




Abstract
Chloroplast is a crucial site for photosynthesis in plants. Determining the location and distribution of proteins in subchloroplasts is significant for studying the energy conversion of chloroplasts and regulating the utilization of light energy in crop production. However, the prediction accuracy of the currently developed protein subcellular site predictors is still limited due to the complex protein sequence features and the scarcity of labeled samples. We propose DaDL-SChlo, a multi-location protein subchloroplast localization predictor, which addresses the above problems by fusing pre-trained protein language model deep learning features with traditional handcrafted features and using generative adversarial networks for data augmentation. The experimental results of cross-validation and independent testing show that DaDL-SChlo has greatly improved the prediction performance of protein subchloroplast compared with the state-of-the-art predictors. Specifically, the overall actual accuracy outperforms the state-of-the-art predictors by 10.7% on 10-fold cross-validation and 12.6% on independent testing. DaDL-SChlo is a promising and efficient predictor for protein subchloroplast localization. The datasets and codes of DaDL-SChlo are available at https://github.com/xwanggroup/DaDL-SChlo.
INTRODUCTION
Chloroplasts are critical organelles in the photosynthetic cells of plant kingdom and some algae [1]. In addition to fixing light energy for the synthesis of sugars, photosynthesis plays a vital role in synthesizing phytochromes, fatty acids, amino acids and proteins [2–5]. Chloroplast proteins must be at specific subchloroplast locations to exert these effects. The chloroplast can be subdivided into five subchloroplast locations: stroma, envelope, plastoglobuli, thylakoid lumen and thylakoid membrane. The prediction of protein subchloroplast localization can accelerate the understanding and utilization of chloroplast protein functions, thus, providing a theoretical basis for adjusting the utilization rate of photosynthesis to improve the production of crop yield.
Various published studies have demonstrated the applications and importance of different bioinformatics predictors regarding prediction of protein targeting in different sub-cellular compartments including nucleus, chloroplast, cytoplasm, mitochondria, endoplasmic reticulum, etc. [7–13]. The rapid development of sequencing technology has led to an explosion in the number of protein sequences in various biological databases. Most traditional experimental methods accurately labeling protein locations, such as using immunofluorescence or sucrose density gradient centrifugation, are time-consuming and expensive, resulting in a small percentage of labeled protein sequence samples. Therefore, utilizing fast and inexpensive machine learning methods to predict protein subchloroplast locations is significant and meaningful. Researchers have also made some achievements in recent years [6]. For example, Du et al. [14] designed the first predictor of protein subchloroplast localization using machine learning methods for the first time. In the following years, based on various feature representations, such as Pseudo-Amino Acid Composition, Gene Ontology and Profile-Alignment feature, several protein subchloroplast localization prediction methods have been developed using machine learning methods, such as K-Nearest Neighbor, Adaboost and Support Vector Machine [15–18]. In 2013, Huang et al. [19] made the first attempt to address this problem by building a system to single- and multi-label protein chloroplasts localization using an improved KNN algorithm. Subsequently, researchers constructed multi-label protein subchloroplast localization databases [20, 22]. They developed several multi-label predictors based on transductive learning and other methods, taking protein subchloroplast localization prediction to a new level [20–22]. Recently, Bankapur et al. [23] applied deep learning to chloroplast subcellular localization for the first time and proposed a deep-neural-network-based skipped-grams of evolutionary profiles approach, which significantly improved prediction accuracy.
However, there are still many issues in the field, one of the most prominent issues is insufficient protein sequences with subchloroplast location annotations verified by experiments [6, 24]. This issue leads to predictors’ inadequate training, limiting and severely hindering the development of high-performance predictors. Furthermore, protein sequences express complex semantic information that is challenging for a traditional single feature extraction method. How to reasonably supplement the data to expand the size of the training feature samples and construct the training set using multiple feature methods fusion to acquire a more complete and varied protein sequence feature expression are the challenges for the current research.
Recently, deep learning has a wide range of applications in bioinformatics [25–28]. For example, a multi-module deep learning framework utilized for predicting the sites of protein post-translational modification [29], utilizing graph convolutional network to predict gene function [30], discovering anticancer peptides with the Wasserstein autoencoder model [31], etc. Generative adversarial networks (GANs), a novel method of generative model, was initially used in image processing and later achieved great achievement in data augmentation [32]. For example, Wan et al. [33] based on protein sequences utilized GAN to generate high-quality biophysical features successfully. Li et al. [34] expanded data using GAN, alleviating data scarcity and improving the accuracy of phage–host interaction prediction. These works demonstrate that GANs can create synthetic samples with desired structures or properties as original data supplements, effectively addressing difficult problems in current research. In addition, some works have shown that using unsupervised methods to train and develop protein pretrained language models based on large protein databases has been shown to extract more diverse and comprehensive protein sequence features [35, 36]. Driven by the latest development of GAN, pre-trained model and other deep learning methods, we apply them in the prediction of protein subcellular localization field for further research.
