




Abstract
Beta-lactamases (BLs) are enzymes localized in the periplasmic space of bacterial pathogens, where they confer resistance to beta-lactam antibiotics. Experimental identification of BLs is costly yet crucial to understand beta-lactam resistance mechanisms. To address this issue, we present DeepBL, a deep learning-based approach by incorporating sequence-derived features to enable high-throughput prediction of BLs. Specifically, DeepBL is implemented based on the Small VGGNet architecture and the TensorFlow deep learning library. Furthermore, the performance of DeepBL models is investigated in relation to the sequence redundancy level and negative sample selection in the benchmark dataset. The models are trained on datasets of varying sequence redundancy thresholds, and the model performance is evaluated by extensive benchmarking tests. Using the optimized DeepBL model, we perform proteome-wide screening for all reviewed bacterium protein sequences available from the UniProt database. These results are freely accessible at the DeepBL webserver at http://deepbl.erc.monash.edu.au/.
Introduction
Since the discovery of the beta-lactam penicillin (Fleming, 1928) and its subsequent clinical usage, a growing set of beta-lactam antibiotics have been discovered, developed and prescribed regularly in clinical practice [1, 2]. Unfortunately, a drastic increase in antibiotic usage has led to the rapid spread of antibiotic resistance that poses a significant global public health threat. Beta-lactamases (BLs) are enzymes expressed by bacteria that hydrolyze beta-lactam antibiotics, giving rise to beta-lactam resistance [3, 4]. The first BL, named penicillinase, was discovered and isolated from Escherichia coli in 1940 by Abraham and Chain, even before penicillin was used clinically [5]. Since then, the number of identified BLs has dramatically increased, with 4326 BLs currently included in beta-lactamase database (BLDB) [6]. Many of these BLs are plasmid encoded and can be shared across bacterial species by lateral transfer, contributing to the rapid spread of drug resistance [7]. BLs are the main propagators of acquired antibiotic resistance, with multidrug resistance in clinical strains being reported frequently [8]. Increasingly, infections caused by organisms that express extended-spectrum beta-lactamases (ESBLs) [9] are difficult to treat both due to challenges in detecting ESBLs and reporting inconsistencies [10]. The same applies to bacteria that express carbapenemases, a category of BLs that inactivates carbapenems, which are often considered to be last resort beta-lactam antibiotics in clinical treatment [10–12]. To make matters worse, a growing number of Enterobacteriaceae, species highly adapted for growth in human tissues, have been shown to express multiple BLs, often of various classes [12, 13]. Accurately classifying BLs is an essential step to guide BL inhibitor design and to deepen our understanding of the mechanisms involved in beta-lactam antibiotic resistance. To that effect, two classification schemes are currently used: (i) molecular classification as classes A–D, based on sequence homology [14] or (ii) functional classification as groups 1–4, based on substrate and inhibitor profile [15]. In this study, we adopt the molecular classification scheme to develop a far-reaching detection tool.
To classify and identify BLs from various organisms after antibiotic susceptibility testing, a variety of BL-specific screening tests can be conducted [13, 16]. However, these methods are resource costly and time consuming. Thus, an alternative solution to identify BL classes is to computational approaches [17], which can rapidly identify potential BLs and narrow down BL candidates for experimental validation. A method like this is feasible due to the rapid accumulation of well-annotated genome sequences. Highly reliable prediction models can be used to not only perform high-throughput screening but also discover potential BLs that are challenging to be identified based on sequence similarity search. Along these lines, it is worth noting that several approaches have been implemented to identify BLs. These approaches can be divided into three major categories:
(i) Knowledge databases: Several web-based BLDBs are available, which provide information resources for BLs. These include BL-specific databases [6, 18, 19], as well as antibiotic multidrug resistance-related databases [20–23], which also includes annotations on BLs.
