




Abstract
Various microbes have proved to be closely related to the pathogenesis of human diseases. While many computational methods for predicting human microbe-disease associations (MDAs) have been developed, few systematic reviews on these methods have been reported. In this study, we provide a comprehensive overview of the existing methods. Firstly, we introduce the data used in existing MDA prediction methods. Secondly, we classify those methods into different categories by their nature and describe their algorithms and strategies in detail. Next, experimental evaluations are conducted on representative methods using different similarity data and calculation methods to compare their prediction performances. Based on the principles of computational methods and experimental results, we discuss the advantages and disadvantages of those methods and propose suggestions for the improvement of prediction performances. Considering the problems of the MDA prediction at present stage, we discuss future work from three perspectives including data, methods and formulations at the end.
Introduction
Microbial communities, which are composed of bacteria, archaea, fungi, viruses and protozoa [1], ubiquitously colonize in the human body. Microorganisms are usually beneficial to human beings. For instance, probiotics in guts are good for fermenting undigested carbohydrates so as to manufacture nutrition which is needed by the human beings [2, 3]. They also make great contributions to the maturity of the immune system [4, 5]. In addition, microbial communities are essential to guarantee that the homeostasis of extracellular fluid and intracellular environment are stable [6]. Hydrogen peroxide and lactic acid, the product of the vaginal Lactobacillus species, protect female vaginas from invasion of pathogens [7, 8]. The microbiota is also able to activate the repair process of the damaged physiological functions as well, such as fixing the intestinal epithelium through the MyD88-dependent process [9].
A group of balanced microorganisms keep the human body away from physiological disorders, while the unusual growth or decline of microorganism population is possibly related to the occurrence of disease. More clinical trials and advanced sequencing technologies make it possible to study intricate microbe-disease associations (MDAs) [10, 11]. For example, infections of facial follicles are typically caused by the massive reproduction of Staphylococcus aureus [12]. It is also known that many regions inside the human body are suitable habitats for various microorganisms, such as the oral cavity. Several studies have implicated that changes in the composition of oral microbiota contribute to periodontitis [13, 14]. The gut microbiota is greatly involved in host metabolic and immunomodulatory activities, forming the most complex microecosystem in human body. Abnormal host–gut microbiota interactions greatly affect host physical health and possibly lead to diseases. For example, the dysbiosis of gut microbiome is a prominent contributor to the chronic inflammation in inflammatory bowel disease [15] and hypertension [16] patients. The colonic mucosa is cumulatively exposed to diet-induced microbial carcinogenic metabolites, promoting colorectal cancer [17]. With regard to obesity that has been proved to correlate with the gut microbiota, a strategy for identifying the pathogenic agent in the gut microbiota has been already proposed, combining with a spectrum of microbiome-wide association studies [18].
As mentioned previously, studies on pathomicrobiology open up promising perspectives. Some small-scale databases focusing on genomic information of specific microbes have been established [19, 20]. Other comprehensive microbial databases such as SILVA [21], IMG/M [22], Pfam [23], M3D [24], MiST [25] and TCDB [26], covering diverse branches of microbiology (e.g. genome, metagenome, proteome, transcription and metabolism), have also been created. Meanwhile, some researchers develop and adopt computational methods to detect microbial influences on human diseases. For example, Coelho et al. have proposed a computational method to predict the impact of microbial proteins on human biological events, which takes the relationship between microbial and human proteins into consideration [27]. Another famous instance related to the microbe project is the Human Microbiome Project launched in 2007 [28].
Discovering MDAs would be truly useful in disease-related areas (e.g. pathogenic genes and drugs) [29]. Taking drug repositioning as an example, type 2 diabetes shares a high similarity with colorectal carcinoma based on their associations with microbes, which infers that these two diseases could be treated with the same drug. This hypothesis has been tested and verified [30, 31]. Moreover, discoveries of MDAs provide plenty of perspectives on disease mechanisms. Accurate prediction of associations narrows down the MDA potential search space, which reduces the time, effort and cost of wet labs’ projects. Figure 1 depicts an entire process that includes data collection, computational prediction, clinical validation and pathology inference.
