




Abstract
Interactions between DNA and transcription factors (TFs) play an essential role in understanding transcriptional regulation mechanisms and gene expression. Due to the large accumulation of training data and low expense, deep learning methods have shown huge potential in determining the specificity of TFs-DNA interactions. Convolutional network-based and self-attention network-based methods have been proposed for transcription factor binding sites (TFBSs) prediction. Convolutional operations are efficient to extract local features but easy to ignore global information, while self-attention mechanisms are expert in capturing long-distance dependencies but difficult to pay attention to local feature details. To discover comprehensive features for a given sequence as far as possible, we propose a Dual-branch model combining Self-Attention and Convolution, dubbed as DSAC, which fuses local features and global representations in an interactive way. In terms of features, convolution and self-attention contribute to feature extraction collaboratively, enhancing the representation learning. In terms of structure, a lightweight but efficient architecture of network is designed for the prediction, in particular, the dual-branch structure makes the convolution and the self-attention mechanism can be fully utilized to improve the predictive ability of our model. The experiment results on 165 ChIP-seq datasets show that DSAC obviously outperforms other five deep learning based methods and demonstrate that our model can effectively predict TFBSs based on sequence feature alone. The source code of DSAC is available at https://github.com/YuBinLab-QUST/DSAC/.
Introduction
Transcription factors (TFs) regulate the gene transcription process by specifically binding DNA fragments of the regulatory region, and these binding recognition sites are called transcription factor binding sites (TFBSs) [1]. Transcription is the main stage of gene regulation, influencing gene expression through specific binding of TFs and DNA [2, 3]. Identification of TFBSs has gained significant importance in the field of bioinformatics, attracting increasing researchers’ attention due to the frequent association with transcription and the key to understanding of the transcriptional regulation mechanism. With the accumulation of large-scale DNA sequencing results and the development of high-throughput experimental techniques, intensive research has been done and one of them is Chromatin Immunoprecipitation-sequence (ChIP-seq), [4] which is used to directly obtain unbiased binding sites from sequenced genomes by combining ChIP with ultra-high-throughput massively parallel sequencing. Even though ChIP-seq has achieved good performance, there are still some problems. On the one hand, the large amount of data and bias generated during the experiment makes the noise exist consistently [5, 6]. On the other hand, ChIP-seq assays require a high amount of experimental materials that are difficult to obtain. For these reasons, new decent solutions need to be discovered.
Computational approaches have been applied to solve many bioinformatics and computational biology problems [7–9], owing to the fact that they do not require specific experimental materials. Conventional methods, such as machine learning methods are developed to improve the prediction. Founding work to predict protein–DNA bindings by computational methods involved the use of Hidden Markov model (HMM) [10],support vector machines(SVM) [11] and random forest [12, 13]. However, a current limitation with all traditional machine-learning algorithms is that for the ever-increasing amount of DNA sequence data, the improvement of the forecast progress and the full utilization of the experimental data cannot be met at the same time.
Since the advent of deep learning, computer vision and natural language processing have gained substantial advancements [14–16]. In recent years, methods based on deep learning, especially the convolutional neural networks (CNNs) [17], have been subsequently applied to the prediction of TFBSs successfully [18–22]. For example, DeepBind [23] utilizes CNNs to ascertain sequence specificities from experimental data and DeepSEA [24] leverages CNNs to predict the noncoding-variant effects de novo from sequence. In addition to these CNN-based methods, there are some networks that try to combine CNNs with other algorithms, such as DanQ [25] is a hybrid framework that combines CNNs and bi-directional long short-term memory (Bi-LSTM) units. The deep learning methods are innately proficient in retrieving the positional relationship between the sequence signals while decreasing computational complexity, which not only improves the prediction accuracy, but also has obvious performance improvement compared with machine learning, making the application of deep learning in this field is more and more extensive and flexible.
