




Abstract
As one of the most important tasks in protein structure prediction, protein fold recognition has attracted more and more attention. In this regard, some computational predictors have been proposed with the development of machine learning and artificial intelligence techniques. However, these existing computational methods are still suffering from some disadvantages. In this regard, we propose a new network-based predictor called ProtFold-DFG for protein fold recognition. We propose the Directed Fusion Graph (DFG) to fuse the ranking lists generated by different methods, which employs the transitive closure to incorporate more relationships among proteins and uses the KL divergence to calculate the relationship between two proteins so as to improve its generalization ability. Finally, the PageRank algorithm is performed on the DFG to accurately recognize the protein folds by considering the global interactions among proteins in the DFG. Tested on a widely used and rigorous benchmark data set, LINDAHL dataset, experimental results show that the ProtFold-DFG outperforms the other 35 competing methods, indicating that ProtFold-DFG will be a useful method for protein fold recognition. The source code and data of ProtFold-DFG can be downloaded from http://bliulab.net/ProtFold-DFG/download
Introduction
Different tertiary structures of proteins lead to various protein functions. A fundamental step to study protein structures is to identify the protein folds [1]. In this regard, different computational methods are proposed to solve the protein fold recognition problem. They can be split into three categories: (i) alignment methods; (ii) machine-learning-based methods; (iii) network-based methods.
The alignment methods directly calculate the similarity between two proteins, based on which the folds can be predicted. For example, the CLUSTAL W [2] is based on the gap penalties. HHsearch [3] is based on the Hidden Markov Model-Hidden Markov Model (HMM-HMM) alignments. The PSI-BLAST [4] considers the evolutionary information by Position-Specific Score Matrices (PSSMs). ACCFold [5] combines the Autocross-covariance (ACC) transformation and PSSMs. Because the residue–residue contacts contain the predicted structure information [6], the residue–residue contacts are incorporated into the computational method so as to further improve the predictive performance [7]. The local or nonlocal interactions among residues or motifs are incorporated into the predictors as well [8–10]. For example, MotifCNN-fold [11] combines the motif-based kernel with Convolutional Neural Network (CNN) and Deep Convolutional Neural Network (DCNN) to extract the fold-specific features.
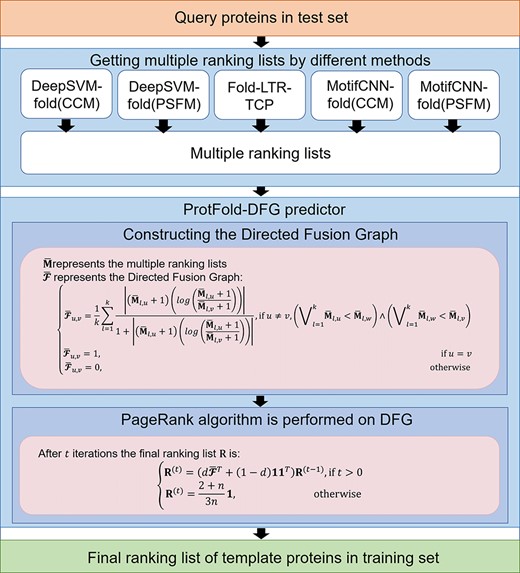
The flowchart of the ProtFold-DFG predictor for protein fold recognition.
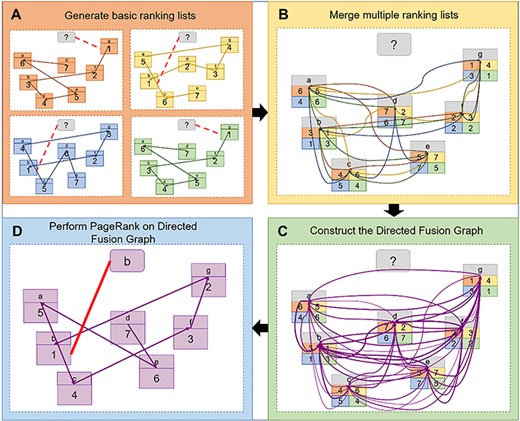
The process of generating the DFG. (A) The basic ranking lists are generated by four methods. (B) The four ranking lists are merged. (C) The Directed Fusion Graph is constructed based on the combined ranking list. (D) The PageRank is performed on the Directed Fusion Graph to recognize the protein folds.
Based on the aforementioned features, some machine-learning-based methods have been proposed [12]. For example, FOLDpro [13] combines different alignment methods and structural information with Support Vector Machines (SVMs) [14]. The ensemble classifier [15] fuses nine basic OET-KNN classifiers into a more accurate classifier. The deep learning techniques [16, 17] have been also applied to this field. For example, DeepFRpro [10] employs the CNN to generate the fold-specific features, based on which a random forest model is constructed to predict the folds. MV-fold [18] combines different features via multiview modeling.
The relationships among proteins can be considered as a protein similarity network. Some network-based methods extract the global interactions among proteins from the protein similarity network to recognize the protein folds. For example, the enrichment of network topological similarity is performed on the protein similarity network to detect the protein folds [19]. CMsearch [20] based on the cross-model learning learns the sequence–structure correlation. Fold-LTR-TCP [21] is a re-rank model via combining the Triadic Closure Principle (TCP) algorithm with Learning to Rank (LTR) [22].