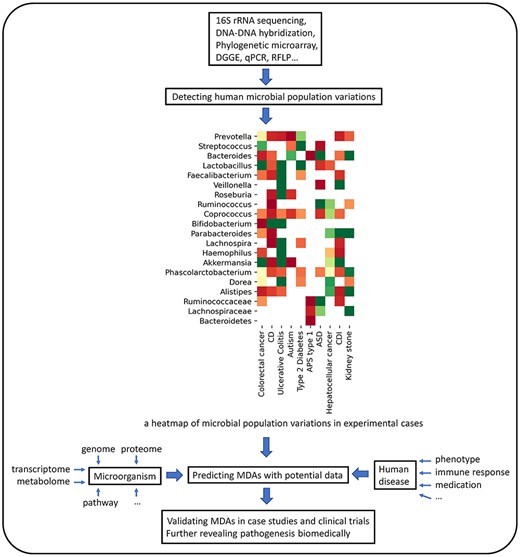
Three main procedures of exploring the relationship between microbes and diseases: (i) Differences between the diseased group and healthy group in microbial populations are captured from metagenome samples based on sequencing technologies. Researchers normalize quantitative differences from reported cases and curate the MDA dataset. (ii) High-confidence MDAs are derived by the computational prediction based on the MDA dataset, microbe-centric data and disease-centric data. (iii) Biologists screen the candidates and seek biomedical interpretation.
However, there is no overarching survey so far regarding the MDA prediction. Therefore, we try to comprehensively review computational methods for the MDA prediction in this study. We divide the computational methods into five categories [32, 33] as shown in Table 1. The following itemized list briefly describes their nature:
Path-based methods: In heterogeneous networks, path-based methods make the prediction by computing path-based scores between microbe nodes and disease nodes.
Random Walk methods: A walker randomly walks in the transition probability network consisting of microbe and disease nodes. These methods search for a potential association by measuring the probability of a random walker that has completed a path starting a node from a side of the association and ending a node from another side.
Bipartite local models (BLMs): BLMs compute the prediction scores of MDAs from two perspectives: diseases and microbes. Prediction scores of both sides are integrated, which is regarded as final prediction score.
Matrix factorization methods: An association matrix is factorized into two low-dimensional matrices where one represents features of diseases while another represents features of microbes. The product of two low-dimensional matrices is the final predicted matrix.
Other methods: Some methods could not be classified into the above-mentioned categories, and thus these methods are grouped into ‘other methods’.
Different methods to predict MDAs in each category
| Category | Method | Description |
|---|---|---|
| Path-based methods | KATZHMDA [40], PBHMDA [63], MDPH_HMDA [40], BWNMHMDA [60],WMGHMDA [47] | Path-based methods generally take into account numbers and weighted scores of various types of paths between two nodes. |
| Random walk methods | RWRH [51], BiRWHMDA [58], PRWHMDA [49], NTSHMDA [65], BDSILP [53], BiRWMP [69], BRWMDA [68], NBLPIHMDA [67], RWHMDA [66] | Random walk methods construct graph-based transition probability matrix for iterative walking. |
| BLMs | LRLSHMDA [72], NGRHMDA [70], Wang et al. [36], NCPHMDA [71], KATZBNRA [57] | BLMs perform independent predictions from both microbe and disease sides. |
| Matrix factorization methods | CMFHMDA [80], GRNMFHMDA [59], NMFMDA [56], KBMF [84], MDLPHMDA [82], mHMDA [86] | Matrix factorization methods optimize two latent informative matrices, whose multiplication approximates the association matrix with different constraint terms. |
| Other methods | ABHMDA [87], BMCMDA [88], MCHMDA [89] | These methods mainly include ensemble learning and matrix completion. |
| Category | Method | Description |
|---|---|---|
| Path-based methods | KATZHMDA [40], PBHMDA [63], MDPH_HMDA [40], BWNMHMDA [60],WMGHMDA [47] | Path-based methods generally take into account numbers and weighted scores of various types of paths between two nodes. |
| Random walk methods | RWRH [51], BiRWHMDA [58], PRWHMDA [49], NTSHMDA [65], BDSILP [53], BiRWMP [69], BRWMDA [68], NBLPIHMDA [67], RWHMDA [66] | Random walk methods construct graph-based transition probability matrix for iterative walking. |
