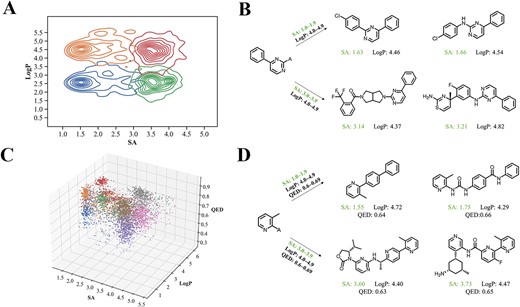
We used two or three properties simultaneously to examine the ability of CMGN to control multiple inputs (Figure 8A). Clusters centered at given ranges of SA and LogP were observed (Figure 8B). Based on the same starting fragment, same LogP values and different given SA values, the generated molecules maintained the given fragment and showed the same ranges of LogP values as predefined. Different SA values caused the model to generate molecules of different complexity, whose SA values were also in the ranges of conditions. Further, SA, LogP and QED were input to the model, and the distribution of properties was depicted in Figure 8C, which also showed separated clusters. The example shown in Figure 8D demonstrated the well controlling power of the CMGN model over three properties.
Examples of generated molecules conditioned on different ranges of (A) SA and (B) LogP.
Evaluation of the effectiveness of the TL strategy and property-guided fragment growing methods of CMGN for target-specific task
To evaluate the effectiveness of the TL strategy and the property control of CMGN in target specific task, a BTK inhibitor reported in our previous study was used as a target molecule [27]. Notably, the target molecule and its derivatives were not included in the datasets of DL, PKI and BTK. Initially, only one fragment was input into CMGN-DL, CMGN-PKI and CMGN-BTK (Figure 9A). Molecules generated by CMGN-BTK were most similar to the target molecule, with Tanimoto similarity coefficient (Tc) values of 0.71, 0.67 and 0.66, but the target molecule was not recovered. Next, the properties of the target molecule were calculated and the property ranges that contain the properties of the target as the input were used as input. To our delight, the target molecule was recovered by CMGN-BTK along with a similar molecule with a Tc of 0.81 (Figure 9B). Additionally, molecules generated by CMGN-DL and CMGN-PKI with properties as input were more complex than molecules generated without properties. This finding may reflect the guiding effect of input properties. The results showed that the TL strategy we applied made the generated molecules more focused and the CMGN had the ability to generate molecules with desired properties in a real drug discovery process.
Distribution and example of properties of generated molecules conditioned on different ranges of (A, B) SA + LogP and (C, D) SA + LogP + QED.
Fragment-to-lead case study
To further verify the utility of CMGN in real drug discovery process, a successful fragment-to-lead case reported in 2020, which belongs to fragment-based drug discovery (FBDD), was employed. In this case. Zhang M et al. identified 1H-indazole-3-carboxamide derivatives as potential PAK1 inhibitors with IC50 values of 5 μM, using a fragment-based screening approach [28, 29] (Figure 10). By linking fragments to the original compound, they successfully obtained a representative compound with an IC50 of 9.8 nM. To evaluate the ability of CMGN to perform fragment-to-lead studies, we trained a CMGN-PAK model to mimic the optimization process, and the similar molecules of the target molecule were all removed from the training set. Here, starting from the Fragment 1 and accompanied by the property range of target molecules, we generated 100 candidates and the most similar molecule had a Tc value of 0.58. It was noted that Fragment 1 was not generated by RDKit and might be unfamiliar to CMGN-PAK. Thus, Fragment 2, which was generated by RDKit, was used for generation. The most similar generated molecule showed a Tc of 0.78, whose only difference was the position of the N atom on pyridine. Further, Fragment 3 was input to optimize the pyridine moiety, and the native molecule was successfully recovered. Ortho- and meta-pyridine were also generated. In the generation process of our model in this case, CMGN-PAK generated molecules based on user-defined and machine-defined fragments. With two rounds of molecule generation based on machine-defined fragments and property ranges of target molecules, CMGN-PAK generated the compounds with the best activity.