The recent progress of natural language processing has been driven in part by the self-attention mechanism [26–28]. The self-attention mechanism relies on the correlation information between different positions in a single sequence to model the features of the sequence, and each position is computed as the weighted sum of all positions. Sequence models can be enhanced to effectively capture long-distance dependencies [29, 30], addressing a major shortcoming of convolutional and recurrent networks. Considering the complementary properties of CNN and self-attention, better results can be prophetically achieved by integrating these two modules [31–34]. The problems in natural language processing tasks can be approximated as sequence problems, and consequently, these methods are applicable to the identification of associations in DNA or RNA sequences. Ullah F and Ben-Hur A [35] proposed a self-attention based deep learning model, which mainly contains a CNN layer and a multi-head self-attention layer to capture regulatory element interactions in genomic sequence. SAResNet proposed by Shen et al. [36] combines the self-attention mechanism and residual network structure and the spatial information and the local information are incorporated to improve the learning ability of the network. These remarkable works show that the self-attention layer is particularly useful for tasks in the detection of potential motifs and can capture a global landscape of interactions between regulatory elements in a given sequence.
In this work, we propose DSAC, a dual-branch network model, merging global representations extracted by self-attention branch and local features extracted by CNN branch for TF binding prediction. The performance of DSAC is evaluated on 165 ChIP-seq datasets, and the results demonstrate that feature extraction in an interactive way at the early stage and the dual-branch architecture at the later stage can tremendously enhance the representation learning of DNA sequences and make significant contributions to improving the model performance.
Materials and methods
Datasets
To evaluate the predictive ability of our model, we chose the same datasets that were used in the work conducted by Zhang et al. [37]. In detail, the 165 ChIP-seq datasets were collected from 690 ChIP-seq datasets produced by the Encyclopedia of DNA Elements (ENCODE) project [38], where positive sets consist of 101 bp DNA sequences that were experimentally confirmed to bind to a given DNA-binding protein and negative sets consist of shuffled positive sequences that maintain dinucleotide frequency. These datasets contain 29 TFs from various cell lines and a detailed description of them is shown in Supplementary Table S1.
Performance evaluation metrics
Accuracy (ACC) [39] is used to simply indicate the percentage of correct predictions for all TFBSs. ROC-AUC represents the area under the receiver operating characteristic (ROC) curve and PR-AUC means the area under the precision-recall curve. Previous studies have focused on ROC-AUC as a measure of performance, even though this is a good assessment of classification performance, it is overly influenced by false positive predictions. When the sequence data are unbalanced, it will favor the side of the large sample, while PR-AUC provides a better assessment of false positives [40, 41]. Hence, we use ACC, ROC-AUC and PR-AUC as the evaluation metrics.
Model architecture
We will start the discussion in three parts. The first two parts describe the structure of the convolution module and the self-attention module in detail and the last one summarizes the overall architecture, showing how these two modules interact to jointly construct the model.
CNN module
Self-attention module
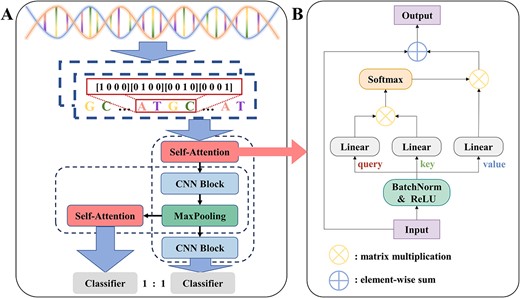
The illustrative diagram of DSAC. (A) Block diagram of the network architecture of DSAC. (B) self-attention module.
In our work, two self-attention modules with dropout [48] were used in the first and final stages of the DSAC architecture, respectively. A dropout rate of 0.2 in the first stage and a dropout rate of 0.7 in the final stage were utilized to effectively alleviate the occurrence of overfitting. We performed worse on the other two sets of dropout values, (0.3, 0.7) and (0.4, 0.6), suggesting that dropout value of (0.2, 0.7) is better at taking advantage of the model's ability to extract information and prevent overfitting.
Model overview
From a global perspective, we use the global context obtained from the self-attention module as feature maps to strengthen the global perception capability of the CNN branch, and local features from the convolution layer are sustainability embed to reinforce the self-attention branch's grasp of local particulars. In this way, there is a flexible interaction between the two branches. In addition, this dual-branch structure also improves the generalization ability of the model and speeds up the model training speed.