| BLMs | LRLSHMDA [72], NGRHMDA [70], Wang et al. [36], NCPHMDA [71], KATZBNRA [57] | BLMs perform independent predictions from both microbe and disease sides. |
| Matrix factorization methods | CMFHMDA [80], GRNMFHMDA [59], NMFMDA [56], KBMF [84], MDLPHMDA [82], mHMDA [86] | Matrix factorization methods optimize two latent informative matrices, whose multiplication approximates the association matrix with different constraint terms. |
| Other methods | ABHMDA [87], BMCMDA [88], MCHMDA [89] | These methods mainly include ensemble learning and matrix completion. |
Different methods to predict MDAs in each category
| Category | Method | Description |
|---|---|---|
| Path-based methods | KATZHMDA [40], PBHMDA [63], MDPH_HMDA [40], BWNMHMDA [60],WMGHMDA [47] | Path-based methods generally take into account numbers and weighted scores of various types of paths between two nodes. |
| Random walk methods | RWRH [51], BiRWHMDA [58], PRWHMDA [49], NTSHMDA [65], BDSILP [53], BiRWMP [69], BRWMDA [68], NBLPIHMDA [67], RWHMDA [66] | Random walk methods construct graph-based transition probability matrix for iterative walking. |
| BLMs | LRLSHMDA [72], NGRHMDA [70], Wang et al. [36], NCPHMDA [71], KATZBNRA [57] | BLMs perform independent predictions from both microbe and disease sides. |
| Matrix factorization methods | CMFHMDA [80], GRNMFHMDA [59], NMFMDA [56], KBMF [84], MDLPHMDA [82], mHMDA [86] | Matrix factorization methods optimize two latent informative matrices, whose multiplication approximates the association matrix with different constraint terms. |
| Other methods | ABHMDA [87], BMCMDA [88], MCHMDA [89] | These methods mainly include ensemble learning and matrix completion. |
| Category | Method | Description |
|---|---|---|
| Path-based methods | KATZHMDA [40], PBHMDA [63], MDPH_HMDA [40], BWNMHMDA [60],WMGHMDA [47] | Path-based methods generally take into account numbers and weighted scores of various types of paths between two nodes. |
| Random walk methods | RWRH [51], BiRWHMDA [58], PRWHMDA [49], NTSHMDA [65], BDSILP [53], BiRWMP [69], BRWMDA [68], NBLPIHMDA [67], RWHMDA [66] | Random walk methods construct graph-based transition probability matrix for iterative walking. |
| BLMs | LRLSHMDA [72], NGRHMDA [70], Wang et al. [36], NCPHMDA [71], KATZBNRA [57] | BLMs perform independent predictions from both microbe and disease sides. |
| Matrix factorization methods | CMFHMDA [80], GRNMFHMDA [59], NMFMDA [56], KBMF [84], MDLPHMDA [82], mHMDA [86] | Matrix factorization methods optimize two latent informative matrices, whose multiplication approximates the association matrix with different constraint terms. |
| Other methods | ABHMDA [87], BMCMDA [88], MCHMDA [89] | These methods mainly include ensemble learning and matrix completion. |
In following sections, we firstly introduce types of data including MDAs and other data for the similarity calculation. Then similarity calculation methods for MDAs and other data are presented. Next, we describe each prediction method with its classification shown in Table 1. A simplified flowchart of predicting MDAs is shown in Figure 2. After that, we evaluate the methods by comparing prediction performances. Finally, we make recommendations for future work of the MDA prediction.
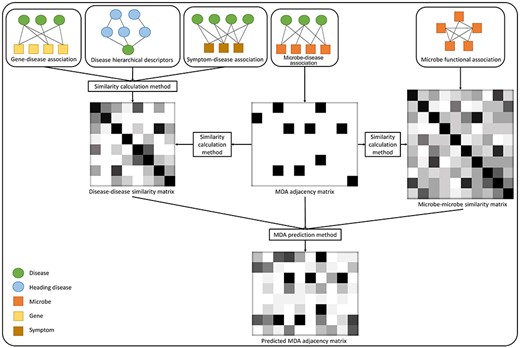
A typical working pattern of computational MDA prediction methods.
Materials
Association and similarity data are usually the inputs of computational methods. There are two types of similarity data. One is computed based on original MDAs, and the other is computed based on other data. A description of all raw data used in the MDA prediction is shown in Table 2.