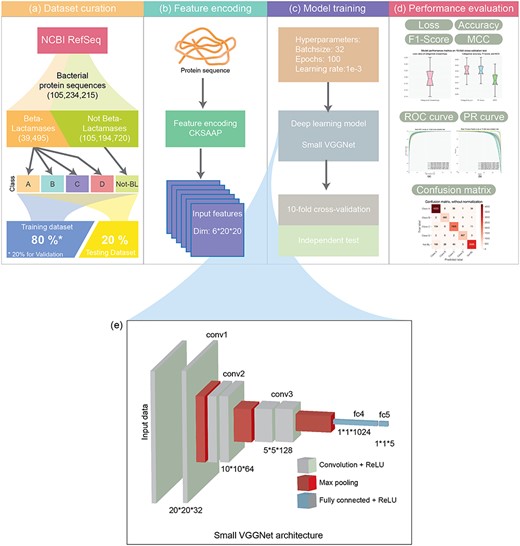
The architecture of the DeepBL methodology. The development of DeepBL involves four major stages, including (A) data curation, where all ‘BL’ sequences annotated in the NCBI RefSeq database are extracted and used as the positive samples, whereas 0.015% of all ‘Not BL’ sequences are randomly chosen as the negative samples to constitute the ‘Not-BL’ subset; (B) feature encoding, where the sequence encoding scheme CKSAAP is applied to encode the sequence of proteins in the benchmark dataset; (B) model training, where the model architecture is built, model hyperparameters are optimized and training strategies are compared and (D) performance evaluation, where the performance of DeepBL models is assessed by performing 10-fold cross-validation and independent tests. (E) shows the detailed architecture of the Small VGGNet deep learning framework.
(ii) Sequence similarity-based methods: These methods apply a general hypothesis that proteins that share similar sequences usually perform similar biochemical functions. Among sequence similarity-based methods, BLAST [24, 25] and HMMER [26] can be used to identify BL classes by querying a sequence against the profile constructed based on the known BLs. Subsequently, the class- or family-specific conserved motifs can be identified. To provide an example, Srivastava et al. proposed a motif-based BL family prediction method using the MEME/MAST suit to identify family-specific motifs or patterns [23, 27, 28].
(iii) Machine learning-based methods: Several machine learning-based algorithms have been proposed to construct the prediction models for BL classification, such as Bayes [29], support vector machine (SVM) [30, 31] and convolutional neural network (CNN) [32].
Despite the progress achieved, there are intrinsic drawbacks and limitations associated with the current methods. These include: (i) sequence similarity-based methods can only be used to annotate a given sequence based on its sequence similarity to already known BL sequences. As such, they cannot identify novel BLs; (ii) the majority of machine learning-based methods [30–32] are developed based on the datasets that had <4000 BL sequences and <800 negative BL sequences, which are small and limited in the size and scale; (iii) despite the relatively high performance achieved by these methods, their predictive performance and generalization ability remain to be validated on large-scale independent test datasets and (iv) another related issue is the selection of a reliable negative dataset. According to the reference sequence (RefSeq) dataset, BL sequences accounted for a small proportion (39 495 out of 105 194 720, curated in March 2019) of the collected bacterial sequences. As such, it is critical to build a reliable and representative negative BL dataset and develop a robust classification model to identify BL sequences with a low false-positive rate.
To address these issues, we develop DeepBL, a high-throughput deep learning-based approach for identifying BLs and their classes using protein sequences. DeepBL is developed based on a well-annotated large dataset containing 39 495 BLs extracted from the National Center for Biotechnology Information (NCBI) RefSeq database (downloaded in March 2019) [33, 34]. Here, we formulate the BL prediction task as a multiclass classification problem. We carefully curate a reliable negative BL dataset by extending the number of sequences and assess the model performance by performing multiple times of undersampling. We then build the prediction model of DeepBL using a simplified version of the VGGNet deep learning architecture [35], namely as the Small VGGNet. Then, we demonstrate this framework outperforms the other four commonly used machine learning algorithms when evaluated on an independent test dataset containing >10 000 sequences. Moreover, we also examine the effect of varying sequence redundancy levels on the model performance. We further apply the optimized model of DeepBL to the entire set of reviewed bacterial sequences, identify potential BL sequences and make this computational compendium publicly accessible at the DeepBL website.
Materials and methods
DeepBL is developed based on the deep learning technique. It is a high-throughput and multiclass classification approach for the identification of BLs and their classes from protein sequence data. The architecture of the DeepBL methodology is illustrated in Figure 1. As can be seen, its development involves four major steps, including data curation, feature encoding, model training and performance evaluation. At the first step, dataset curation, a high-quality dataset was collected from the NCBI RefSeq [33]. At the second step, the classical feature encoding scheme, composition of k-spaced amino acid pairs (CKSAAP) [36, 37] was employed to extract the sequence features. At the third step, a customized neural network, i.e. the Small VGGNet [35] was applied to train the prediction model of DeepBL. At the final step, 10-fold cross-validation and independent tests were performed to rigorously evaluate the performance of DeepBL models in terms of several performance metrics.