In this paper, we propose a novel multi-location protein subchloroplast localization predictor, DaDL-SChlo. The predictor utilizes GANs and protein pre-training language model to address the current challenges in protein subchloroplast localization. DaDL-SChlo has three modules, including feature extraction, data enhancement and multi-label prediction. First, based on protein sequences, the deep learning features extracted by pre-trained model ProtBERT are fused with handcrafted features of Position-Specific Score Matrix with Automatic Cross-Covariance (ACC-PSSM) to construct a fusion feature set. Then, utilizing the fusion feature set trains Wasserstein GAN with gradient penalty (WGAN-gp) to synthesize feature samples and supplement the original fusion feature set. Finally, a hybrid neural network of transformer encoder and convolutional neural network is used to predict subchloroplast locations based on the extended feature set after supplementation. The entire prediction process of current predictor DaDL-SChlo is shown in Figure 1 through a flow chart which uses three steps: feature extraction, data augmentation and multi-location prediction. Experimental results demonstrate that DaDL-SChlo exceeds the state-of-the-art protein subchloroplast prediction methods with significant improvements in all prediction performance metrics.
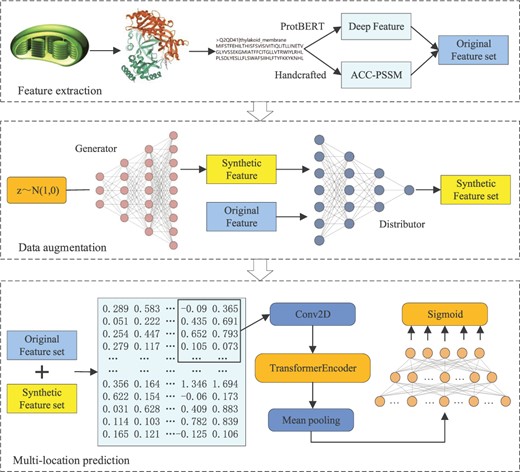
The overall flow chart of the protein subchloroplast predictor DaDL-SChlo we proposed. It includes three steps: feature extraction, data augmentation and multi-location prediction.
MATERIALS AND METHODS
Datasets
To ensure the quality of training data and better evaluate model performance, we used two publicly available multi-location protein subchloroplast datasets, MSchlo578 [20] and Novel [22]. The protein sequence data in both datasets were retrieved from UniProtKB/Swiss-Prot database. These sequences are distributed in five subchloroplast locations verified by experiments. The MSchlo578, which was used as a benchmark dataset, collected proteins added to the UniProtKB/Swiss-Prot database before 31 May 2013. The protein sequence homology in this dataset is less than 40% and only contains the well-annotated complete protein sequences. The protein subchloroplast predictor DaDL-SChlo proposed in this paper was only trained on MChlo578 throughout the training process. In addition, the Novel, which was used as an independent test set, collected chloroplast proteins added to the UniProtKB/Swiss-Prot database from 1 June 2013 to 11 November 2015, ensuring that the training set and independent testing in this study do not have the same sequences.
In addition, MSchlo578 and Novel contain 578 and 122 protein sequences, respectively. They locate at stroma, envelope, plastoglobule, thylakoid lumen and thylakoid membrance. The detailed protein sequences distribution of each subchloroplast location in the datasets can be found in Table 1. Specifically, in addition to the protein sequences with one labeled subchloroplast location, MSchlo578 contains 21 protein sequences with two labeled subchloroplast locations and one protein sequence with three labeled subchloroplast locations, and Novel contains nine protein sequences with two subchloroplast locations. The datasets are available at https://github.com/xwanggroup/DaDL-SChlo.
Feature extraction
Deep learning feature
ProtBERT [37] is a powerful high-performance protein language model obtained by self-supervised training on UniRef and BFD databases containing 393 billion amino acids. Each layer of the model contains a multi-headed attention sublayer and a fully connected feedforward layer. It is a multi-layer bidirectional transformer encoder, which is leveraged for unsupervised deep learning feature extraction in this paper. As shown in Figure 2, first, we use ‘CLS’ tokening each protein subchloroplast sequence and add positional encoding to each amino acid. Then, the characterization vectors generated in the first step are fed as input to the ProtBERT model creating context-aware embeddings for each input (i.e. each amino acid). These embeddings, i.e. features, are concatenated and merged according to the length dimension, and a global average pool is applied to obtain fixed-size features that can be directly input to the downstream prediction module.
The specific distribution of protein sequences at each subchloroplast location in datasets of MSchlo578 and Novel.
| Datasets | Stroma | Envelope | Plastoglobule | Thylakoid lumen | Thylakoid membrane | Overall |
|---|---|---|---|---|---|---|
| MSchlo578 | 105 | 199 | 30 | 34 | 233 | 578 |
| Novel | 26 | 61 | 0 | 5 | 39 | 122 |
| Datasets | Stroma | Envelope | Plastoglobule | Thylakoid lumen | Thylakoid membrane | Overall |
|---|---|---|---|---|---|---|
| MSchlo578 | 105 | 199 | 30 | 34 | 233 | 578 |
| Novel | 26 | 61 | 0 | 5 | 39 | 122 |
The specific distribution of protein sequences at each subchloroplast location in datasets of MSchlo578 and Novel.
| Datasets | Stroma | Envelope | Plastoglobule | Thylakoid lumen | Thylakoid membrane | Overall |
|---|---|---|---|---|---|---|
| MSchlo578 | 105 | 199 | 30 | 34 | 233 | 578 |
| Novel | 26 | 61 | 0 | 5 | 39 | 122 |
| Datasets | Stroma | Envelope | Plastoglobule | Thylakoid lumen | Thylakoid membrane | Overall |
|---|---|---|---|---|---|---|
| MSchlo578 | 105 | 199 | 30 | 34 | 233 | 578 |
| Novel | 26 | 61 | 0 | 5 | 39 | 122 |
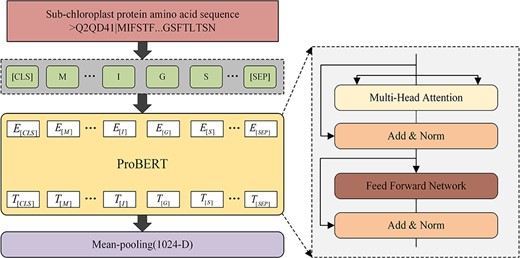
Details of feature extraction by ProtBERT. (i) Using ‘CLS’ tokening each protein sequence and adding positional encoding. (ii) Passing through the ProtBERT model and creating context aware embeddings. (iii) concatenating and merging embedding. (iv) applying a global average pool to obtain features.