As discussed above, the network-based methods achieve the state-of-the-art performance. However, two bottleneck problems still exist in the network-based methods, which should be further explored: (i) how to incorporate the relationships among different proteins into the protein similarity network? (ii) how to accurately recognize the proteins sharing the same fold with the query protein based on the network? To answer these two questions, in this study we propose the Directed Fusion Graph (DFG) to incorporate the relationships among proteins detected by different methods. The PageRank algorithm [23] is then performed on the DFG for protein fold recognition by considering the global relationships among proteins. The new computational predictor is called ProtFold-DFG.
Materials and methods
Benchmark data set
In order to facilitate the fair performance comparison with other competing methods, the ProtFold-DFG is evaluated on a widely used and rigorous benchmark data set, LINDAL data set [24] with 321 proteins covering 38 protein folds extracted from the SCOP database. In order to rigorously simulate the protein fold recognition task, these proteins are divided into two subsets sharing no proteins from the same family or superfamily. Based on these two subsets, the 2-fold cross-validation is used to evaluate the performance of various methods. For more information on the benchmark data set, please refer to [24].
The framework of ProtFold-DFG
Previous studies show that different methods for protein fold recognition are complementary [18, 25]. Therefore, it is possible to further improve the predictive performance by fusing these complementary methods. The protein fold recognition task can be treated as an information retrieval problem. For each query protein, it is searched against the data set by a computational method to generate the corresponding ranking list. The query protein is more likely to share the same fold with the top hit in the ranking list. For multiple ranking lists, the common top hits in different ranking lists are more likely to be in the same fold as the query protein. In this regard, we propose the DFG to incorporate these characteristics.
In this study, we fuse different ranking lists generated by five computational methods. The DFG is constructed to reflect the relationships among proteins in the network based on the ranking lists generated by the different methods. Finally, the PageRank algorithm [23] is performed on the DFG to re-rank the proteins in DFG so as to generate the final ranking list. The proposed new computational method is called ProtFold-DFG, whose overall flowchart is shown in Figure 1. In the following sections, we will introduce the detailed information of each step.

The pseudo-code of ProtFold-DFG predictor by combining PageRank and DFG.
Generate the basic ranking lists
As shown in Figure 2A, ranking lists are generated by different computational methods to reflect the relationships between the query protein and the template proteins, including DeepFR [10], DeepSVM-fold (CCM) [7], DeepSVM-fold (PSFM) [7], MotifCNN-fold (CCM) [11], MotifCNN-fold (PSFM) [11] and Fold-LTR-TCP [21]. Among these methods, the DeepFR, DeepSVM-fold (CCM), DeepSVM-fold (PSFM), MotifCNN-fold (CCM) and MotifCNN-fold (PSFM) are based on the deep learning techniques to extract the fold-specific features to improve the predictive performance, whereas Fold-LTR-TCP is based on the LTR [22].
DeepFR [10] extracts the fold-specific features by using DCNN, based on which the proteins are converted into feature vectors. The cosine similarity between two feature vectors is used to measure the similarity between two proteins.
DeepSVM-fold [7] calculates the feature vectors of proteins based on the fold-specific features generated using convolutional neural network–bidirectional long short-term memory (CNN-LSTM), and then the similarity between two proteins is calculated by cosine between their corresponding feature vectors. DeepSVM-fold (CCM) based on reside–residue contact (CCM) and DeepSVM-fold (PSFM) based on Position-Specific Frequency Matrix (PSFM) are employed in this study.
MotifCNN-fold [11] are based on the structural motif kernels to construct the motif-based CNNs. In this study the MotifCNN-fold (CCM), MotifCNN-fold (PSFM) are used, which are constructed based on the CCM and PSFM, respectively.
Fold-LTR-TCP [21] searches the query protein against the template proteins so as to find the template proteins in the same fold with the query protein in a supervised manner. The ranking list generated by Fold-LTR-TCP without TCP is used in this study.
Merge multiple ranking lists
Construct the DFG
This updated digraph G represents the reachability among vertices, incorporating the relationships among proteins in the basic ranking lists.
Performance of the ProtFold-DFG predictors based on different ranking lists on the LINDAHL data set evaluated by using 2-fold cross-validation
| Method | Accuracy (%) |
|---|---|
| ProtFold-DFGa | 77.88 |
| ProtFold-DFGb | 78.82 |
| ProtFold-DFGc | 71.03 |
| ProtFold-DFGd | 76.65 |
| ProtFold-DFGe | 76.65 |
| ProtFold-DFGf | 81.93 |
| ProtFold-DFGg | 80.69 |
| Method | Accuracy (%) |
|---|---|
| ProtFold-DFGa | 77.88 |
| ProtFold-DFGb | 78.82 |
| ProtFold-DFGc | 71.03 |
| ProtFold-DFGd | 76.65 |
| ProtFold-DFGe | 76.65 |
| ProtFold-DFGf | 81.93 |
| ProtFold-DFGg | 80.69 |
aFusing the rank lists generated by DeepSVM-fold (CCM), DeepSVM-fold (PSFM) and Fold-LTR-TCP.
bFusing the rank lists generated by MotifCNN-fold (CCM), MotifCNN-fold(PSFM) and Fold-LTR-TCP.
cFusing the rank lists generated by DeepFR and Fold-LTR-TCP.
dFusing the rank lists generated by DeepSVM-fold (CCM), DeepSVM-fold (PSFM), DeepFR and Fold-LTR-TCP.
eFusing the rank lists generated by MotifCNN-fold (CCM), MotifCNN-fold (PSFM), DeepFR and Fold-LTR-TCP.
fFusing the rank lists generated by DeepSVM-fold (CCM), DeepSVM-fold (PSFM), MotifCNN-fold (CCM), MotfiCNN-fold (PSFM) and Fold-LTR-TCP.
gFusing the rank lists generated by DeepSVM-fold (CCM), DeepSVM-fold (PSFM), MotifCNN-fold (CCM), MotifCNN-fold (PSFM), DeepFR and Fold-LTR-TCP.