A description of all data used in the MDA prediction
| Data | Source | Original form | Size/coverage in HMDAD | Similarity process |
|---|---|---|---|---|
| MDA data [29] | HMDAD contains evidence of the perturbation of microorganism populations associated with diseases from PubMed (http://www.cuilab.cn/hmdad). | 483 deregulatory (increase/decrease) evidence of 292 microbes associated with 39 diseases from published studies | 292 microbes mainly at genus and species level, 39 diseases | They are simplified as 450 de-duplicated known MDAs and then used for GIP kernel, Cosine and Spearman correlation similarity calculation. |
| Gene-based disease data [34, 35] | DisGeNET contains GDAs from UNIPROT, CGI, ClinGen, Genomics England, CTD (human subset), PsyGeNET, Orphanet and those obtained by text mining MEDLINE abstracts (https://www.disgenet.org). | 628 685 GDAs, covering GDA scores, diseases specificity index for genes, PMID evidence and so on, between 17 549 genes and 24 166 diseases | 37 mapped diseases, 1850 genes, 2715 GDAs | Neighbor-based similarity method uses GDA scores to calculate supplementary similarities among a subset of diseases. |
| Symptom-based disease data [37, 38] | HSDN integrates large-scale medical bibliographic records of disease–symptom relationships from PubMed (https://www.nature.com/articles/ncomms5212). | Counts and TF-IDF weighted values of co-occurrence between 322 symptoms and 4442 diseases, including 147 978 connections | 22 mapped diseases, 269 symptoms, 1858 symptom-disease connections | TF-IDFs of co-occurrence between one disease and all symptoms serve to calculate the symptom-based disease similarity. |
| Semantics-based disease data [41, 42] | The National Library of Medicine contains MeSH trees to define diseases hierarchically (https://meshb.nlm.nih.gov/search). | Systematically organized disease categories represented by hierarchy trees | 33 mapped diseases | Two disease trees consisting of hierarchical descriptors are used for calculating their DAG-based semantic similarity. |
| Protein family-based microbe data [44] | STRING database contains protein–protein interactions and protein-related knowledge from many data sources (https://string-db.org). | 11 362 951 Species-COG mappings and gene neighbor scores between 62 816 502 pairs of COGs | 1391 mapped microbes at species level, gene neighbor scores of 932 370 pairs of COGs | The gene neighbor score defines whether an edge between two COGs exists or not. The ratio of edges across two microorganisms to those within either measures their microbe functional similarity and is averaged for genus level. |
| Data | Source | Original form | Size/coverage in HMDAD | Similarity process |
|---|---|---|---|---|
| MDA data [29] | HMDAD contains evidence of the perturbation of microorganism populations associated with diseases from PubMed (http://www.cuilab.cn/hmdad). | 483 deregulatory (increase/decrease) evidence of 292 microbes associated with 39 diseases from published studies | 292 microbes mainly at genus and species level, 39 diseases | They are simplified as 450 de-duplicated known MDAs and then used for GIP kernel, Cosine and Spearman correlation similarity calculation. |
| Gene-based disease data [34, 35] | DisGeNET contains GDAs from UNIPROT, CGI, ClinGen, Genomics England, CTD (human subset), PsyGeNET, Orphanet and those obtained by text mining MEDLINE abstracts (https://www.disgenet.org). | 628 685 GDAs, covering GDA scores, diseases specificity index for genes, PMID evidence and so on, between 17 549 genes and 24 166 diseases | 37 mapped diseases, 1850 genes, 2715 GDAs | Neighbor-based similarity method uses GDA scores to calculate supplementary similarities among a subset of diseases. |
| Symptom-based disease data [37, 38] | HSDN integrates large-scale medical bibliographic records of disease–symptom relationships from PubMed (https://www.nature.com/articles/ncomms5212). | Counts and TF-IDF weighted values of co-occurrence between 322 symptoms and 4442 diseases, including 147 978 connections | 22 mapped diseases, 269 symptoms, 1858 symptom-disease connections | TF-IDFs of co-occurrence between one disease and all symptoms serve to calculate the symptom-based disease similarity. |
| Semantics-based disease data [41, 42] | The National Library of Medicine contains MeSH trees to define diseases hierarchically (https://meshb.nlm.nih.gov/search). | Systematically organized disease categories represented by hierarchy trees | 33 mapped diseases | Two disease trees consisting of hierarchical descriptors are used for calculating their DAG-based semantic similarity. |