Handcrafted feature
Position Specific Scoring Matrix (PSSM) [38] contains rich information about protein evolution, and it is widely used in biological research, such as protein subcellular location prediction, protein structure prediction, etc. Position-Specific Score Matrix with Automatic Cross-Covariance (ACC-PSSM) is the PSSM processed by the automatic cross variance transformation (ACC) method. First, using the PSSM extracts evolutionary information from protein sequences. The PSSM for a protein sequence of length L can be expressed as formula 1.
where |$L$| denotes the length of the protein sequence, |$S_{p,q}$| denotes the statistical score of the amino acid mutation to type |$q$| at position |$p$| during the evolution of the protein.
Due to protein sequence length differences, the generated PSSM matrix cannot be used as the input of the downstream deep learning prediction module directly. Therefore, the PSSM matrix is then converted into fixed length feature vectors using the ACC, which generates two variables, the self-covariance AC between the same attributes and the cross-covariance CC between different attributes. AC variable measures the correlation between two residues of the same property and the distance of |$lg$| along the sequence interval. It can be calculated according to formula 2.
where |$i$| is one of the residues and |$\bar{S_{i}}$| is the average score of amino acid |$i$| along the whole sequence.
CC variable measures the correlation of two different properties between two residues separated by lg along the sequence. It can be calculated by formula 3.
where |$i_{1}$| and |$i_{2}$| are two different amino acids, and |$S_{i_{1}}$|, |$S_{i_{2}}$| is the average value of amino acid |$i_{1}$| and |$i_{2}$| in the sequence. Thus, the protein sequence can be calculated to generate a |$400^{*}lg$| dimensional vector by ACC-PSSM.
Feature selection and fusion
The performance of protein subchloroplast localization predictors depends heavily on the quality of the protein sequence features. Fusing features extracted from different methods can obtain a more comprehensive description of protein sequences, allowing downstream prediction module better to learn the distribution features of different subchloroplast locations. This paper utilizes the complementarity between deep learning features extracted by ProtBERT and the widely used ACC-PSSM handcrafted features to construct a fused feature set.
We used a serial feature fusion strategy to fuse the deep features extracted by the pre-trained protein language model ProtBERT and the handcrafted features extracted by ACC-PSSM. And then, we performed feature selection by Xtreme Gradient Boosting (XGBoost) method. As a feature selection method, XGBoost has been successfully used in the field of bioinformatics. It can directly and effectively obtain the most discriminative features and accelerating the training of downstream prediction modules.
Specificly, we fused 1024-D deep learning features extracted by ProtBERT with 4000-D handcrafted features by ACC-PSSM and then used machine learning method XGBoost to select the 2000-D most representative and differentiated protein sequence features. Then, we reshape the 1D vector with 2000 elements into a 200*10 feature matrix and then feed that into the hybrid deep neural network for prediction.
Data augmentation
GANs [39] are a novel deep learning generative method to complement data. WGAN-gp [40], with the penalty gradients, is a more stable and better generative diversity GAN. The objective function is shown in formula 4, which contains the original critic loss and the gradient penalty term.
In this study, utilizing WGAN-gp learns the underlying distribution of the original feature samples and generates synthetic feature samples addressing the scarcity issue of training samples. Firstly, setting the original feature samples as |$I={p_{1},p_{2},\cdots ,p_{n}}$|, |$p_{i}$| denotes the feature sample of the i-th subchloroplast protein sequence and n represents the number of subchloroplast protein sequences. The original feature samples set |$I$| is fed into the GAN data augmentation module training to generate appropriate synthetic feature samples. An adversarial training paradigm is used by GAN. Among them, the generator network and the discriminator network mutual ‘battle’ learn the real distribution of original feature samples. The generator network with five fully connected layers (formula 5) is used to learn the original protein subchloroplast feature distribution to generate synthetic feature samples. The discriminator network with four fully connected layers (formula 6) trying to distinguish whether the input feature sample is an original feature sample or not. The two networks are trained stronger and stronger on the tasks of generation and discrimination, respectively. When a balance is reached, i.e. the discriminator can no longer distinguish whether a given feature sample is a synthetic sample or not, and the generator can no longer generate higher quality feature samples, we choose the synthetic feature samples in the above cases as a supplement training set to the original feature samples.
where |$I$| denotes the original feature sample set, |$I^{^{\prime}}$| denotes the generated synthetic feature sample set, |$O_{g}$| is the generator output and |$O_{d}$| is the discriminator output. |$L_{t}$| denotes the linear layer with tanh activation function, |$L_{r}$| denotes the linear layer with ReLU activation function and |$L_{lr}$| denotes the linear layer with LeakyReLU activation function.