Performance of the ProtFold-DFG predictors based on different ranking lists on the LINDAHL data set evaluated by using 2-fold cross-validation
| Method | Accuracy (%) |
|---|---|
| ProtFold-DFGa | 77.88 |
| ProtFold-DFGb | 78.82 |
| ProtFold-DFGc | 71.03 |
| ProtFold-DFGd | 76.65 |
| ProtFold-DFGe | 76.65 |
| ProtFold-DFGf | 81.93 |
| ProtFold-DFGg | 80.69 |
| Method | Accuracy (%) |
|---|---|
| ProtFold-DFGa | 77.88 |
| ProtFold-DFGb | 78.82 |
| ProtFold-DFGc | 71.03 |
| ProtFold-DFGd | 76.65 |
| ProtFold-DFGe | 76.65 |
| ProtFold-DFGf | 81.93 |
| ProtFold-DFGg | 80.69 |
aFusing the rank lists generated by DeepSVM-fold (CCM), DeepSVM-fold (PSFM) and Fold-LTR-TCP.
bFusing the rank lists generated by MotifCNN-fold (CCM), MotifCNN-fold(PSFM) and Fold-LTR-TCP.
cFusing the rank lists generated by DeepFR and Fold-LTR-TCP.
dFusing the rank lists generated by DeepSVM-fold (CCM), DeepSVM-fold (PSFM), DeepFR and Fold-LTR-TCP.
eFusing the rank lists generated by MotifCNN-fold (CCM), MotifCNN-fold (PSFM), DeepFR and Fold-LTR-TCP.
fFusing the rank lists generated by DeepSVM-fold (CCM), DeepSVM-fold (PSFM), MotifCNN-fold (CCM), MotfiCNN-fold (PSFM) and Fold-LTR-TCP.
gFusing the rank lists generated by DeepSVM-fold (CCM), DeepSVM-fold (PSFM), MotifCNN-fold (CCM), MotifCNN-fold (PSFM), DeepFR and Fold-LTR-TCP.
Performance of the ProtFold-DFG predictors with and without the transitive closure on the LINDAHL data set evaluated by using 2-fold cross-validation
Performance of the ProtFold-DFG predictors with and without the transitive closure on the LINDAHL data set evaluated by using 2-fold cross-validation
Perform PageRank on DFG
The PageRank algorithm is able to find the most cited literature from global perspective [29]. It has also been successfully applied to protein remote homology detection [30]. The PageRank algorithm assumes that the accuracy of a ranking is positively correlated with the relationships among proteins in the graph. Therefore, as shown in Figure 2D in this study the proteins are considered as the literature, and the proteins in the same fold can be considered as the most relevant literature. Based on these similarities, the PageRank is performed on the DFG for protein fold recognition.
Performance of the ProtFold-DFG predictors using KL divergence and out-degree on the LINDAHL data set evaluated by using 2-fold cross-validation
| Method | Accuracy (%) |
|---|---|
| ProtFold-DFG (out-degree)a | 80.06 |
| ProtFold-DFG (out-degree)b | 7.47 |
| ProtFold-DFG (KL divergence)c | 81.93 |
| ProtFold-DFG (KL divergence)d | 81.93 |
| Method | Accuracy (%) |
|---|---|
| ProtFold-DFG (out-degree)a | 80.06 |
| ProtFold-DFG (out-degree)b | 7.47 |
| ProtFold-DFG (KL divergence)c | 81.93 |
| ProtFold-DFG (KL divergence)d | 81.93 |
aThe evaluation score between two proteins is the reciprocal of the vertex’s out-degree and the initial value is the average of basic methods.
bThe evaluation score between two proteins is the reciprocal of the vertex’s out-degree and the initial value is |${\mathbf{R}}^{(0)}$| (Equation 8).
cThe evaluation score between two proteins is calculated by the KL divergence and the initial value is the average of basic methods.
dThe evaluation score between two proteins is calculated by the KL divergence and the initial value is |${\mathbf{R}}^{(0)}$| (Equation 8).