Feature representation and model implementation
Results and discussion
Model variants
In order to specifically evaluate the performance and show the improvement of our model, we will make a discussion from two aspects: features and structures. For feature analysis, the discussion mainly includes dissection of the local features extracted from CNN module and the global representations extracted from the self-attention mechanism. In this part, we designed a model without CNN branch and self-attention branch to compare with the original model, which can effectively reflect the influence of local features and global features on the prediction effect of the model. For structure analysis, we designed two model variants. The one is to simply stack the CNN module and the self-attention module linearly and is dubbed as SACSAC, which means that CNN modules and self-attention modules are interleaved and finally fed into one classifier. And the other variant, SCAF is to cancel the architecture of the two classifiers based on the original model structure. After the execution of the CNN branch and the self-attention branch, the refined feature maps obtained by the two branches from the sequence profiles are concatenated along the first dimension and features are reintegrated by a fully connected layer. Finally, a sigmoid classifier is used to obtain the output classification probabilities. These architectures of the model variants were designed to be compared with the model of the dual structure, thereby demonstrating the superiority of our model structure. A detailed description of the model variants with different structures is shown in Figure 2. All the model variants were trained on 165 ChIP-seq benchmark data sets, which are same to the proposed model DSAC, and the same hyperparameter settings were used in all experiments.
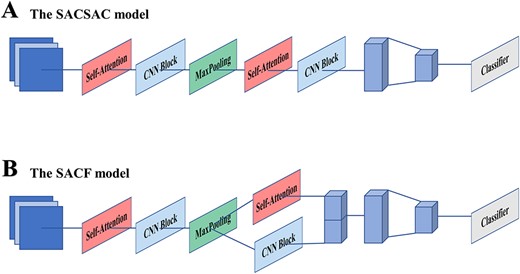
The brief illustration of variant models: (A) the SACSAC model. (B) the SACF model.
Analysis on features
Without the CNN branch, when the local features, which are extracted through the previous convolution layers, are embedded in the self-attention module and the model will lay special emphasis on capturing global information about DNA sequences rather than local details. Therefore, through this comparison, it can indicate that an independent self-attention branch does not pay enough attention to local features, resulting in poor final prediction effects. Without the self-attention branch, additionally the stacking degree of the convolution is not enough, only the initial self-attention mechanism cannot obtain sufficient global features. Therefore, the model lacks the extraction of multi-level features, resulting in a decrease in the performance of the model. Figure 3 illustrates the performance of DSAC and model variants under the average ACC, ROC-AUC and PR-AUC metrics, and it can be seen that our model outperforms the model without CNN branch and the model without self-attention branch on all the aforementioned metrics. Specifically, the average ROC-AUC of DSAC was 0.887, which was 0.8% higher than the model without self-attention branch (0.879) and 2.1% higher than the model without CNN branch (0.866). And we also observed that the relative improvement of DSAC (0.891) overed the model without CNN branch (0.885) was 0.6% and the model without self-attention branch (0.869) was 2.2% in terms of PR-AUC scores. It can also be seen from Figure 4 that DSAC significantly outperforms the variant models under the ROC-AUC and PR-AUC distributions. This indicates that our model can extract the more informative features and better in TFBSs prediction. Furthermore, the detailed local features gradually supplied by the CNN branch and the global representations steadily learned by the self-attention branch are not isolated, but applied to our model in a complementary and interactive manner, which inherits the advantages of convolutional network and self-attention mechanism.
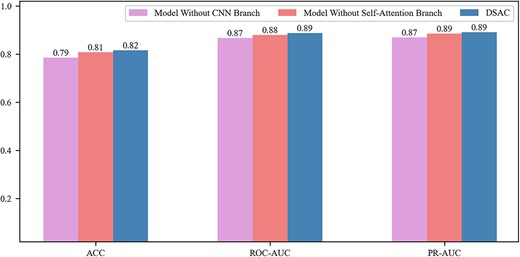
Comparison of DSAC with the model without CNN branch and the model without self-attention branch on 165 ChIP-seq datasets under the average ACC, ROC-AUC and PR-AUC metrics.