| Protein family-based microbe data [44] | STRING database contains protein–protein interactions and protein-related knowledge from many data sources (https://string-db.org). | 11 362 951 Species-COG mappings and gene neighbor scores between 62 816 502 pairs of COGs | 1391 mapped microbes at species level, gene neighbor scores of 932 370 pairs of COGs | The gene neighbor score defines whether an edge between two COGs exists or not. The ratio of edges across two microorganisms to those within either measures their microbe functional similarity and is averaged for genus level. |
A description of all data used in the MDA prediction
| Data | Source | Original form | Size/coverage in HMDAD | Similarity process |
|---|---|---|---|---|
| MDA data [29] | HMDAD contains evidence of the perturbation of microorganism populations associated with diseases from PubMed (http://www.cuilab.cn/hmdad). | 483 deregulatory (increase/decrease) evidence of 292 microbes associated with 39 diseases from published studies | 292 microbes mainly at genus and species level, 39 diseases | They are simplified as 450 de-duplicated known MDAs and then used for GIP kernel, Cosine and Spearman correlation similarity calculation. |
| Gene-based disease data [34, 35] | DisGeNET contains GDAs from UNIPROT, CGI, ClinGen, Genomics England, CTD (human subset), PsyGeNET, Orphanet and those obtained by text mining MEDLINE abstracts (https://www.disgenet.org). | 628 685 GDAs, covering GDA scores, diseases specificity index for genes, PMID evidence and so on, between 17 549 genes and 24 166 diseases | 37 mapped diseases, 1850 genes, 2715 GDAs | Neighbor-based similarity method uses GDA scores to calculate supplementary similarities among a subset of diseases. |
| Symptom-based disease data [37, 38] | HSDN integrates large-scale medical bibliographic records of disease–symptom relationships from PubMed (https://www.nature.com/articles/ncomms5212). | Counts and TF-IDF weighted values of co-occurrence between 322 symptoms and 4442 diseases, including 147 978 connections | 22 mapped diseases, 269 symptoms, 1858 symptom-disease connections | TF-IDFs of co-occurrence between one disease and all symptoms serve to calculate the symptom-based disease similarity. |
| Semantics-based disease data [41, 42] | The National Library of Medicine contains MeSH trees to define diseases hierarchically (https://meshb.nlm.nih.gov/search). | Systematically organized disease categories represented by hierarchy trees | 33 mapped diseases | Two disease trees consisting of hierarchical descriptors are used for calculating their DAG-based semantic similarity. |
| Protein family-based microbe data [44] | STRING database contains protein–protein interactions and protein-related knowledge from many data sources (https://string-db.org). | 11 362 951 Species-COG mappings and gene neighbor scores between 62 816 502 pairs of COGs | 1391 mapped microbes at species level, gene neighbor scores of 932 370 pairs of COGs | The gene neighbor score defines whether an edge between two COGs exists or not. The ratio of edges across two microorganisms to those within either measures their microbe functional similarity and is averaged for genus level. |
| Data | Source | Original form | Size/coverage in HMDAD | Similarity process |
|---|---|---|---|---|
| MDA data [29] | HMDAD contains evidence of the perturbation of microorganism populations associated with diseases from PubMed (http://www.cuilab.cn/hmdad). | 483 deregulatory (increase/decrease) evidence of 292 microbes associated with 39 diseases from published studies | 292 microbes mainly at genus and species level, 39 diseases | They are simplified as 450 de-duplicated known MDAs and then used for GIP kernel, Cosine and Spearman correlation similarity calculation. |
| Gene-based disease data [34, 35] | DisGeNET contains GDAs from UNIPROT, CGI, ClinGen, Genomics England, CTD (human subset), PsyGeNET, Orphanet and those obtained by text mining MEDLINE abstracts (https://www.disgenet.org). | 628 685 GDAs, covering GDA scores, diseases specificity index for genes, PMID evidence and so on, between 17 549 genes and 24 166 diseases | 37 mapped diseases, 1850 genes, 2715 GDAs | Neighbor-based similarity method uses GDA scores to calculate supplementary similarities among a subset of diseases. |
| Symptom-based disease data [37, 38] | HSDN integrates large-scale medical bibliographic records of disease–symptom relationships from PubMed (https://www.nature.com/articles/ncomms5212). | Counts and TF-IDF weighted values of co-occurrence between 322 symptoms and 4442 diseases, including 147 978 connections | 22 mapped diseases, 269 symptoms, 1858 symptom-disease connections | TF-IDFs of co-occurrence between one disease and all symptoms serve to calculate the symptom-based disease similarity. |