Performance of the ProtFold-DFG predictors using KL divergence and out-degree on the LINDAHL data set evaluated by using 2-fold cross-validation
| Method | Accuracy (%) |
|---|---|
| ProtFold-DFG (out-degree)a | 80.06 |
| ProtFold-DFG (out-degree)b | 7.47 |
| ProtFold-DFG (KL divergence)c | 81.93 |
| ProtFold-DFG (KL divergence)d | 81.93 |
| Method | Accuracy (%) |
|---|---|
| ProtFold-DFG (out-degree)a | 80.06 |
| ProtFold-DFG (out-degree)b | 7.47 |
| ProtFold-DFG (KL divergence)c | 81.93 |
| ProtFold-DFG (KL divergence)d | 81.93 |
aThe evaluation score between two proteins is the reciprocal of the vertex’s out-degree and the initial value is the average of basic methods.
bThe evaluation score between two proteins is the reciprocal of the vertex’s out-degree and the initial value is |${\mathbf{R}}^{(0)}$| (Equation 8).
cThe evaluation score between two proteins is calculated by the KL divergence and the initial value is the average of basic methods.
dThe evaluation score between two proteins is calculated by the KL divergence and the initial value is |${\mathbf{R}}^{(0)}$| (Equation 8).
Performance of the ProtFold-DFG predictors based on three link analysis algorithms on the LINDAHL data set evaluated by using 2-fold cross-validation
| Method | Link analysis algorithm | Accuracy (%) |
|---|---|---|
| ProtFold-DFGa | Hypertext Induced Topic Selection | 80.37 |
| ProtFold-DFGb | Triadic Closure Principle | 79.43 |
| ProtFold-DFGc | PageRank | 81.93 |
| Method | Link analysis algorithm | Accuracy (%) |
|---|---|---|
| ProtFold-DFGa | Hypertext Induced Topic Selection | 80.37 |
| ProtFold-DFGb | Triadic Closure Principle | 79.43 |
| ProtFold-DFGc | PageRank | 81.93 |
aThe Hypertext Induced Toxic Selection (HITS) algorithm is performed on the DFG.
bThe Triadic Closure Principle (TCP) algorithm is performed on the DFG.
cThe PageRank algorithm is performed on the DFG.
Performance of the ProtFold-DFG predictors based on three link analysis algorithms on the LINDAHL data set evaluated by using 2-fold cross-validation
| Method | Link analysis algorithm | Accuracy (%) |
|---|---|---|
| ProtFold-DFGa | Hypertext Induced Topic Selection | 80.37 |
| ProtFold-DFGb | Triadic Closure Principle | 79.43 |
| ProtFold-DFGc | PageRank | 81.93 |
| Method | Link analysis algorithm | Accuracy (%) |
|---|---|---|
| ProtFold-DFGa | Hypertext Induced Topic Selection | 80.37 |
| ProtFold-DFGb | Triadic Closure Principle | 79.43 |
| ProtFold-DFGc | PageRank | 81.93 |
aThe Hypertext Induced Toxic Selection (HITS) algorithm is performed on the DFG.
bThe Triadic Closure Principle (TCP) algorithm is performed on the DFG.
cThe PageRank algorithm is performed on the DFG.
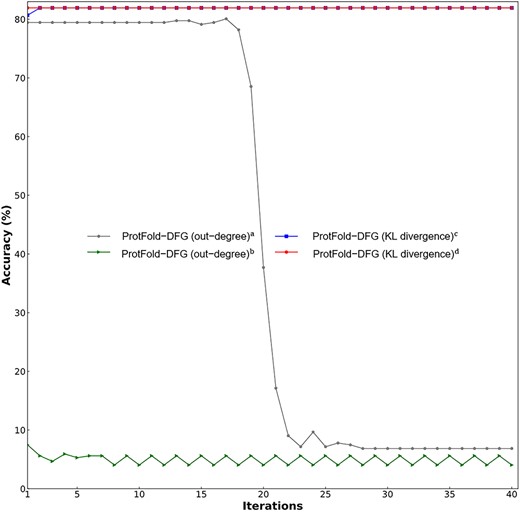
The performance of four ProtFold-DFG predictors based on the out-degree and KL divergence with different iterations. For the meanings of a, b, c and d, please refer to the footnote of Table 3.