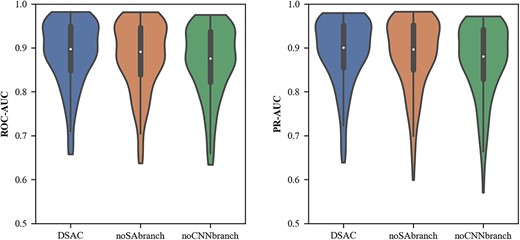
The ROC-AUC distribution and PR-AUC distribution of DSAC and variant models. The white point and the thick black bar in the middle indicate the median and interquartile range, respectively. The black line represents the interval from the minimum non-outlier value to the maximum non-outlier value.
Analysis on structure
Among the connection methods of the CNN module and the self-attention module, many researchers have proposed that self-attention can be used to enhance the convolutional networks. The significant defect of the convolution operation is that it only works on a local neighborhood, thus missing the global information. While the self-attention module has the capability of obtaining more relational information in the network, which provides the possibility to optimize the performance of convolutional networks. Another way is to improve self-attention networks with the CNN modules. The self-attention mechanism captures global contextual information at each location, thus losing the ability to focus on a specific region, and additional convolution modules make up for this shortcoming by adding local information to the self-attention network. Instead, our model structure can make self-attention and convolution mutually promote each other, making the best use of the advantages of both networks. From Figure 5, it is evident that DSAC is substantially better than the two model variants, SACSAC and SACF. As can be seen from the comparison with SACSAC, the dual-branch design structure of our model is better than the architecture of interleaved stacking of CNN and self-attention modules. It is because information between modules cannot be effectively transitioned, as a result, the simple stacked structure cannot make the CNN or self-attention handle the results from the previous layer well, and naturally cannot fully utilize the complementarity of the two modules. SACF also adopts the dual-branch architecture, but the prediction effect of using features after concatenation and reintegration is far less than that without concatenation and reintegration. It can also be seen from Figure 5 that DSAC performs better on the vast majority of datasets than SACF. This shows that both branches have the ability of independent prediction, and the structure of SACF makes the integrated features have a lot of redundant information, thus reducing the prediction accuracy. In summary, the design of our model architecture enables the two branches to extract various feature information at the same time, and subsequently can specialize in the features obtained by themselves after obtaining complementary information from each other, which greatly enhances the model’s ability to capture DNA sequence information.
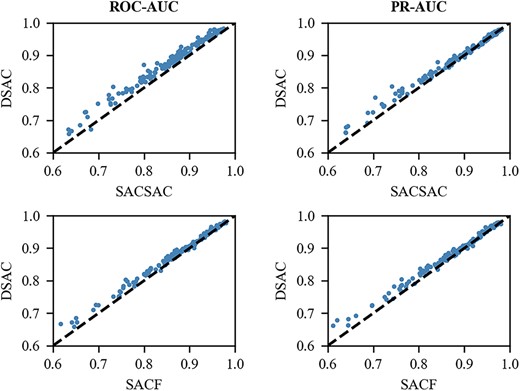
Comparison of DSAC with SACSAC and SACF on 165 ChIP-seq datasets under the ROC-AUC and PR-AUC metrics.
Comparison with other methods
To further evaluate the performance of DSAC, we compare it with various deep-learning-based models, including DeepBind [23], DanQ [25], CRPTS [51], DLBSS [52] and D-SSCA [37]. As in the previous experiments, 165 ChIP-seq experimental datasets were utilized to evaluate our model and all the competing methods. Figure 6 shows the comparison results (ACC, ROC-AUC and PR-AUC scores across all the test sets) between DSAC and other predictors. Among all the evaluation metrics calculated in our work, the maximum values were higher than the models except DLBSS, even though the maximum values were not higher than DLBSS, the minimum values were substantially improved compared with other competing approaches, which shows that our model achieved a surprisingly good generalization ability. These results indicate that our model performs better than all other methods, revealing that the interactive feature extraction and the dual-branch network architecture contribute to improving the prediction performance of the model. From Table 1, we can clearly observe that DSAC achieved a statistically significant performance improvement in terms of ACC, ROC-AUC and PR-AUC scores. To be specific, the average ACC, ROC-AUC and PR-AUC scores of DSAC were 0.816, 0.887 and 0.891, respectively, which were 2.3, 2.% and 2.0% higher than the suboptimal model (0.793, 0.867 and 0.871, respectively).