Benchmark dataset
To construct a benchmark dataset for training and validating the model of DeepBL, a nonredundant bacterial protein sequence dataset was originally downloaded from the NCBI RefSeq database [33]. This allowed us to collect >100 million bacterial protein sequences. The RefSeq [33] database provides a comprehensive, nonredundant and well-annotated collection of reference sequences. It is periodically augmented with newly published sequence data. The sequences of BLs were extracted by searching the database with the keyword combination of ‘BL’ and the respective class of A, B, C and D. As the BL sequences only account for a smaller portion of the whole-sequence dataset, we performed a random selection of the not-beta-lactamases (Not-BL) sequences. We define the Not-BL sequences as those without the annotation of ‘BL’. To retrieve such a reliable negative dataset, we selected the sequences from RefSeq without the annotation of ‘BL’ and resulted negative datasets had a reasonable scale (i.e. no more than 10 times larger than that of the smallest BL class) compared with the positive BL sequence dataset. To enable statistical significance test, we randomly selected the negative dataset for five times and generated five negative datasets for the model training and performance evaluation. The data distribution of the datasets is shown in Figure 2 and Table S1. The sequence redundancy in the datasets was removed with CD-HIT [38] using the sequence identity cutoff thresholds ranging from 1.0 to 0.7 with the step size of 0.05. The resulting datasets were used to assess the model performance and compare with four other machine learning algorithms. Finally, a total of 35 datasets clustered with different sequence identity thresholds were obtained. All these datasets can be downloaded from the webserver of DeepBL at http://deepbl.erc.monash.edu.au/download.html/.
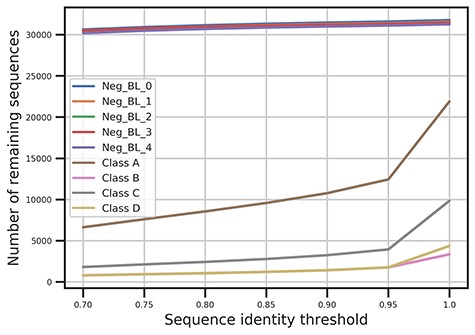
The relationship of the number of remaining sequences in the BL sequence datasets clustered by CD-HIT in accordance with different sequence identity cutoff thresholds, which ranged from 0.7 to 1.0.
Composition of k-spaced amino acid pair
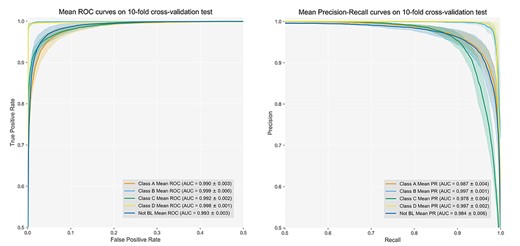
ROC curves and PR curves on the 10-fold cross-validation test. The AUC values of the ROC curves and PR curves are calculated with average and SD values.
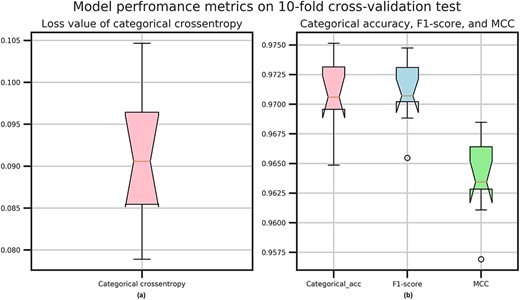
Boxplots of performance results on the 10-fold cross-validation tests. Performance metrics monitored during model training included (a): Loss value, (b): Categorical accuracy, F1-Score and MCC.
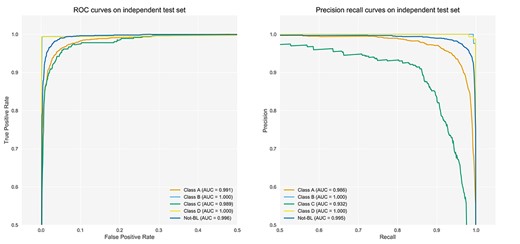
ROC and PR curves for the prediction of Classes A, B, C, D and Not-BLs on the independent test datasets.
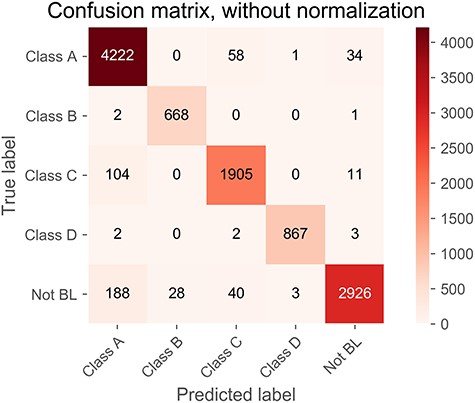
Confusion matrix of predicted results on the independent test. The matrix represents the distribution of the outputs for each of the five BL classes. Correctly predicted numbers are highlighted and shown on the diagonal line.