As the distribution of feature samples differs for each subchloroplast location, we separately train five GAN data augmentation models corresponding to five locations. In detail, the generator receives a random noise input of the original feature vector dimension and outputs a synthetic feature sample of the same dimension. And, an Adam optimizer with a learning rate of 5e-4 is used to optimize the networks of discriminator and generator, respectively. In this case, a total of 10 000 iterations are completed for each subchloroplast location GAN model, saving every 200 iterations of model output to visualize changes in the synthetic feature samples distribution. When the synthetic feature samples work better, we reduce the number of iterations to capture the model under the optimal iteration as the final generated model. Finally, we mix the original and synthetic feature samples at each location in different proportions to form an extended feature dataset for the training of the prediction module.
Multi-location prediction
A hybrid deep neural networks is constructed by the transformer encoder and the convolutional neural network. It is used for predicting multi-location protein subchloroplast localizations based on the extended feature dataset. First, the subchloroplast location label set uses one-hot encoding. N subchloroplast location states are encoded with N-bit status registers, each with its independent register bits. Using 0, 1 to indicate whether the protein sequence is present at this subchloroplast location, e.g. the subchloroplast location labeled envelope is represented as (1, 0, 0, 0, 0), stroma as (0, 1, 0, 0, 0) and the subchloroplast location labeled both envelope and thylakoid membrane as (1, 0, 1, 0, 0). As protein sequence feature samples often have complex long- and short-term dependencies, the feature vectors are reshaped into a feature matrix |$M_{P}$| by placing values in rows to facilitate the neural network to capture the complex relationships.
The hybrid deep neural network is designed to exploit the local feature samples capture capability of the convolutional neural network [41] with the dynamic attention mechanism of the transformer encoder [42] so that the feature samples retain the most comprehensive and valid information before entering the fully connected classification prediction. The feature matrix |$M_{P}$| is fed into the convolutional layer (Conv2D), then dimensionally compressed and fed into the transformer encoder layer containing the mean-pooling to obtain the final feature matrix |$M_{p^{^{\prime}}}$| (formula 7). |$M_{p^{^{\prime}}}$| is as input to the fully connected layer for classification, and the five neurons in the final layer (corresponding to the five positions) are each connected to a sigmoid activation function for multi-label prediction.
where |$L_{s}$| means the multi-layer perceptron neural networks with Sigmoid activation function and |$L_{r}$| means the multi-layer perceptron neural networks with ReLU activation function.
Performance assessment
Consistent with the current state-of-the-art protein subchloroplast predictors, this paper evaluates the performance of the predictor using multi-label evaluation metrics (formulae 9–14) commonly used in the field of bioinformatics, i.e. Recall, Precision, F1-score (F1), Accuracy, Overall Actual Accuracy (OAA) and Grand Mean [23]. Let |$AL(P_{i})$| denote the actual label sets for the i-th protein sequence, and |$PL(P_{i})$| denote the predicted sets for the i-th protein sequence. The above metrics can be formulated as follows:
OAA is a multi-label accuracy metric, strictly querying for each location of the subchloroplast, and if one location is incorrectly predicted, its corresponding prediction contribution is 0.
Accuracy is also a multi-label accuracy metric but more lenient, and if one location is correctly predicted, it will obtain a prediction value.
Precision is used to describe the average frequency of correct prediction of subchloroplast locations.
Recall is used to describe the average rate of correct prediction of subchloroplast locations.
F1 is used to describe the sum-average of multi-label recall and the multi-label precision.
Additionally, Grand mean donates the average of Recall, Precision, F1-score (F1), Accuracy, Overall Actual Accuracy (OAA).
RESULTS AND DISCUSSION
Hybrid deep neural network parameter optimization
In the prediction module, the performance of the hybrid deep neural network model is affected by the structure and parameters. Therefore, we used grid search to select the best model hyperparameters according to the average result of 10-fold cross-validation of the model. All the models are trained using the Adam optimizer. We have considered the number of filters (set to 32, 64, 128, respectively) and the size (set to 1*5, 1*10, 1*15, respectively) in the setting of the convolution layer. In the setting of TransformerEncoderLayer, the number of the expected features in the input (set to 100, 200, 400, respectively) and the heads in the multi-head attention models (set to 2, 4, 6, respectively) are considered. In addition, we also considered the learning rate (set to 0.001, 0.0001, 0.0001, respectively) to determine the best parameters. According to the experimental results, we have selected 64 filters with the size of 1*10, and the TransformerEncoderLayer input is set to 200 and uses a two-headed attention model finally.
Selecting optimal number of iterations for WGAN-gp synthetic feature samples
To verify that the synthetic protein subchloroplast feature samples are indistinguishable from original feature samples, we use the t-SNE [43] technique to visualize the distribution of synthetic and original feature samples in two dimensions at five subchloroplast locations. From Figure 3, we can see that the generated protein synthetic feature samples blend almost perfectly with the original feature samples.
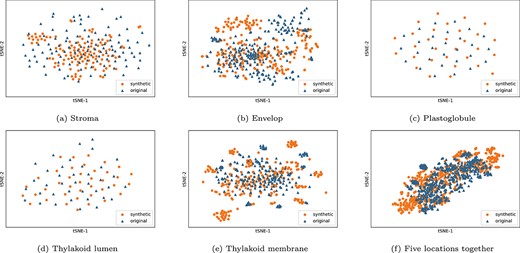
The original feature samples and optimal synthetic feature samples visual distribution charts of subchloroplast locations (stroma, envelope, plastoglobule, thylakoid lumen and thylakoid membrane) and five subchloroplast locations together.