Performance of 36 computational methods for protein fold recognition on LINDAHL data set via 2-fold cross-validation
| Methods | Categorya | Accuracy (%) | Source |
|---|---|---|---|
| PSI-BLAST | Alignment | 4 | [4] |
| HMMER | Alignment | 4.4 | [33] |
| SAM-T98 | Alignment | 3.4 | [34] |
| BLASTLINK | Alignment | 6.9 | [24] |
| SSEARCH | Alignment | 5.6 | [35] |
| SSHMM | Alignment | 6.9 | [35] |
| THREADER | Alignment | 14.6 | [36] |
| FUGUE | Alignment | 12.5 | [37] |
| RAPTOR | Alignment | 25.4 | [38] |
| SPARKS | Alignment | 24.3 | [39] |
| SP3 | Alignment | 28.7 | [51] |
| FOLDpro | Machine learning | 26.5 | [13] |
| HHpred | Alignment | 25.2 | [40] |
| SP4 | Alignment | 30.8 | [41] |
| SP5 | Alignment | 37.9 | [42] |
| BoostThreader | Machine learning | 42.6 | [43] |
| SPARKS-X | Alignment | 45.2 | [44] |
| FFAS-3D | Machine learning | 35.8 | [45] |
| RF-Fold | Machine learning | 40.8 | [46] |
| DN-Fold | Machine learning | 33.6 | [47] |
| RFDN-Fold | Machine learning | 37.7 | [47] |
| DN-FoldS | Machine learning | 33.3 | [47] |
| DN-FoldR | Machine learning | 27.4 | [47] |
| HH-fold | Machine learning | 42.1 | [48] |
| TA-fold | Machine learning | 53.9 | [48] |
| dRHP-PseRA | Machine learning | 34.9 | [49] |
| MT-fold | Machine learning | 59.1 | [18] |
| DeepFR (strategy1) | Machine learning | 44.5 | [10] |
| DeepFR (strategy2) | Machine learning | 56.1 | [10] |
| DeepFRpro (strategy1) | Machine learning | 57.6 | [10] |
| DeepFRpro (strategy2) | Machine learning | 66.0 | [10] |
| DeepSVM-fold | Machine learning | 67.3 | [7] |
| MotifCNN-fold | Machine learning | 72.6 | [11] |
| Fold-LTR-TCP | Network | 73.2 | [21] |
| FoldRec-C2C | Network | 77.88 | [50] |
| ProtFold-DFG b | Network | 81.93 | This study |
| Methods | Categorya | Accuracy (%) | Source |
|---|---|---|---|
| PSI-BLAST | Alignment | 4 | [4] |
| HMMER | Alignment | 4.4 | [33] |
| SAM-T98 | Alignment | 3.4 | [34] |
| BLASTLINK | Alignment | 6.9 | [24] |
| SSEARCH | Alignment | 5.6 | [35] |
| SSHMM | Alignment | 6.9 | [35] |
| THREADER | Alignment | 14.6 | [36] |
| FUGUE | Alignment | 12.5 | [37] |
| RAPTOR | Alignment | 25.4 | [38] |
| SPARKS | Alignment | 24.3 | [39] |
| SP3 | Alignment | 28.7 | [51] |
| FOLDpro | Machine learning | 26.5 | [13] |
| HHpred | Alignment | 25.2 | [40] |
| SP4 | Alignment | 30.8 | [41] |
| SP5 | Alignment | 37.9 | [42] |
| BoostThreader | Machine learning | 42.6 | [43] |
| SPARKS-X | Alignment | 45.2 | [44] |
| FFAS-3D | Machine learning | 35.8 | [45] |
| RF-Fold | Machine learning | 40.8 | [46] |
| DN-Fold | Machine learning | 33.6 | [47] |
| RFDN-Fold | Machine learning | 37.7 | [47] |
| DN-FoldS | Machine learning | 33.3 | [47] |
| DN-FoldR | Machine learning | 27.4 | [47] |
| HH-fold | Machine learning | 42.1 | [48] |
| TA-fold | Machine learning | 53.9 | [48] |
| dRHP-PseRA | Machine learning | 34.9 | [49] |
| MT-fold | Machine learning | 59.1 | [18] |
| DeepFR (strategy1) | Machine learning | 44.5 | [10] |
| DeepFR (strategy2) | Machine learning | 56.1 | [10] |
| DeepFRpro (strategy1) | Machine learning | 57.6 | [10] |
| DeepFRpro (strategy2) | Machine learning | 66.0 | [10] |
| DeepSVM-fold | Machine learning | 67.3 | [7] |
| MotifCNN-fold | Machine learning | 72.6 | [11] |
| Fold-LTR-TCP | Network | 73.2 | [21] |
| FoldRec-C2C | Network | 77.88 | [50] |
| ProtFold-DFG b | Network | 81.93 | This study |
a‘Alignment’, ‘Machine learning’ and ‘Network’ mean that the corresponding predictors are alignment methods, machine-learning-based methods and network-based methods respectively according to the three categories introduced in the introduction section.
bThe ProtFold-DFG predictor is based on the KL-divergence, PageRank and the DFG generated by DeepSVM-fold (CCM), DeepSVM-fold (PSFM), MotifCNN-fold (CCM), MotifCNN-fold (PSFM) and Fold-LTR-TCP.