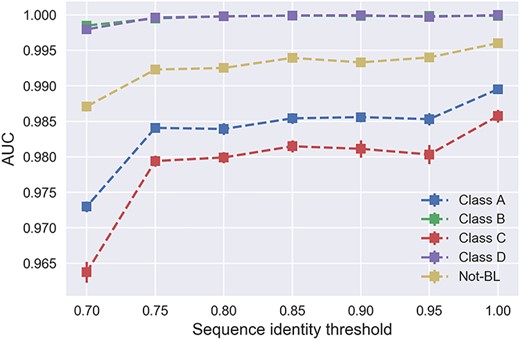
Relationship between the resulting AUC values and varying sequence identity thresholds on the benchmark datasets.
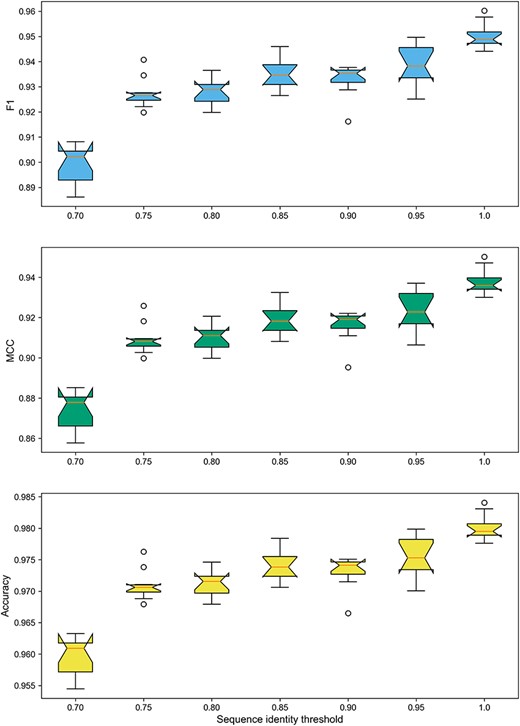
Boxplots of F1-Score, MCC and ACC on the datasets with varying sequence identity thresholds.
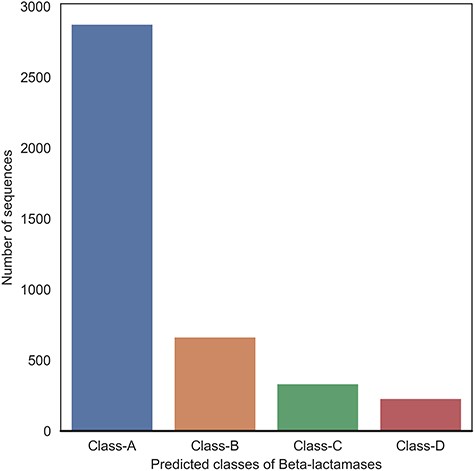
Statistics of proteome-wide prediction of BLs and their respective classes by applying the optimized DeepBL model.
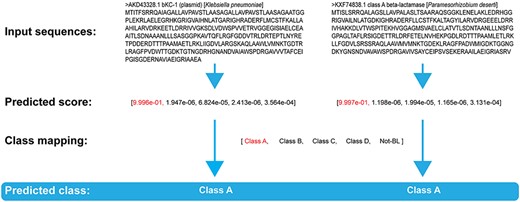
Prediction outputs of the two novel BLs in the BKC-1, PAD-1 case study.
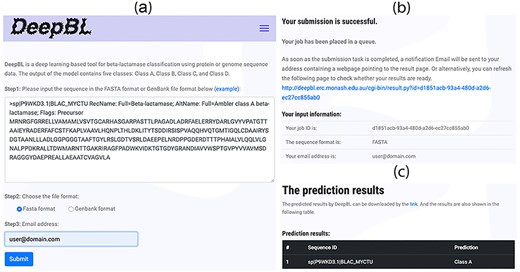
Screenshot of the web pages of the DeepBL webserver. (A) The input page of DeepBL; (B) the submission page of DeepBL and (C) the prediction output interface of DeepBL.
Architecture of the deep learning networks
In view of the dimension and scale of the dataset, we employed the Small VGGNet deep learning framework to train the prediction model. Small VGGNet is a VGGNet-like architecture for deep learning neural networks, derived from the VGGNet [42] architecture. The VGGNet was proposed in Large Scale Visual Recognition Challenge 2014 and won the subtask of image classification and localization. Afterward, many well-established variations of VGGNet are implemented for specific applications. A VGGNet-like neural network has the following features: (i) using 3 × 3 convolutional layers stacked on top of the other layers in increasing network depth; (ii) using the max-pooling layers to reduce the volume size and (iii) stacking a fully connected layer prior to the SoftMax or the sigmoid layer at the end of the network. For our network architecture, five convolution layers were adopted along with the ReLU layers, max-pooling layers, batch-normalization layers and dropout layers, as shown in Figure 1E. Furthermore, the Small VGGNet was customized to fit our input feature dimensions and the five-class classification problem. Lastly, the model architecture was implemented using python and the Keras library [43] with the TensorFlow [44] framework as the backend.