In the initial iterations, as the generator has not yet started to learn the feature sample distribution, the discriminator can clearly distinguish the synthetic feature samples and the original feature samples, and the accuracy of discriminator is 1.0. With the number of training iterations increased, the generator learns more and more effectively, and the feature samples distribution gradually converges. After 4900, 5100, 5300, 2000 and 1900 iterations, respectively, the distributions of synthetic protein feature samples for the stroma, envelop, thylakoid, thylakoid lumen and plastoglobule also become similar to the corresponding real ones, because the accuracies of discriminators reache 0.5.
We have used t-SNE to visualize the sample distribution of WGAN-gp data augmentation model at different training periods in the whole iteration process. Since the feature samples at each position are basically consistent during the iterators, we used the plastoglobule location with the smallest number of samples and the envelope location with the largest number of samples as representatives. As shown in Figure 4 a–d, when WGAN-gp started training and completed the first iteration, the distribution distance between the synthetic sample and the real sample of plastoglobule is large. There is a great difference between the synthetic sample and the real sample. At this time, the accuracy of the discriminator is 1, and the discriminator can clearly identify the authenticity of the samples. After 500 iterations, the model began to learn the distribution of original feature samples, and generated synthetic samples similar to the original sample. With further training, after 1000 iterations, we can see from the distribution that the overlapping area of the synthetic sample and the original sample gradually increases, and the accuracy of the discriminator is also gradually decreasing. Finally, after 1900 times of training, the synthetic feature sample of the plastoglobule location almost coincides with the original feature sample, and the discriminator has obtained an accuracy of 0.501. In other words, the training of the site is balanced, and the quality of the synthetic feature sample at this period can be used as a supplement to the original sample. In Figure 4 e–h, the feature sample synthesis process of envelope location is similar to the plastogloble, the difference being there are many original samples. To fully learn the original sample distribution, WGAN-gp needs more training time and iteration times to balance the generator and discriminator. In addition, we iterate individually to train multi-labeled sequence feature samples to obtain high-quality multi-labeled samples to supplement the training data.
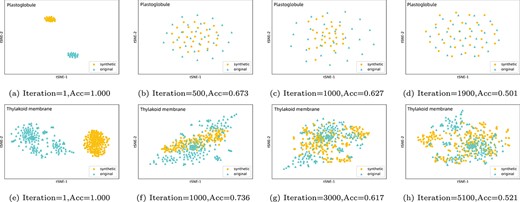
Comparing the distribution of samples synthesized by early iteration and later iteration with the original samples.
GAN-based data augmentation significantly improves prediction performance
In this section, we analyze the effect of data augmentation to improve prediction performance of protein chloroplast localization. We train a DaDL-SChlo variant without data augmentation module and DaDL-SChlo on the benchmark dataset to compare the prediction results. As shown in Table 2, DaDL-SChlo, which contains supplementary data of synthetic feature samples, is significantly improved in various indicators than the variant trained only with original feature samples. Among them, OAA and Accuracy increased by 7.7% and 6.9%, respectively. It is obvious that the predictor after data augmentation achieves better performance.
DaDL-SChlo compare with a DaDL-SChlo variant without data augmentation
| OAA | Accuracy | Precision | Recall | F1 | Grand Mean | |
|---|---|---|---|---|---|---|
| the variant | 0.758 | 0.874 | 0.906 | 0.874 | 0.873 | 0.857 |
| DaDL-SChlo | 0.835 | 0.943 | 0.951 | 0.943 | 0.945 | 0.923 |
| OAA | Accuracy | Precision | Recall | F1 | Grand Mean | |
|---|---|---|---|---|---|---|
| the variant | 0.758 | 0.874 | 0.906 | 0.874 | 0.873 | 0.857 |
| DaDL-SChlo | 0.835 | 0.943 | 0.951 | 0.943 | 0.945 | 0.923 |
DaDL-SChlo compare with a DaDL-SChlo variant without data augmentation
| OAA | Accuracy | Precision | Recall | F1 | Grand Mean | |
|---|---|---|---|---|---|---|
| the variant | 0.758 | 0.874 | 0.906 | 0.874 | 0.873 | 0.857 |
| DaDL-SChlo | 0.835 | 0.943 | 0.951 | 0.943 | 0.945 | 0.923 |
| OAA | Accuracy | Precision | Recall | F1 | Grand Mean | |
|---|---|---|---|---|---|---|
| the variant | 0.758 | 0.874 | 0.906 | 0.874 | 0.873 | 0.857 |
| DaDL-SChlo | 0.835 | 0.943 | 0.951 | 0.943 | 0.945 | 0.923 |
It suggests that the downstream prediction task requires massive data for sufficiently training. Using GAN synthesizes high-quality feature samples to supplement the original feature sample set can effectively increase the small sample data and solve the problems of insufficient training, stability and low prediction performance of the predictor.