| Semantics-based disease data [41, 42] | The National Library of Medicine contains MeSH trees to define diseases hierarchically (https://meshb.nlm.nih.gov/search). | Systematically organized disease categories represented by hierarchy trees | 33 mapped diseases | Two disease trees consisting of hierarchical descriptors are used for calculating their DAG-based semantic similarity. |
| Protein family-based microbe data [44] | STRING database contains protein–protein interactions and protein-related knowledge from many data sources (https://string-db.org). | 11 362 951 Species-COG mappings and gene neighbor scores between 62 816 502 pairs of COGs | 1391 mapped microbes at species level, gene neighbor scores of 932 370 pairs of COGs | The gene neighbor score defines whether an edge between two COGs exists or not. The ratio of edges across two microorganisms to those within either measures their microbe functional similarity and is averaged for genus level. |
MDA data
A publicly accessible database, Human Microbe-disease Association Database (HMDAD), provides major data for prediction methods [29]. There are currently hundreds of microorganisms and dozens of diseases sorted out from scraps of published studies in HMDAD by Ma et al. [29]. If there exists a known association between a microbe-disease pair, the corresponding entity in the association matrix is equal to 1, otherwise 0. Furthermore, known associations can be formatted into a bipartite graph that is composed of microbe nodes, disease nodes and edges (associations) connecting them.
Supplementary data for similarity calculation
For the disease similarity:
Gene-based disease data: DisGeNET integrates the massive human gene-disease association (GDA) information from expert-curated repositories [34]. MEDLINE (i.e. an international comprehensive bibliographic database of integrated biomedical information) stores quite a few GDAs [35]. Diseases documented in both HMDAD and human GDA databases were selected and used for the similarity calculation methods [36].
Symptom-based disease data: Human symptom-disease associations have been collected to construct the human symptoms-disease network (HSDN) by Zhou et al. from PubMed [37, 38]. Meanwhile, they used term frequency-inverse document frequency (TF-IDF) [39] to measure the symptom-based disease similarity based on the co-occurrence frequency between a disease and a symptom. Based on these data, Chen et al. extracted those symptom-based similarities of common diseases from HMDAD [40].
Semantics-based disease data: The National Library of Medicine records Medical Subject Headings (MeSHs) describe a given disease in hierarchical descriptor [41], and thus the overlap among parental descriptors for a pair of diseases could be used to measure how semantically similar the pair is. In addition, Disease Ontology is an intuitive scheme to encapsulate the structure of disease and disease-related concepts using terminologies with standards of MeSH, ICD and so on [42]. These efforts enable a sequence of hierarchical descriptors to be viewed as a directed acyclic graph (DAG) where a descriptor corresponds to a node as in Figure 3. The disease similarity can be measured based on two DAGs [43].

Two types of semantic DAG of liver cirrhosis.
For the microbe similarity:
Protein family-based microbe data: The STRING database consists of a considerable variety of protein–protein interactions, the presence/absence of clusters of orthologous groups (COGs) in species, relationships among COGs (e.g. neighborhood, fusion, co-occurrence and co-expression) and so on, currently involving more than 5000 organisms [44]. Each COG holds a group of proteins (i.e. a protein family) having common ancestry and related function (be orthologous across at least three lineages), which is useful for studying evolutionarily interspecific relationships. Based on the STRING database, Kamneva defined the microbe-microbe functional association index [45]. It is measured by the scale of edges (defined based on the neighborhood score between two COGs) which are distributed in the transboundary network of functionally linked protein families of a microbe-microbe pair. Moreover, although microbes used for the quantification of functional association index are at the species level, Fan et al. picked up those species-level microorganisms affiliated to the genus-level microorganisms in HMDAD [46]. Then, they averaged those functional association indexes as the microbe functional similarity at the genus level. Long et al. additionally provided a simple example to show how microbe functional similarity is calculated [47].