Model training
Two different model training strategies were adopted with Keras [43] based on the curated benchmark dataset: (i) model training for 10-fold cross-validation and (ii) model training for the independent test. As for the model training for 10-fold cross-validation, the model performance could be thoroughly evaluated on the given benchmark dataset and mitigate the bias caused by the data split. As for the model training for independent test, the larger independent test dataset could be used to evaluate the generalization of the trained model using the training dataset. To avoid the potential bias, we performed the model training for 10 times and accordingly calculated the mean and SD of the performance result of the 10 times. These two model training strategies shared the same hyperparameters including the number of epochs, batch size, learning rate, dropout rate and validation ratio (with corresponding values 100, 32, 1e−3, 0.5 and 0.2). To avoid model overfitting, an early stopping setting and dropout layers were used during model training. By combining the hyperparameters and the settings, model loss values and accuracies on both the training and validation datasets gradually changed and stabilized at specific values, as shown in Figure S1. The final optimal hyperparameters were selected based on the F1-score and accuracy on the validation dataset and used for performance evaluation and comparison.
Performance evaluation metrics
In particular, the ROC curves and Precision–Recall (PR) curves are plotted on 10-fold cross-validation and the independent test. The corresponding AUC values are calculated as an essential metric to evaluate the performance of the trained models and compared with different methods.
Performance evaluation at different sequence identity levels
When using conventional machine learning algorithms, sequence redundancy removal plays an essential role in sequence analysis and adoption of different sequence identity thresholds to remove the sequence redundancy would influence the model performance. After removing highly similar sequences from the benchmark datasets, the classifiers are trained using the representative sequences, which could enhance the robustness and generalization of the trained model and also reduce the risk of overfitting. Traditional machine learning algorithms can often achieve relatively good performance even on relatively small training datasets. However, compared with the traditional machine learning algorithms, deep learning algorithms have different requirements. They typically rely on a large amount of data for training the models to achieve good performance. Use of limited training data to train deep learning models may lead to the overfitting of the model; in some scenarios, data augmentation is employed to generate more similar training samples to enhance the model performance [45]. Moreover, a series of mechanisms are in place to prevent the overfitting in deep learning, including the ReLU activation function, the dropout rate and the pooling layer. In this study, in order to evaluate the effect of sequence redundancy removal on the performance of deep learning models, we benchmark the model performance on the datasets clustered using varying sequence identity thresholds by the CD-HIT program.
Performance comparison with different machine learning algorithms
To illustrate the effectiveness of DeepBL based on the Small VGGNet architecture, we assessed and compared the performance of DeepBL using the same dataset split (i.e. training dataset: 80%, independent testing dataset: 20%) and feature encoding scheme (CKSAAP) and that of four other popular machine learning algorithms, including Naïve Bayes (NB), Logistical Regression (LR), SVM and Random Forest (RF). In particular, the ‘one versus the rest’ strategy was used to train the multiclass classification models for the four conventional machine learning algorithms. The four machine learning algorithms were implemented using the Scikit-learn package [46] of Python.
Results and discussion
Performance evaluation on 10-fold cross-validation
We evaluateddation on the benchmark datasets. The mean ROC curves and mean PR curves on the 10-fold cross-validation test are displayed in Figure 3. As can be seen, DeepBL achieved a remarkable performance, with an AUC score of >0.99 for all the five classes. Among the five classes, DeepBL performed the best for predicting Class B with an AUC-ROC score of 0.999 ± 0.0 and an AUPRC score of 0.997 ± 0.001, respectively.
In Figure 4A, we also show the loss values of categorical cross-entropy of model training on 10-fold cross-validation test. As can be seen, the loss values range from 0.14 to 0.26. The performance results in terms of ACC, MCC and F1-Score are shown as box plots in Figure 4B. Altogether, these results demonstrate that DeepBL achieved a high performance on 10-fold cross-validation test when evaluated on the whole curated benchmark dataset.