To find the most appropriate proportion between original and synthetic feature samples, we train the predictor with a mixture of original and synthetic feature samples in five different ratios (i.e. 1:0.5, 1:1, 1:1.5, 1:2, 1:3). As can be seen in Figure 5, the predictor trained using the feature set supplemented with synthetic feature samples performs significantly better than the original feature samples. As the number of training feature samples increases, the ability of the predictor becomes stronger. At the same time, the prediction results stabilize when the ratio of original to synthetic feature samples increases to 1:1.5. Therefore, we generated 157 stroma feature samples, 298 envelope feature samples, 45 plastoglobule feature samples, 51 thylakoid lumen feature samples and 349 thylakoid membrane feature samples based on five different chloroplast sites. A total of 900 synthetic samples were mixed with the original feature samples as supplementary data, and finally 1478 training samples were obtained. Compared with the original feature samples set, the optimal augmented feature samples combination (i.e. 1:1.5 combination of original and synthetic feature samples) improves by 7.7% at OAA. The experiments show that utilizing a suitable proportion of synthetic feature samples to supplement the data allows the predictor to be trained more adequately, thus successfully improving the prediction performance and generalization of protein subchloroplast localization predictor.
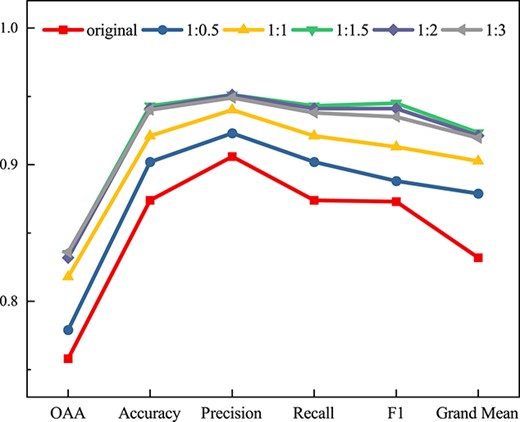
Comparing the prediction performance of different proportions of original and synthetic samples combinations.
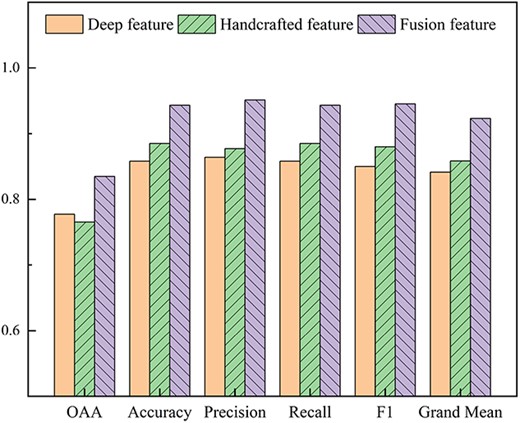
Comparing the prediction performance between single feature samples and fusion feature samples.
Comparison with current state-of-the-art methods on MSchlo578 (benchmark dataset)
| Methods | OAA | Accuracy | Precision | Recall | F1 | Grand Mean |
|---|---|---|---|---|---|---|
| MultiP-SChlo | 0.555 | 0.633 | 0.641 | 0.711 | 0.674 | 0.643 |
| EnTrans-Chlo | 0.6 | 0.66 | 0.673 | 0.711 | 0.68 | 0.665 |
| LNP-Chlo | 0.66 | 0.709 | 0.726 | 0.744 | 0.725 | 0.712 |
| Sanjay et al. | 0.728 | 0.89 | 0.869 | 0.89 | 0.851 | 0.851 |
| DaDL-SChlo | 0.835 | 0.943 | 0.951 | 0.943 | 0.945 | 0.923 |
| Methods | OAA | Accuracy | Precision | Recall | F1 | Grand Mean |
|---|---|---|---|---|---|---|
| MultiP-SChlo | 0.555 | 0.633 | 0.641 | 0.711 | 0.674 | 0.643 |
| EnTrans-Chlo | 0.6 | 0.66 | 0.673 | 0.711 | 0.68 | 0.665 |
| LNP-Chlo | 0.66 | 0.709 | 0.726 | 0.744 | 0.725 | 0.712 |
| Sanjay et al. | 0.728 | 0.89 | 0.869 | 0.89 | 0.851 | 0.851 |
| DaDL-SChlo | 0.835 | 0.943 | 0.951 | 0.943 | 0.945 | 0.923 |
Comparison with current state-of-the-art methods on MSchlo578 (benchmark dataset)
| Methods | OAA | Accuracy | Precision | Recall | F1 | Grand Mean |
|---|---|---|---|---|---|---|
| MultiP-SChlo | 0.555 | 0.633 | 0.641 | 0.711 | 0.674 | 0.643 |
| EnTrans-Chlo | 0.6 | 0.66 | 0.673 | 0.711 | 0.68 | 0.665 |
| LNP-Chlo | 0.66 | 0.709 | 0.726 | 0.744 | 0.725 | 0.712 |
| Sanjay et al. | 0.728 | 0.89 | 0.869 | 0.89 | 0.851 | 0.851 |
| DaDL-SChlo | 0.835 | 0.943 | 0.951 | 0.943 | 0.945 | 0.923 |
| Methods | OAA | Accuracy | Precision | Recall | F1 | Grand Mean |
|---|---|---|---|---|---|---|
| MultiP-SChlo | 0.555 | 0.633 | 0.641 | 0.711 | 0.674 | 0.643 |
| EnTrans-Chlo | 0.6 | 0.66 | 0.673 | 0.711 | 0.68 | 0.665 |
| LNP-Chlo | 0.66 | 0.709 | 0.726 | 0.744 | 0.725 | 0.712 |
| Sanjay et al. | 0.728 | 0.89 | 0.869 | 0.89 | 0.851 | 0.851 |
| DaDL-SChlo | 0.835 | 0.943 | 0.951 | 0.943 | 0.945 | 0.923 |
Comparison with current state-of-the-art methods on Novel (independent test set)
| Methods | OAA | Accuracy | Precision | Recall | F1 | Grand Mean |
|---|---|---|---|---|---|---|
| MultiP-SChlo | 0.27 | 0.328 | 0.353 | 0.367 | 0.347 | 0.332 |
| EnTrans-Chlo | 0.361 | 0.463 | 0.485 | 0.549 | 0.499 | 0.471 |
| LNP-Chlo | 0.549 | 0.574 | 0.598 | 0.574 | 0.582 | 0.575 |
| Sanjay et al. | 0.628 | 0.82 | 0.802 | 0.82 | 0.81 | 0.775 |
| DaDL-SChlo | 0.754 | 0.862 | 0.877 | 0.86 | 0.864 | 0.842 |
| Methods | OAA | Accuracy | Precision | Recall | F1 | Grand Mean |
|---|---|---|---|---|---|---|
| MultiP-SChlo | 0.27 | 0.328 | 0.353 | 0.367 | 0.347 | 0.332 |