Performance of 36 computational methods for protein fold recognition on LINDAHL data set via 2-fold cross-validation
| Methods | Categorya | Accuracy (%) | Source |
|---|---|---|---|
| PSI-BLAST | Alignment | 4 | [4] |
| HMMER | Alignment | 4.4 | [33] |
| SAM-T98 | Alignment | 3.4 | [34] |
| BLASTLINK | Alignment | 6.9 | [24] |
| SSEARCH | Alignment | 5.6 | [35] |
| SSHMM | Alignment | 6.9 | [35] |
| THREADER | Alignment | 14.6 | [36] |
| FUGUE | Alignment | 12.5 | [37] |
| RAPTOR | Alignment | 25.4 | [38] |
| SPARKS | Alignment | 24.3 | [39] |
| SP3 | Alignment | 28.7 | [51] |
| FOLDpro | Machine learning | 26.5 | [13] |
| HHpred | Alignment | 25.2 | [40] |
| SP4 | Alignment | 30.8 | [41] |
| SP5 | Alignment | 37.9 | [42] |
| BoostThreader | Machine learning | 42.6 | [43] |
| SPARKS-X | Alignment | 45.2 | [44] |
| FFAS-3D | Machine learning | 35.8 | [45] |
| RF-Fold | Machine learning | 40.8 | [46] |
| DN-Fold | Machine learning | 33.6 | [47] |
| RFDN-Fold | Machine learning | 37.7 | [47] |
| DN-FoldS | Machine learning | 33.3 | [47] |
| DN-FoldR | Machine learning | 27.4 | [47] |
| HH-fold | Machine learning | 42.1 | [48] |
| TA-fold | Machine learning | 53.9 | [48] |
| dRHP-PseRA | Machine learning | 34.9 | [49] |
| MT-fold | Machine learning | 59.1 | [18] |
| DeepFR (strategy1) | Machine learning | 44.5 | [10] |
| DeepFR (strategy2) | Machine learning | 56.1 | [10] |
| DeepFRpro (strategy1) | Machine learning | 57.6 | [10] |
| DeepFRpro (strategy2) | Machine learning | 66.0 | [10] |
| DeepSVM-fold | Machine learning | 67.3 | [7] |
| MotifCNN-fold | Machine learning | 72.6 | [11] |
| Fold-LTR-TCP | Network | 73.2 | [21] |
| FoldRec-C2C | Network | 77.88 | [50] |
| ProtFold-DFG b | Network | 81.93 | This study |
| Methods | Categorya | Accuracy (%) | Source |
|---|---|---|---|
| PSI-BLAST | Alignment | 4 | [4] |
| HMMER | Alignment | 4.4 | [33] |
| SAM-T98 | Alignment | 3.4 | [34] |
| BLASTLINK | Alignment | 6.9 | [24] |
| SSEARCH | Alignment | 5.6 | [35] |
| SSHMM | Alignment | 6.9 | [35] |
| THREADER | Alignment | 14.6 | [36] |
| FUGUE | Alignment | 12.5 | [37] |
| RAPTOR | Alignment | 25.4 | [38] |
| SPARKS | Alignment | 24.3 | [39] |
| SP3 | Alignment | 28.7 | [51] |
| FOLDpro | Machine learning | 26.5 | [13] |
| HHpred | Alignment | 25.2 | [40] |
| SP4 | Alignment | 30.8 | [41] |
| SP5 | Alignment | 37.9 | [42] |
| BoostThreader | Machine learning | 42.6 | [43] |
| SPARKS-X | Alignment | 45.2 | [44] |
| FFAS-3D | Machine learning | 35.8 | [45] |
| RF-Fold | Machine learning | 40.8 | [46] |
| DN-Fold | Machine learning | 33.6 | [47] |
| RFDN-Fold | Machine learning | 37.7 | [47] |
| DN-FoldS | Machine learning | 33.3 | [47] |
| DN-FoldR | Machine learning | 27.4 | [47] |
| HH-fold | Machine learning | 42.1 | [48] |
| TA-fold | Machine learning | 53.9 | [48] |
| dRHP-PseRA | Machine learning | 34.9 | [49] |
| MT-fold | Machine learning | 59.1 | [18] |
| DeepFR (strategy1) | Machine learning | 44.5 | [10] |
| DeepFR (strategy2) | Machine learning | 56.1 | [10] |
| DeepFRpro (strategy1) | Machine learning | 57.6 | [10] |
| DeepFRpro (strategy2) | Machine learning | 66.0 | [10] |
| DeepSVM-fold | Machine learning | 67.3 | [7] |
| MotifCNN-fold | Machine learning | 72.6 | [11] |
| Fold-LTR-TCP | Network | 73.2 | [21] |
| FoldRec-C2C | Network | 77.88 | [50] |
| ProtFold-DFG b | Network | 81.93 | This study |
a‘Alignment’, ‘Machine learning’ and ‘Network’ mean that the corresponding predictors are alignment methods, machine-learning-based methods and network-based methods respectively according to the three categories introduced in the introduction section.
bThe ProtFold-DFG predictor is based on the KL-divergence, PageRank and the DFG generated by DeepSVM-fold (CCM), DeepSVM-fold (PSFM), MotifCNN-fold (CCM), MotifCNN-fold (PSFM) and Fold-LTR-TCP.
Evaluation methodology
Results and discussion
The influence of the different combinations of the basic ranking lists on the performance of the ProtFold-DFG predictors
In this section, we investigate the influence of the basic predictors on the predictive performance. As introduced in the method section, six computational methods are selected to generate the basic ranking lists. There are two reasons for selecting these predictors: (i) they are the state-of-the-art methods in the field of protein fold recognition. (ii) They are based on different theories and techniques, and therefore, they are complementary.
The results of the ProtFold-DFG predictors by fusing different ranking lists are shown in Table 1, from which we can see the followings: (i) the performance of ProtFold-DFG can be improved when adding more complementary ranking lists. (ii) The ProtFold-DFG predictor based on the five ranking lists generated by DeepSVM-fold (CCM), DeepSVM-fold (PSFM), MotifCNN-fold (CCM), MotfiCNN-fold (PSFM) and Fold-LTR-TCP achieves the best performance. (iii) The ProtFold-DFG predictor achieves lower performance when adding the ranking list generated by DeepFR. The reason is that Fold-LTR-TCP is also based on the DeepFR, and their predictive results are not complementary. Noise information will be introduced when incorporating the predictive results of DeepFR into the ProtFold-DFG predictor. This conclusion is further supported by the fact that the ProtFold-DFG predictor based on DeepFR and Fold-LTR-TCP can only achieve an accuracy of 71.03%.