Performance comparison between DeepBL and the other algorithms
To illustrate the effectiveness and robustness of DeepBL, we further evaluated the predictive performance of DeepBL models trained using the optimally tuned hyperparameters on the independent test dataset. The corresponding ROC and PR curves of DeepBL on the independent test are shown in Figure 5. Again, these results on the independent test clearly show that DeepBL achieved a remarkable performance. Figure 5 also shows that both Class B and Class D obtained the best performance, with an AUC value of 1.0 and an AURPC value of 0.999, followed by the Not-BL class with an AUC value of 0.996 and an AURPC value of 0.995, respectively. In addition, Class A and Class C achieved an AUC value of 0.991 and 0.989 and an AURPC value of 0.986 and 0.932, respectively.
Figure 6 shows the normalized confusion matrix of the prediction of five classes of BLs, which further demonstrates the outstanding performance achieved by DeepBL. Moreover, we also benchmarked the performance of DeepBL against four other commonly used machine learning algorithms (i.e. SVM, RF, NB and LR). The ROC curves of these four compared algorithms are provided in Figures S2–S5. Altogether, the performance comparison results show DeepBL clearly outperformed the other conventional machine learning algorithms.
Taken together, we conclude that DeepBL achieved the remarkable performance due to the following two primary reasons: (i) the CNN employed by DeepBL offers an attractive advantage of automatically extracting useful features from the input data compared with other conventional machine learning algorithms without the need of complex feature engineering and extraction and (ii) the class imbalance and sequence redundancy in the benchmark dataset would significantly influence the model performance of conventional machine learning algorithms, but for the deep learning framework of DeepBL, multiple strategies and settings are in place during the deep learning model training to avoid the overfitting. These included use of the dropout layer, max-pooling layer as well as the early stopping strategy. Furthermore, the use of large-scale training data could also help to improve the generalization and robustness of the deep learning model of DeepBL.
Effect of sequence identity level on the predictive performance
To evaluate the potential effect of sequence redundancy removal on the model performance, we generated the benchmark datasets at seven different sequence identity thresholds (ranging from 0.7 to 1.0). A global test dataset was extracted from dataset after sequence redundancy removal with the threshold of 0.7. To reduce the bias of model performance comparison among different sequence identities, we further removed the sequence redundancy between each training dataset with the global test dataset using the threshold of 0.7. Finally, 35 training datasets and one global test dataset were prepared. Then, we trained models on the 35 training datasets and tested the model performance using the global test dataset. During this procedure, we trained the models for 10 times and calculated the average AUC, F1-Score, MCC and Accuracy, and the results are shown in Figures 7 and 8 and Figures S6–S13. As shown in Figures 7 and 8, the model performance expectedly tends to increase in accordance with the larger sequence identity thresholds. In Figure 7, the model trained on datasets without any sequence redundancy removal achieved the overall best performance across all the five classes, with the AUCs ranging from 0.985 to 1.0 depending on the particular BL class. As a comparison, the models trained on datasets with the sequence identity threshold of 0.7 achieved the lowest performance. Among these models, the largest performance difference was >0.02 for the prediction of Class-C. In addition, we also observed that nearly all models achieved stable AUCs. In addition, only 40% sequences of Class-C were retained after the sequence redundancy removal at the sequence identity cutoff threshold of 0.95 (Figure 2). This indicates that the Class-C BL sequences had the highest homology levels, which may explain the relatively larger variation of AUC for Class-C BL prediction compared with the other classes.
As shown in Figure 8, the DeepBL models trained on the datasets without any sequence redundancy removal achieved the highest F1-Score, MCC and ACC with the mean values from 0.93 to 0.98, whereas the models trained on the datasets with sequence redundancy removal at the sequence identity threshold of 0.7 attained the worst performance with the mean values of 0.88–0.96. We can see that the model performance was relatively stable with respect to the three metrics. Altogether, the results indicate that researchers should practice with caution regarding the necessity to perform sequence redundancy removal, especially for training deep learning-based models. Before proceeding to carry out the sequence redundancy removal, the need to do so should be carefully evaluated in lieu of the research task itself.
In an effort to computationally characterize a complete repertoire of novel putative BLs, we further applied the optimized DeepBL model to perform the proteome-wide screening using the bacterial proteome data. We downloaded all reviewed bacterial protein sequences from the UniProt database, which contained 334 009 bacterial protein sequences in the release dated on 16 May 2019. As a result, DeepBL identified 2876 Class-A, 665 Class-B, 335 Class-C and 231 Class-D BLs, respectively. And data visualization is shown in Figure 9. A full list of all predicted BL sequences is available at the DeepBL webserver (http://deepbl.erc.monash.edu.au/download/proteomics_scan/screening_results.zip).