Similarity calculation methods
Based on the microbe and disease data mentioned in Section ‘Materials’, many similarity calculation methods have been designed and adopted for MDA prediction. We summarize these similarity calculation methods that are tailored to the MDA prediction with the mechanistic explanations. We then divide them into two categories: one is aiming at calculating both microbe and disease similarity based on MDAs and the other is based on the supplementary data.
Calculating similarity based on MDAs
These methods take the MDA matrix A as the input, inter-microbe matrix Sm and inter-disease similarity matrix Sd as output. The processes of deriving the two similarity matrices are similar. Therefore, we only present disease similarity here as an example.
The result is then projected into |$[0,1]$| by the min–max normalization.
Calculating similarity based on supplementary data
There are two similarity calculation methods publicly reported in the MDA studies based on other data, which are gene-based disease data (GDAs) and semantics-based disease data (the disease hierarchical descriptors represented by a DAG).
Similarity adjustment
Based on the above similarity calculation methods, several strategies have been proposed to improve their results. According to the analysis of [54], the similarity value of a pair of diseases below a threshold tends to reflect a weak relationship, while the similarity value greater than a threshold indicates a strong relationship. The logistic function, which serves as an activation function to address this issue, has firstly been applied to GDAs [55] and then introduced to MDAs [40, 56–58].
Another approach to adjust the similarity distribution uses the topological structure of similarity networks. There is a decay multiplier imposed on the value of similarity between a pair of diseases or microbes when they do not mutually belong to the k-nearest neighbors of each other [56, 59, 60].
Methods
In this section, we give detailed description of some state-of-the-art prediction methods in Table 1. All methods in the following subsections take three types of data as input including an association matrix |$A\in{\mathrm{\mathbb{R}}}^{n_d\times{n}_m}$|, a microbe similarity matrix |${S}_m\in{\mathrm{\mathbb{R}}}^{n_m\times{n}_m}$| and a disease similarity matrix |${S}_d\in{\mathrm{\mathbb{R}}}^{n_d\times{n}_d}$|.
Path-based methods
Path-based methods make use of the path information among three kinds of networks. These methods generally measure the weight of a potential path as the score of an unknown association by considering indirect paths across networks.
KATZ measure
Weighted Path
HeteSim measure
The relationships of a microbe-disease path were simplified in MDPH_HMDA based on path-based HeteSim scores by Fan et al. [46]. They chose two types of paths with the length of 3: microbe-disease-disease paths and microbe-microbe-disease paths. Microbe similarities could be served as microbe–microbe relationships, while disease similarities could be served as disease–disease relationships. Moreover, six types of customized paths called meta-graphs were defined in WMGHMDA [47]. These meta-graphs are composed of different combinations of adjacency matrices. Both MDPH_HMDA and WMGHMDA take the GIP kernel similarity and integrate them with the symptom-based disease similarity and the microbe functional similarity, respectively, to ensure more informative homogeneous paths.
Random walk methods
The random walk methods predict an unknown association by measuring the probability that the walker arrives the final node (one end of the association) from the seed node (the other end of the association). There are several subcategories based on the random walk, such as the random walk on a heterogeneous network, the bi-random walk and the graph inference.
Random walk on a heterogeneous network
Bi-random walk
Similarly, probability matrix Pm that walks on the microbe similarity network will finally converge in this way. In NBLPIHMDA [67], the iterative formula to calculate Pt + 1 is rewritten so that restarting the initial state, P0, is replaced by restarting the previous state, Pt. When Pt + 1 converges, P0, P1 … Pt + 1 average the final probabilities rather than Pt + 1. Yan et al. made an improvement by proposing BRWMDA that combines the similarity network fusion (SNF) process with the bi-random walk [68]. The SNF method performs an effective integration of the GIP kernel similarity and the symptom-based disease similarity by using the k-nearest neighbor and the iterative fusion operation.
Graph inference
Note that both Sd and Sm need to be normalized.