![The ProtFold-DFG prediction visualization. The best ProtFold-DFG predictor based on the five basic ranking lists is analyzed. The ranking lists generated by DeepSVM-fold (CCM), DeepSVM-fold (PSFM), Fold-LTR-TCP, MotifCNN-fold (CCM) and MotifCNN-fold (PSFM) are visualized in (A–E), respectively. Based on the basic ranking lists, the corresponding DFG is generated (F), and then the PageRank is performed on the DFG to detect the template proteins in DFG sharing the same protein fold with the query protein (G). For these figures, the blue lines and gray lines represent the relationships among proteins. The relationship between two proteins linked by blue line is closer than that linked by gray line. The red lines indicate the final prediction results: the query proteins and the template proteins are predicted to be in the same protein folds. These figures are plotted by using the software tool Gephi [52].](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/bib/22/3/10.1093_bib_bbaa192/1/m_bbaa192f5.jpeg?Expires=1749184017&Signature=iULHbDesmg8ItpLhVt7j0weynY1TYW34ljBVk4MShj5zjGSb03nUTGdMSkDBzkSYV2oBsLTDj4l3jEqlWn0fnfiO4~KmlkI7p9QLGZm~FkRB7CxCNOAVa6LuvxcIvCULQIVlGQnKCuei5ll9vdVBA6nGbfHXtM95t91KGe0RKIHZYe-YOA4cQFALzXYQqaMBtxweRxdbyIC54yBMf0sEANWeMcgZL7JQZBbBZaTMVGK8D2DuAXU1dbtZ9bkQv4IVLTSRMz~j2nIlOVU-WoPgdaPruC0bngEy8azVWnbgV~4f3r95rknOn-xdkIj-dNnuh5A0OIwOSvGs4J7q3Wruvg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
The ProtFold-DFG prediction visualization. The best ProtFold-DFG predictor based on the five basic ranking lists is analyzed. The ranking lists generated by DeepSVM-fold (CCM), DeepSVM-fold (PSFM), Fold-LTR-TCP, MotifCNN-fold (CCM) and MotifCNN-fold (PSFM) are visualized in (A–E), respectively. Based on the basic ranking lists, the corresponding DFG is generated (F), and then the PageRank is performed on the DFG to detect the template proteins in DFG sharing the same protein fold with the query protein (G). For these figures, the blue lines and gray lines represent the relationships among proteins. The relationship between two proteins linked by blue line is closer than that linked by gray line. The red lines indicate the final prediction results: the query proteins and the template proteins are predicted to be in the same protein folds. These figures are plotted by using the software tool Gephi [52].
The impact of the transitive closure on the performance of ProtFold-DFG
The main contribution of this study is to propose the DFG to fuse different ranking lists so as to model the global interactions among proteins. Different from the Google matrix [23], the DFG incorporates more interactions among proteins in the ranking lists by using the transitive closure [26]. In this section, we will explore the influence of the transitive closure on the performance of ProtFold-DFG, and the results are listed in Table 2, from which we can obviously see that the transitive closure can obviously improve the performance of ProtFold-DFG. The reason for its better performance is that when using transitive closure, the DFG cannot only consider the relationships between adjacent proteins in the ranking lists, but it also can incorporate the local or global interactions among proteins in the ranking lists, which will provide valuable information for the PageRank to re-rank the ranking list so as to accurately recognize the protein folds.
ProtFold-DFG based on KL divergence can improve its generalization ability
In this study, we employ the KL divergence [27] to assign weights to the edges in the DFG |$\overline{\mathbf{\mathcal{F}}}$|. Besides KL divergence, out-degree [29] can also calculate the weight for each edge. The performance of ProtFold-DFG predictors based on these two methods with different node initial values is shown in Table 3 and Figure 4, from which we can see the followings: (i) the two ProtFold-DFG predictors based on the KL divergence can achieve stable performance, indicating that the DFG constructed by KL divergence is insensitive with the node initial values and the iteration times t (Equation 8). (ii) In contrast, the two ProtFold-DFG predictors based on the out-degree achieve lower performance, and the initial value and the iteration times obviously impact their performance.
As discussed above, the PageRank updates the ranking list during each iteration. Therefore, a suitable node initial value helps the PageRank to find an optimum ranking list. However, the initial value has little impact on the results with more iteration times. Compared with the out-degree, the KL divergence considers the subtle interactions among proteins in the ranking list. Therefore, the two ProtFold-DFG predictors based on the KL divergence achieve higher and more stable performance than that of the two ProtFold-DFG predictors based on the out-degree.
The performance of three link analysis algorithms performed on the DFG
Inspired by the Google matrix, we propose the DFG to fuse different ranking lists and incorporate the local and global interactions among proteins. The link analysis algorithm is then performed on the DFG for protein fold recognition. In this section, the performance of three link analysis algorithms is explored, including PageRank [23], Hyperlink Induced Topic Selection (HITS) [32] and Triadic Closure Principle (TCP) [21]. The predictive results of the three ProtFold-DFG predictors based on the three link analysis algorithms are shown in Table 4. The ProtFold-DFG predictor based on PageRank achieves the best performance. The reason is that PageRank is able to search the related proteins in the DFG globally, making full use of the mutual recommendation relationships among all the proteins in the DFG. In contrast, HITS only models the protein local interactions, and TCP only considers the pairwise interactions between two proteins.