Case study
To further examine the capability of DeepBL in identifying novel BLs, we performed an independent case study by predicting two rarely reported BLs that were excluded from the training dataset of our DeepBL model. The first BL, BKC-1 (accession number: AKD43328.1), was reported as a novel BL from a Klebsiella pneumoniae clinical isolate and characterized as a carbapenemase [47]. Purified BKC-1 can not only hydrolyze carbapenems but also penicillins, cephalosporins and monabactams. DeepBL successfully predicted BKC-1 with the predicted probability score of 0.9996 and classified it as a Class-A BL. The second BL, PAD-1 (accession number: KXF74838.1), was isolated from the desert soil bacterium, Paramesorhizobium deserti and classified as a novel Class-A environmental carbapenemase with an unusual substrate profile [48]. DeepBL was correctly able to predict this BL and its class with the predicted probability score of 0.9997. The prediction outputs of these two case study BLs are provided in Figure 10. Together, these results highlight the capability of DeepBL in identifying and discovering novel BLs from the sequence information.
Webserver and user guide
In order to facilitate experimental researchers’ work, the DeepBL webserver has been developed and freely available at http://DeepBL.erc.monash.edu/. The user interface of DeepBL is shown in Figure 11. The DeepBL server was mainly implemented using Python on the base of Apache and configured in the Ubuntu OS on a 4-core server with 32 GB memory and a 1 TB hard disk. The server uses the optimal model to identify the BL sequences for the submitted tasks. According to the benchmarking test, DeepBL can complete the prediction of 100 protein sequences in approximately 10 s, which is presumably due to the application of effective feature encoding schemes and small deep learning architecture like the small VGGNet framework. Moreover, the curated benchmark datasets and the bacterial proteome-wide prediction results can be downloaded from the DeepBL webserver as well.
Researchers can submit bacterial protein sequences as the input. Upon the job submission, the DeepBL webserver will process the submitted tasks, predict and return the BL protein sequences and their class information using the optimized deep learning model. The prediction results can be directly visualized within the webserver, whereas the results with all predicted scores can be downloaded for users’ follow-up analysis. With the implementation and availability of the webserver, DeepBL will be maintained and made publicly available for at least 5 years. We plan to regularly update the model and webserver when more sequence data become available in the future. It is anticipated that DeepBL will be exploited as an indispensable tool for in silico discovery of novel BLs.
Conclusion
In this study, we have proposed DeepBL, a powerful deep learning-based approach for identifying BLs and their corresponding classes from the sequence data. More specifically, DeepBL was developed using the Small VGGNet architecture based on the large-scale and reliable curated datasets extracted from the NCBI RefSeq database. Through extensive benchmarking experiments, we demonstrated the generalization and robustness of DeepBL based on strictly and comprehensively conducted performance assessments on both 10-fold cross-validation and independent tests. Built on the deep CNN architecture, the performance of DeepBL benefitted from the use of large-scale datasets with less influence by data imbalance and sequence redundancy. Through the performance comparison of the models trained using various datasets with sequence redundancy removed at different sequence identity thresholds, we obtained the best-performing model of DeepBL on the dataset without any sequence redundancy removal at all. In terms of the feature encoding scheme, we only used CKSAAP as it can convert sequences of any length into the features of the same size. We further applied the optimized model to perform in silico screening and construct a whole-proteome complete catalogue of BLs. DeepBL is a generalized and useful tool for BL identification presumably due to the use of significantly enlarged benchmark datasets. We anticipate DeepBL will be a valuable tool for the wider research community and be exploited to shed light on the identification and functional annotation of putative BLs and their classes in the future.
Accurate identification of beta-lactamases (BLs) and their specific subclasses is an essential step to guide BL inhibitor design and improve our understanding of the mechanisms involved in beta-lactam antibiotic resistance.
We present DeepBL, a deep learning-based approach by incorporating sequence-derived features to enable high-throughput prediction of BLs.
Specifically, DeepBL is implemented based on the Small VGGNet architecture and the TensorFlow deep learning library.
The performance of DeepBL models is assessed with respect to the sequence redundancy level and negative sample selection in the benchmark datasets.
Using the optimized DeepBL model, we perform proteome-wide screening for all reviewed bacterium protein sequences available from UniProt and accordingly construct a whole-proteome catalogue of putative BLs and their subclasses.
These proteome-wide prediction results and the web server of DeepBL is freely available at http://deepbl.erc.monash.edu.au/.
Conflict of Interest
The authors declare no conflict of interest.
Funding
National Health and Medical Research Council of Australia (NHMRC) (APP1127948 and APP1144652); the Australian Research Council (ARC) (DP120104460); the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (R01 AI111965); a Postgraduate Scholarship from the Monash–Newcastle Alliance; a Major Inter-Disciplinary Research (IDR) project awarded by Monash University and the Collaborative Research Program of Institute for Chemical Research, Kyoto University; Informatics Institute of the School of Medicine at UAB (to T.T.M.L. and A.L.). T.L. is an ARC Laureate Fellow.