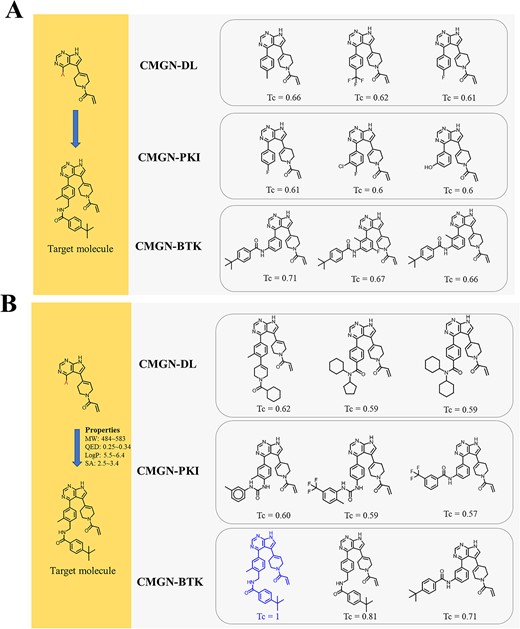
Molecular generation by CMGN-DL, CMGN-PKI and CMGN-BTK based on fragment and properties: (A) molecular generation based only on a fragment; (B) molecular generation based on fragment and properties of the target molecule.
Multi-objective parameter optimization of BTK inhibitors
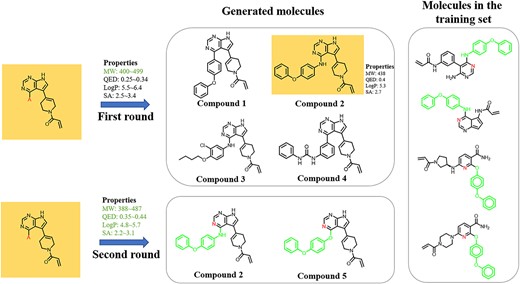
As shown in the test results above, the CMGN had the ability to effectively manage one or more parameters of molecules, which was absolutely critical in lead optimization because it was necessary to consider upfront the ‘druggability’ of compounds in drug discovery. The target molecule in our previous study exhibited acceptable potency, but its MW exceeded 500. Since CMGN had been demonstrated to have the ability to control the properties of generated molecules based on the same starting fragment, CMGN was applied to generate molecules with MWs between 400 and 500. Therefore, the MW range of the input properties was adjusted to 400–500. CMGN-BTK and CMGN-PKI were applied to generate molecules, and four representative compounds were shown in Figure 11. The binding pattern of the four molecules was predicted using covalent docking module and compound 2 exhibited the highest docking score (Figure S3). The details of the docking methods are shown in Table S4. For compound 2, the MW, LogP and SA values were in or near the ranges of input properties, and its QED showed significant improvement. To further generate molecules with optimized QED and SA values, the property ranges of compound 2 were input to CMGN, which resulted in compound 5. In order to analyze the reason why the model could generate compounds 2 and 5 bearing 4-phenoxyphenol and 4-phenoxyphylline moiety, we found the molecules containing these two fragments in the training set. As shown in the right side of Figure 11, the 4-phenoxyphenol and 4-phenoxyphylline moiety of the molecules all lied in the ortho-position of the N atom of pyridine or pyrimidine, which was similar to compounds 2 and 5. Thus, CMGN recognized a connection between different molecular fragments to generate new molecules. Notably, we synthesized compounds 2 and 5 and their analogs, and tested their biological activity (Table S3). All compounds showed high potency against BTK.
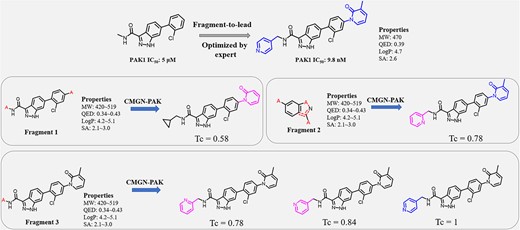
Fragment-to-lead case study.
Overall, we used a ‘hit’ from our previous study, and performed two rounds of optimization by setting preferred molecular properties. CMGN generated property-controlled molecules, and the corresponding analogs showed substantial inhibitory activity against BTK.
BMS-986142 is a noncovalent BTK inhibitor that has completed Phase II clinical trials for treating rheumatoid arthritis (Figure 12) [30]. The MW of this compound is high, and its SA exceeds 4, indicating difficult synthesis. Moreover, its QED is only 0.33, even lower than that of ibrutinib, which bears an unstable moiety and QED of 0.47. Since it had been proved that TL strategy used in CMGN could help chemists in ligand-based drug design, CMGN was used to explore structural modification of BMS-986142. First, BMS-986142 was fragmented, and the core Fragment 1 was extracted. We also defined Fragment 2, which changed the position of one anchor compared with Fragment 1. The fragments were then input into CMGN without defined properties. The model generated noncovalent molecules (compounds 6 and 7) with similar structures to that of BMS-986142. Further, CMGN generated covalent molecules (compounds 8 and 9). This result was, to the best of our knowledge, the first recognition of covalent inhibitors based on the BMS-986142 scaffold. Covalent docking studies with compounds 8 and 9 suggested that compound 9 may be more likely to bind covalently to BTK (Figure S4). This compound still had a low QED (0.27).