Performance comparison with 35 other related methods
In order to evaluate the performance of the ProtFold-DFG, it is compared with other 35 computational methods for protein fold recognition, including PSI-BLAST [4], HMMER [33], SAM-T98 [34], BLASTLINK [24], SSEARCH [35], SSHMM [35], THREADER [36], FUGUE [37], RAPTOR [38], SPARKS [39], SP3 [39], FOLDpro [13], HHpred [40], SP4 [41], SP5 [42], BoostThreader [43], SPARKS-X [44], FFAS-3D [45], RF-Fold [46], DN-Fold [47], RFDN-Fold [47], DN-FoldS [47], DN-FoldR [47], HH-fold [48], TA-fold [48], dRHP-PseRA [49], MT-fold [18], DeepFR (strategy 1) [10], DeepFR (strategy 2) [10], DeepFRpro (strategy 1) [10], DeepFRpro (stragegy 2) [10], DeepSVM-fold [7], MotifCNN-fold [11], Fold-LTR-TCP [21] and FoldRec-C2C [50].
The results of the 36 predictors are listed in Table 5, from which we can see the followings: (i) ProtFold-DFG outperforms the compuational methods used for generating the basic ranking lists, including DeepSVM-fold, Fold-LTR-TCP and MotifCNN-fold. These results further confirm that the fusion of the different basic ranking lists by DFG can improve the predictive performance. (ii) The three network-based methods (Fold-LTR-TCP, FoldRec-C2C and ProtFold-DFG) show better performance than the alignment methods and the machine-learning-based methods. The reason is that the network-based methods incorporate the global relationships among proteins into the prediction, which is more efficient than only considering the pairwise protein similarities or the local relationships among proteins. (iii) ProtFold-DFG achieves the best performance among the 36 predictors. The reason is that ProtFold-DFG is able to more accurately mesaure the relationships among proteins by fusing complementary ranking lists, and the link analysis algorithm PageRank is able to globally analyze the relationships among proteins in the DFG to accurately detect the protein folds.
Feature analysis
In order to further explore the reasons why the ProtFold-DFG predictor is able to accurately detect the protein folds, a query protein 1bdo-d1bdo from protein fold 2_59 (SCOP ID) is selected as an example, and the prediction results of ProtFold-DFG are visualized in Figure 5, from which we can see the followings: (i) according to the top hits in the five basic ranking lists shown in Figure 5A–E, the fold type of the query protein is incorrectly predicted by the five computational predictors. (ii) DFG is able to fuse the five ranking lists by considering the local and global relationships among proteins (Figure 5F). (iii) Performed on the DFG, the PageRank algorithm is able to re-rank the proteins in the DFG and correctly predict the fold of the query protein 1bdo-d1bdo as 2_59 (Figure 5G). These results are not surprising, although the top hits in the basic ranking lists of the five predictors are incorrect, these errors can be corrected by fusing these complementary ranking lists and analyzing their relationships comprehensively. ProtFold-DFG treats the proteins as the literature, and finds the most closely linked proteins by combining the PageRank and DFG. These proteins can be considered as in the same protein fold.
Conclusion
Protein fold recognition is one of the key tasks in bioinformatics. In this regard, some computational methods have been proposed. Among these methods, the network-based methods can achieve the state-of-the-art performance, because they can more efficiently model the relationships among proteins with the help of the protein similarity network [50]. In this study, we propose a new network-based computational predictor called ProtFold-DFG. Compared with other existing methods, it has the following advantages: (i) The proposed DFG is able to fuse the complementary ranking lists generated by different methods, and more interactions can be incorporated by transitive closure. (ii) KL divergence can more accurately calculate the weights of the edges in DFG, which is insensitive with the initial values and the literation times. As a result, the performance and generalization ability of ProtFold-DFG can be obviously improved. (iii) The PageRank algorithm can correct the prediction errors in the basic ranking lists by comprehensively considering the protein interactions in the DFG. Because the DFG is able to fuse the results of different ranking methods, other applications in bioinformatics can be anticipated, such as protein remote homology detection [53].
Protein fold recognition is a fundamental task for detecting protein structures and determining the protein functions. More accurate computational predictors are highly required.
We propose the ProtFold-DFG predictor for protein fold recognition. It is based on the DFG generated by different basic ranking methods. In order to globally consider the interactions among proteins in the DFG, the PageRank is performed on DFG to re-rank the ranking lists so as to improve the predictive performance.
Experimental results show that the ProtFold-DFG predictor outperforms the other 35 state-of-the-art computational methods. The source code and data of the ProtFold-DFG can be accessed from http://bliulab.net/ProtFold-DFG/download/
Acknowledgements
The authors are very much indebted to the three anonymous reviewers, whose constructive comments are very helpful in strengthening the presentation of this article.
Funding
The National Key R&D Program of China (No. 2018AAA0100100); National Natural Science Foundation of China (No. 61672184, 61822306, 61702134, 61732012, 61861146002); the Beijing Natural Science Foundation (No. JQ19019); Fok Ying-Tung Education Foundation for Young Teachers in the Higher Education Institutions of China (161063).
Jiangyi Shao is a master student at the School of Computer Science and Technology, Beijing Institute of Technology, China. His expertise is in bioinformatics.
Bin Liu is a professor at the School of Computer Science and Technology, Beijing Institute of Technology, Beijing, China. His expertise is in bioinformatics, natural language processing and machine learning.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}