| EnTrans-Chlo | 0.361 | 0.463 | 0.485 | 0.549 | 0.499 | 0.471 |
| LNP-Chlo | 0.549 | 0.574 | 0.598 | 0.574 | 0.582 | 0.575 |
| Sanjay et al. | 0.628 | 0.82 | 0.802 | 0.82 | 0.81 | 0.775 |
| DaDL-SChlo | 0.754 | 0.862 | 0.877 | 0.86 | 0.864 | 0.842 |
Comparison with current state-of-the-art methods on Novel (independent test set)
| Methods | OAA | Accuracy | Precision | Recall | F1 | Grand Mean |
|---|---|---|---|---|---|---|
| MultiP-SChlo | 0.27 | 0.328 | 0.353 | 0.367 | 0.347 | 0.332 |
| EnTrans-Chlo | 0.361 | 0.463 | 0.485 | 0.549 | 0.499 | 0.471 |
| LNP-Chlo | 0.549 | 0.574 | 0.598 | 0.574 | 0.582 | 0.575 |
| Sanjay et al. | 0.628 | 0.82 | 0.802 | 0.82 | 0.81 | 0.775 |
| DaDL-SChlo | 0.754 | 0.862 | 0.877 | 0.86 | 0.864 | 0.842 |
| Methods | OAA | Accuracy | Precision | Recall | F1 | Grand Mean |
|---|---|---|---|---|---|---|
| MultiP-SChlo | 0.27 | 0.328 | 0.353 | 0.367 | 0.347 | 0.332 |
| EnTrans-Chlo | 0.361 | 0.463 | 0.485 | 0.549 | 0.499 | 0.471 |
| LNP-Chlo | 0.549 | 0.574 | 0.598 | 0.574 | 0.582 | 0.575 |
| Sanjay et al. | 0.628 | 0.82 | 0.802 | 0.82 | 0.81 | 0.775 |
| DaDL-SChlo | 0.754 | 0.862 | 0.877 | 0.86 | 0.864 | 0.842 |
Fusion of pretrained deep learning features and traditional handcrafted features successfully improves prediction accuracy
It is critical for choosing appropriate feature extraction methods in constructing a predictor for protein subchloroplast localization. Comparing the effects of different feature methods on the performance of predictor, this paper uses two single feature sample sets and the fusion feature sample set to train the predictor, respectively. Among them, the single feature sample sets include deep learning features extracted by ProtBERT and ACC-PSSM handcrafted features. All the above feature sample sets have been supplemented with synthetic feature samples. We feed the feature sample sets into the hybrid deep neural prediction network with the leave-one-out cross-validation method. The experimental results of single feature (deep learning, ACC-PSSM) and fusion feature comparison are shown in Figure 6. The OAA of deep learning features and handcrafted features are 0.777 and 0.765, respectively, while the OAA of fusion features reaches 0.835, which is about 6% higher than that of single features. It shows that the fusion feature utilizes the complementarity between deep learning features and handcrafted features, combining the advantages of single features to express protein sequences more comprehensively and effectively. Thus, the predictor performance has been greatly improved.
Comparison of DaDL-SChlo with existing state-of-the-art methods on 10-fold cross-validation and independent test sets
To objectively evaluate the performance of DaDL-SChlo, this paper adopts the same datasets and validation approach as the current state-of-the-art predictors. The predictor trains on MSchlo578 (the benchmark dataset) and its predictive performance is assessed using the leave-one-out cross-validation, as shown in Table 3. Additionally, utilizing Novel (the independent test set) verifies its generalization further, as shown in Table 4.
The comparison results of six performance metrics of the existing multi-label subchloroplast localization predictors, such as MultiP-SChlo [20], EnTrans-Chlo [21], LNP-Chlo [22] and Bankapur et al. [23] proposed, trained on the benchmark datasets are clearly summarized in Table 3. The prediction results on the MSchlo578 demonstrate that in all performance metrics, the DaDL-SChlo exceeds the current state-of-the-art method proposed by Bankapur et al. In details, compared with the methods proposed by Bankapur et al, the value of OAA increases by up to 10.7%, and the F1 and Precision values improve by 9.4% and 8.2%, respectively. Moreover, the Grand Mean, which represents the average of the five evaluation indicators, increased by 7.2%. In general, all the metrics except OAA and Grand Mean exceed 0.9, reaching excellent performance of the predictor. This fully shows that the design of the DaDL-SChlo predictor is very advantageous for protein subchloroplast localization prediction, and its performance is more competitive compared with other methods.