Availability and implementation
To facilitate rapid identification of potential beta-lactamases from protein or genome sequences, for widespread use by the research community, a user-friendly webserver and the curated datasets used in this study have been made publicly available at http://DeepBL.erc.monash.edu.au/.
Yanan Wang is currently a PhD candidate in the Biomedicine Discovery Institute and the Department of Biochemistry and Molecular Biology at Monash University, Australia. He received his Bachelor degree in communication engineering from the University of Shanghai for Science and Technology and his Master degree in control science and technology from Shanghai Jiao Tong University, China. His research interests are bioinformatics, machine learning and data mining.
Fuyi Li received his PhD in Bioinformatics from Monash University, Australia. He is currently a research fellow in the Department of Microbiology and Immunology, The Peter Doherty Institute for Infection and Immunity, The University of Melbourne, Australia. His research interests are bioinformatics, computational biology, machine learning and data mining.
Manasa Bharathwaj is currently a PhD candidate in the Department of Microbiology at the Biomedicine Discovery Institute, Monash University, Australia. She received her Bachelor degree in Biotechnology (Engineering) from SASTRA University, India, and her Masters by Research degree in Infectious diseases from The University of Edinburgh, UK. Her research interests include antibiotic resistance, bacterial protein transport and transcriptional regulation.
Natalia C. Rosas is currently a PhD candidate in the Department of Microbiology at the Biomedicine Discovery Institute, Monash University, Australia. She gained her Bachelor degree in bacteriology and clinical laboratory from the Universidad del Valle, Colombia, and her Master’s degree in Biotechnology from the University of Melbourne, Australia. Her experience and interests are in microbiology, molecular biology and antimicrobial resistance.
André Leier is currently an assistant professor in the Department of Genetics and the Department of Cell, Developmental and Integrative Biology, University of Alabama at Birmingham (UAB) School of Medicine, USA. He is also an associate scientist in the UAB Comprehensive Cancer Center. He received his PhD in Computer Science (Dr. rer. nat.), University of Dortmund, Germany. He conducted postdoctoral research at Memorial University of Newfoundland, Canada, The University of Queensland, Australia and ETH Zürich, Switzerland. His research interests are in biomedical informatics and computational and systems biomedicine.
Tatsuya Akutsu received his DEng degree in Information Engineering in 1989 from University of Tokyo, Japan. Since 2001, he has been a professor in the Bioinformatics Center, Institute for Chemical Research, Kyoto University, Japan. His research interests include bioinformatics and discrete algorithms.
Geoffrey I. Webb received his PhD degree in 1987 from La Trobe University, Australia. He is a professor in the Faculty of Information Technology and director of the Monash Centre for Data Science at Monash University. His research interests include machine learning, data mining, computational biology and user modeling.
Tatiana T. Marquez-Lago is an associate professor in the Department of Genetics and the Department of Cell, Developmental and Integrative Biology, UAB School of Medicine, USA. Her research interests include multiscale modeling and simulations, artificial intelligence, bioengineering and systems biomedicine. Her interdisciplinary lab studies stochastic gene expression, chromatin organization, antibiotic resistance in bacteria and host–microbiota interactions in complex diseases.
Jian Li is a professor and group leader in the Monash Biomedicine Discovery Institute and Department of Microbiology, Monash University, Australia. He is a Web of Science 2015–2017 Highly Cited Researcher in Pharmacology & Toxicology. He is currently an NHMRC Principal Research Fellow. His research interests include the pharmacology of polymyxins and the discovery of novel, safer polymyxins.
Trevor Lithgow is a professor in the Department of Microbiology at Monash University, Australia, and Director of the Centre to Impact AMR. He received his PhD degree in 1992 from La Trobe University. His research interests particularly focus on bacterial molecular cell biology and bioinformatics. His lab develops and deploys multidisciplinary approaches including comparative genomics to understand and image the assembly of proteins into bacterial outer membranes, and to discover and characterize the activity of bacteriophages that kill antibiotic-resistant ‘superbugs’.
Jiangning Song is an associate professor and group leader in the Monash Biomedicine Discovery Institute and Department of Biochemistry and Molecular Biology, Monash University, Melbourne, Australia. He is a member of the Monash Centre for Data Science, Faculty of Information Technology and an associate investigator of the ARC Centre of Excellence in Advanced Molecular Imaging, Monash University. His research interests include bioinformatics, computational biology, machine learning, data analytics and pattern recognition.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}