Property-guided rounds for optimization of BTK inhibitors. Property ranges shown in green are properties different from those of the original molecule. Similar fragments between generated molecules and molecules in the training set were highlighted.
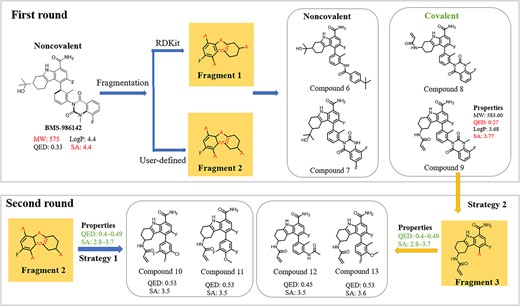
Two rounds of optimization of BMS-986142, based on the core fragment, yielded acceptable QED and SA values.
To further improve the QED value of generated molecules while maintaining the covalent binding mode, we proposed two strategies. First, Fragment 3 was extracted from compound 4 to serve as input fragment. QED of 0.4–0.5 and SA of 2.8–3.7 were fed to CMGN; second, generating molecules based on Fragment 1 with additional optimized property inputs as the first strategy. Both strategies generated covalent binding molecules with QED and SA values in or around the input ranges of properties. Further, the only difference between molecules was in substitutions introduced to the benzene ring. Considering the availability of raw materials, we synthesized compounds s3–s5 with other substitutions on the benzene ring and tested their inhibitory activity (Table S3). Molecules showed moderate potency with IC50 values ~10 nM. Compound s6 bearing a propenamide moiety was synthesized preliminarily to verify covalent binding. The IC50 value was 345.16 nM, significantly weaker than that of compound s3 bearing an acrylamide group. This group in compound s3 formed a covalent bond with BTK. Hence, two rounds of optimization on BMS-986142 led to covalent BTK inhibitors with high QED and low SA values.
The advantages of CMGN
Case studies have demonstrated usefulness of CMGN in FBDD and lead optimization. For FBDD and lead optimization, the diversity and flexibility of fragments that can be input into the model are crucial, which directly affects the practicability of generation methods. Thus, the molecular fragments used for training models and training strategies are important. SyntaLinker [9] and MolGPT [7] are two previously reported generation methods that can generate molecules based on fragments and constraints. Among them, SyntaLinker employs MMP methods to cut molecules into three fragments. The model is trained to generate complete molecules with two fragments as input. Thus, SyntaLinker allows users to input two fragments and the third fragments are generated as the linker. MolGPT uses RDkit toolkit to generate Bemis−Murcko scaffolds, and only one fragment can be extracted (Table 3). The model is trained to generate complete molecules with one fragments and properties as input. Thus, MolGPT allow users to input only one fragment. In contrast, the cutting methods we used can cut molecules into fragments without fixed number, and the fragments of druglike dataset range from 2 to 18. In addition, as shown in Table 3, fragments of CMGN show various sizes, which is consistent with the variable fragment size in actual scenarios. Moreover, our training strategy is different from SyntaLinker and MolGPT. During the training stage, one to three fragments are randomly selected to be input into the model, which result in that we can input one to three fragments in FBDD and lead optimization. In conclusion, CMGN allows users to input one to three fragments with variable size, which cannot be realized by SyntaLinker and MolGPT. The flexible input of CMGN makes it more convenient for users to perform drug design.
Comparison of cutting algorithms used by different generation methods
 | ||||
|---|---|---|---|---|
| Generation methods | Cutting algorithm | Numbers of fragments | Numbers of input fragments | Structures of fragments |
| SyntaLinker | MMP | 3(fixed) | 2(fixed) |  |
| MolGPT | Bemis−Murcko scaffolds | 1(fixed) | 1(fixed) |  |
| CMGN | BRICS + RECAP | 2–18a | 1–3 |  |
| ||||
|---|---|---|---|---|
| Generation methods | Cutting algorithm | Numbers of fragments | Numbers of input fragments | Structures of fragments |
| SyntaLinker | MMP | 3(fixed) | 2(fixed) | |
| MolGPT | Bemis−Murcko scaffolds | 1(fixed) | 1(fixed) | |
| CMGN | BRICS + RECAP | 2–18a | 1–3 | |
aThe numbers of fragments of molecules in the druglike dataset.