The excellent performance of DaDL-SChlo on the benchmark training dataset suggests that the use of data-augmented fusion features and hybrid neural network strategies can effectively improve the performance of the protein subchloroplast localization predictors. To further test whether the predictor has good generalization ability and whether there is overfitting, we further conduct independent tests. As can be seen in Table 4, in Novel (the independent test set), it shows that the four metrics (Accuracy, Precision, Recall and F1) of the predictor still maintain high scores, all above 0.85, reaching 0.862, 0.877, 0.86 and 0.864, respectively. In addition, compared with the predictor proposed by Sanjay et al., the OAA value has greatly improved by 12.6%. As can be seen, DaDL-SChlo achieves the overall best performance in independent tests.
Afterwards, we have also evaluated the single-label prediction performance of the proposed predictor DADL-SChlo using several single-label metrics (the accuracy and Matthews correlation coefficient (MCC) for each class). Up to now, none of the webservers and source codes of the state-of-the-art predictors is accessible, so only the results of the proposed predictor DaDL-SChlo in terms of the single-label metrics are listed. Specifically, the accuracy values for stroma, envelop, thylakoid lumen and thylakoid membrane are 0.84, 0.95, 0.4 and 0.74, respectively. And the MCC values for stroma, envelop, thylakoid lumen and thylakoid membrane are 0.85, 0.72, 0.62 and 0.87, respectively. These results show that the proposed predictor DaDL-SChlo achieves good prediction performance using the single-label metrics.
These experimental results fully demonstrate that the DaDL-SChlo we proposed not only has a higher prediction accuracy, but also has good robustness and generalization ability.
CONCLUSION
Chloroplast is an essential organelle in green plants responsible for performing many functions. Studying protein subchloroplast localization is vital for regulating accelerating chloroplast functional studies and regulating the utilization of light energy in crop production. In this paper, a novel multi-location protein subchloroplast localization predictor, DaDL-SChlo, is proposed. DaDL-SChlo fuses deep learning features extracted by pre-trained model ProtBERT and handcrafted features ACC-PSSM firstly, then utilizes WGAN-gp to generate synthetic feature samples as supplement for the original feature samples and finally predicts the locations of subchloroplast utilizing a hybrid neural network. Among them, the data augmentation module can be used as a generic module to dock to other downstream prediction networks to help solve the data sparsity problem of the original feature samples and enhance the training effect of predictor. In experiments, OAA values reach 0.835 on the benchmark dataset and 0.754 on the independent test set, respectively. These performance comparisons indicate that the prediction performance of DaDL-SChlo we proposed exceeds that of state-of-the-art protein subchloroplast localization methods. Thus, DaDL-SChlo will be a convenient tool to accelerate protein subchloroplast localization prediction and chloroplast research.
In the future, we hope to develop an online protein subchloroplast prediction platform based on DaDL-SChlo to facilitate easier and faster research by researchers. Furthermore, we will attempt to incorporate the label correlation into the multi-label learning algorithm for improving the performance of our proposed predictor and enhance the interpretability of deep learning model for assisting the biologists better understand model decisions.
More comprehensive protein sequence features are obtained from the fusion of deep learning feature extracted by protein pre-trained model ProtBERT and traditional handcrafted feature ACC-PSSM for the first time.
A GAN-based data augmentation module is developed, which can synthesize high-quality feature samples to supplement the original data, helping to solve the issue of scarcity in the number of training samples.
Multi-location prediction utilizing a hybrid deep neural network based on transformer encoder and convolutional neural network effectively improves the prediction performance of subchloroplast locations.
DATA AND CODE AVAILABILITY
The source codes and data for DaDL-SChlo are available at https://github.com/xwanggroup/DaDL-SChlo.
FUNDING
This work was supported in part by funds from the National Natural Science Foundation of China (No. 61906175), the Key Research Project of Colleges and Universities of Henan Province (No. 22A520013, No. 23B520004), the Key Science and Technology Development Program of Henan Province (No. 202102210144), the Training Program of Young Backbone Teachers in Colleges and Universities of Henan Province (No. 2019GGJS132).
ACKNOWLEDGMENTS
The authors thank the anonymous reviewers for their valuable suggestions.
Author Biographies
Xiao Wang is currently an associate professor at the School of Computer and Communication Engineering, Zhengzhou University of Light Industry, Zhengzhou, China. His research interests include bioinformatics, machine learning, deep learning and evolutionary computation.
Lijun Han is currently pursuing the Master’s degree in computer science and technology at the School of Computer and Communication Engineering, Zhengzhou University of Light Industry. Her research interests include bioinformatics, machine learning and deep learning.
Rong Wang is currently a lecturer in the School of Computer and Communication Engineering at Zhengzhou University of Light Industry. Her research interests include bioinformatics, machine learning, evolutionary computing and deep learning. She has published more than 10 refereed papers in journals and conferences.
Haoran Chen received the Ph.D. from the School of Computer Science, Beijing Industry University in 2019. He is currently an assistant professor at the School of Computer and Communication Engineering, Zhengzhou University of Light Industry. His research interests include artificial intelligence, bioinformatics and machine learning.
References

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}