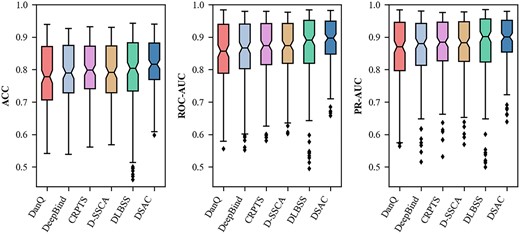
Performance comparison between D-SSCA and other five competing methods on 165 ChIP-seq datasets under the ACC, ROC-AUC and PR-AUC metrics. The middle line inside the box indicates the median and the ends of the box are the upper and lower quartiles. The two lines outside the box are the whiskers extending to the highest and lowest observations and the diamond marks indicate outliers.
Among these competing methods, DeepBind and DLBSS are the models only based on CNNs while DanQ and CRPTS both use a hybrid framework that combines CNNs and BLSTMs. Moreover, D-SSCA mainly uses convolution modules and attention mechanisms. Our model performs better than DeepBind and DLBSS, which shows that the addition of the self-attention module is effective and plays a crucial role in the performance of our presented model. What causes the results is that the model with mutual cooperation of convolution modules and self-attention mechanisms can extract informative features more than the model with the stacking of convolution modules. DeepBind and DLBSS perform worse than our model and it mainly because only using CNNs with a limited receptive field is not enough to extract long-term dependencies and obtain sufficient information. BLSTM is a variant of the recurrent neural network (RNN) that consists of two LSTMs: one taking the input in a forward direction and the other in a backward direction. BLSTMs can effectively increase the amount of information available to the network and capture long-term dependencies like self-attention mechanism. However, Both DanQ and CRPTS are worse than our model, which indicates that self-attention module in our model architecture has a strong learning ability for long-range dependencies in DNA sequences. On the other hand, it shows that BLSTMs, which involved in DanQ and CRPTS can capture global information, but they do not make good use of local information when combined with CNNs. Particularly, in terms of capturing long-range dependencies, the addition of the self-attention mechanism makes our model neither need to apply recurrent networks, which are computationally expensive to train, nor need to use convolution networks with large window sizes.
Performance comparison of different methods
| Method | ACC | ROC-AUC | PR-AUC |
|---|---|---|---|
| DanQ | 0.782 | 0.849 | 0.855 |
| DeepBind | 0.785 | 0.853 | 0.858 |
| CRPTS | 0.793 | 0.862 | 0.867 |
| DLBSS | 0.793 | 0.865 | 0.871 |
| D-SSCA | 0.793 | 0.867 | 0.871 |
| DSAC | 0.816 | 0.887 | 0.891 |
| Method | ACC | ROC-AUC | PR-AUC |
|---|---|---|---|
| DanQ | 0.782 | 0.849 | 0.855 |
| DeepBind | 0.785 | 0.853 | 0.858 |
| CRPTS | 0.793 | 0.862 | 0.867 |
| DLBSS | 0.793 | 0.865 | 0.871 |
| D-SSCA | 0.793 | 0.867 | 0.871 |
| DSAC | 0.816 | 0.887 | 0.891 |
Performance comparison of different methods
| Method | ACC | ROC-AUC | PR-AUC |
|---|---|---|---|
| DanQ | 0.782 | 0.849 | 0.855 |
| DeepBind | 0.785 | 0.853 | 0.858 |
| CRPTS | 0.793 | 0.862 | 0.867 |
| DLBSS | 0.793 | 0.865 | 0.871 |
| D-SSCA | 0.793 | 0.867 | 0.871 |
| DSAC | 0.816 | 0.887 | 0.891 |
| Method | ACC | ROC-AUC | PR-AUC |
|---|---|---|---|
| DanQ | 0.782 | 0.849 | 0.855 |
| DeepBind | 0.785 | 0.853 | 0.858 |
| CRPTS | 0.793 | 0.862 | 0.867 |
| DLBSS | 0.793 | 0.865 | 0.871 |
| D-SSCA | 0.793 | 0.867 | 0.871 |
| DSAC | 0.816 | 0.887 | 0.891 |
In summary, to evaluate the performance improvement of DSAC, we compared with five deep-learning-based models on 165 ChIP-seq experimental datasets. The results show that our model performs superior to other competing models and demonstrate the feasibility of a dual-branch model architecture combining self-attention mechanism and CNNs.