Comparison of cutting algorithms used by different generation methods
| ||||
|---|---|---|---|---|
| Generation methods | Cutting algorithm | Numbers of fragments | Numbers of input fragments | Structures of fragments |
| SyntaLinker | MMP | 3(fixed) | 2(fixed) | |
| MolGPT | Bemis−Murcko scaffolds | 1(fixed) | 1(fixed) | |
| CMGN | BRICS + RECAP | 2–18a | 1–3 | |
| ||||
|---|---|---|---|---|
| Generation methods | Cutting algorithm | Numbers of fragments | Numbers of input fragments | Structures of fragments |
| SyntaLinker | MMP | 3(fixed) | 2(fixed) | |
| MolGPT | Bemis−Murcko scaffolds | 1(fixed) | 1(fixed) | |
| CMGN | BRICS + RECAP | 2–18a | 1–3 | |
aThe numbers of fragments of molecules in the druglike dataset.
CONCLUSION
In this work, we proposed CMGN, a conditional molecular generation model based on TL strategy, to solve the challenging problem of generating target-specific molecules with desired properties. For target-specific molecular generation, the TL strategy was performed by pretraining on large-scale molecules and fine tuning on target-specific dataset. For the ability of generation models to generate molecules with desired properties, molecular fragments and properties were input into the model and the training goal was to recover the original molecules. Evaluations were focused on the verification of the effectiveness of TL strategy and the property-guided power of CMGN. The distribution of the molecules generated by CMGN-BTK demonstrated the ability of our model to navigate the chemical space for a specific target. A subset of GuacaMol was employed to evaluate the control of CMGN over four properties. Among them, MW, SA and logP showed excellent controlling power and CMGN exhibited the ability to effectively manage one or more molecular parameters. Further, CMGN showed good performance in a fragment-to-lead case study and multi-objective parameter optimization of BTK inhibitors. In conclusion, CMGN offers a highly effective way to achieve generating target-specific molecules with desired properties tasks.
A CMGN is proposed for tackling the problem of designing target-specific molecules with desired properties.
CMGN applies large-scale pretraining for molecular understanding and navigates the chemical space for specified targets by fine-tuning with corresponding datasets. Fragments and properties are trained to recover molecules to learn the SPR.
CMGN demonstrated the advantages and utility of our model in fragment-to-lead processes and multi-objective lead optimization.
DATA AVAILABILITY
The codes for generating molecules using CMGN are available from the GitHub repository: https://github.com/WJmodels/CMGN.
ACKNOWLEDGMENTS
We thank Yutong Wu for the support of the studies and thank Dr Yadong Chen for the support of the docking studies. The computing resources were supported by biomedical high-performance computing platform, Chinese academy of medical sciences.
FUNDING
This work was financially supported by the National Natural Science Foundation of China (NSFC no. 82073692), CAMS Innovation Fund for Medical Sciences (CIFMS, no. 2021-I2M-1-028) and Disciplines Construction Project (Grant no. 201920200802).
Author Biographies
Minjian Yang is a PhD candidate at the State Key Laboratory of Bioactive Substances and Functions of Natural Medicines, Department of Medicinal Chemistry, Beijing Key Laboratory of Active Substances Discovery and Druggability Evaluation, Institute of Materia Medica, Peking Union Medical College and Chinese Academy of Medical Sciences. He currently works on artificial intelligence-based drug discovery.
Hanyu Sun, Xue Liu and Xi Xue are graduate students at the State Key Laboratory of Bioactive Substances and Functions of Natural Medicines, Institute of Materia Medica, Peking Union Medical College and Chinese Academy of Medical Sciences. Their research interests mainly lie in the area of drug design based on artificial intelligence.
Yafeng Deng is the CEO of CarbonSilicon AI Technology Co., Ltd. He is currently a PhD candidate at the Innovation Leadership Project of Tsinghua University. His current research interests mainly lie in artificial intelligence-based drug discovery.
Xiaojian Wang is currently a professor at the State Key Laboratory of Bioactive Substances and Functions of Natural Medicines, Department of Medicinal Chemistry, Beijing Key Laboratory of Active Substances Discovery and Druggability Evaluation, Institute of Materia Medica, Peking Union Medical College and Chinese Academy of Medical Sciences. His current research interests mainly lie in artificial intelligence-based drug discovery.
REFERENCES
Zhang M, Fang X, Wang C, et al.
Author notes
Minjian Yang and Hanyu Sun contributed equally to this work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}