Conclusion
In this study, we have presented DSAC, a dual-branch combination network for TF binding prediction. Deep learning frameworks have been extensively applied to bioinformatics due to limited training data and high computational cost, and have shown thrillingly promising potential to mine the complex relationship hidden in large-scale biological data [53]. Our work is a demonstration of one method, which combines global representations extracted by self-attention branch and local features extracted by CNN branch, enhancing feature learning in an interactive fashion. In addition, our model adopts a dual-branch architecture, and the comparison with the model based on the architecture of interleaved stacking of convolutional blocks and self-attention modules and the model that fuses two-branch features can show that our model structure can more effectively utilize the advantages of the CNN branch and the self-attention branch. Moreover, benchmarking experiments show that the performance of DSAC is apparently higher than that of other competing deep learning methods on the 165 ChIP-seq datasets.
Even though DSAC has achieved a good performance, there are still some limitations and we can further improve our model in the following aspects. First, the architecture of DSAC is limited to setting up independent paths for CNN and self-attention modules. Although there is an interaction between the two modules in our model, the internal relationship between them is not fully utilized. Later research can start from the internal structure of the two modules to find better ways to connect with each other. Second, due to the limitation of computing resources, we did not design comparative experiments with different hyperparameters, but only roughly determined the hyperparameters. Third, we only used DNA sequences to extract features and no other information such as DNA shape profiles can be fused to improve the predictive power of TFBSs.
This study proposes a dual-branch network, termed DSAC, which combines the self-attention mechanism and CNN modules, to predict transcription factor binding sites.
The self-attention mechanism can focus on global representation learning and the CNN modules pay more attention to local feature details. The two in our model reached a good cooperation, enhancing the representation learning of DNA sequences.
The dual branch architecture allows better interaction between the self-attention mechanism and the CNN module and also enhances the robustness of the model.
Benchmarking experiments show that the performance of DSAC is apparently higher than that of other competing deep learning methods on the 165 ChIP-seq datasets.
Funding
National Natural Science Foundation of China (62172248, 61932018); Natural Science Foundation of Shandong Province of China (ZR2021MF098).
Author Biographies
Yutong Yu is an undergraduate at the College of Information Science and Technology, Qingdao University of Science and Technology, China. Her research interests are bioinformatics and deep learning.
Pengju Ding is a master student at the College of Information Science and Technology, Qingdao University of Science and Technology, China. Her research interests are bioinformatics and deep learning.
Hongli Gao is a master student at the College of Mathematics and Physics, Qingdao University of Science and Technology, China. Her research interests are bioinformatics and deep learning.
Guozhu Liu is a professor at the College of Information Science and Technology, Qingdao University of Science and Technology, China. His research interests include artificial intelligence, machine learning and algorithms.
Fa Zhang is a professor at the School of Medical Technology, Beijing Institute of Technology, China. His research interests include bioinformatics, high-performance computing and machine learning.
Bin Yu is a professor at the College of Information Science and Technology, School of Data Science, Qingdao University of Science and Technology, China. His research interests include bioinformatics, artificial intelligence and biomedical image processing.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}