Bipartite local models
BLMs work independently on the basis of both sides of a microbe-disease pair and can be combined to yield a definitive prediction result.
Collaborative filtering
Network consistency projection
Laplacian regularized least squares
Inference on bipartite networks
Wang et al. proposed a novel microbe-disease prediction approach regarding bipartite networks where the information on GDAs was firstly introduced for the neighbor-based similarity calculation [36]. To build inference from a bipartite network, several kinds of kernel matrices are computed with the microbe and disease similarity based on multi-source association data. As for diseases, the potential prediction associations scored by the product of a low-dimensional projection matrix and kernel matrices are transformed from the multi-source disease similarity. An analogous process is implemented with regard to microbes. Finally, the inference on the bipartite network is gathered as the predicted outcome.
Matrix factorization methods
Matrix factorization methods are based on the idea that the input matrix decomposes into two low-dimensional matrices, and the product of the two low-dimensional matrices is approximately equal to the input matrix [75, 76]. Two low-dimensional matrices |$W\in{\mathrm{\mathbb{R}}}^{n_d\times k}$| and |$H\in{\mathrm{\mathbb{R}}}^{n_m\times k}$| are trained to meet that |${WH}^T\approx A$|. Referring to [77], columns of W and H contain feature information of diseases and microbes, respectively, and the unknown associations in matrix A could be completed by the multiplication of two feature vectors.
Graph regularized non-negative matrix factorization
In addition, He et al. presented an improved method, namely GRNMFHMDA [59], that incorporates Weighted K Nearest Known Neighbors (WKNKN) [79]. By WKNKN, a binary association matrix turns into a non-zero matrix that takes the pre-estimation of potential associations.
Collaborative matrix factorization
Updating rules of W and H could be obtained by taking partial derivatives of the objective function with respect to W and H. Moreover, it is common that W and H are initialized randomly, but the reasonable initialization can accelerate convergence. For example, the singular value decomposition of A could be used in initializing W and H [80].
Sparse learning method
Kernelized Bayesian matrix factorization
The kernelized Bayesian matrix factorization (KBMF) [84] has been used in previous studies [85]. To solve the complex computation of posterior distribution, the variational approximation is applied to infer the distribution of the low-dimensional subspace for approximating the complicated distribution. Especially, low-dimensional projection matrices Pm and Pd, where the projection parameters correspond to the priors λm and λd, respectively, are constructed. Then, kernel matrices Sm and Sd are projected into a uniform low-dimensional space by projection matrices, and potential associations could be searched among the low-dimensional space containing informative representations of microbes and diseases. Additionally, Wu et al. modified Tikhonov regularization terms in the NMF optimization function [86].
Other methods
Besides methods mentioned above, there are still some methods that do not belong to any of the categories. Hence, we discuss these methods and put them in this subsection.
Ensemble learning with adaptive boosting
The ensemble learning with adaptive boosting was adopted from the prediction method ABHMDA [87]. The decision trees are chosen as weak learners. Because of lacking other types of concrete feature information and property labels, the combination of the GIP kernel microbe similarity and the symptom-based disease similarity is served as the feature vector of a training sample. Due to the lack of known associations regarded as positive samples, unknown associations regarded as negative samples are randomly divided into different parts. The same number of negative samples as the number of positive samples is drawn from each part in order to keep the balance between positive and negative samples during the training of a decision tree. According to the adaptive boosting, the misclassified samples are more critical to inform the subsequent training of weak classifiers, and a weak classifier with less error yields a higher proportion of the final combined output of all weak classifiers.
Matrix completion
Shi et al. proposed a novel method BMCMDA [88] based on the binary matrix completion which assumes that an unobserved microbe-disease pair (i.e. an unknown microbe-disease pair) is likely to be associated at the probability |$f({x}_{i,j})$| or unassociated at the probability |$1-f({x}_{i,j})$|. |$X=\{{x}_{i,j}\}$| is the probability parameter matrix corresponding to association matrix A, and |$f(\cdot )$| denotes the cumulative distribution function. The matrix X is optimized by maximizing the log-likelihood function of the present observation. After that, incomplete matrix A could be recovered with the optimal probability parameters.
In addition, another method, MCHMDA [89], uses the singular value thresholding algorithm [90] to carry out the matrix completion. Specifically, a low-rank heterogeneous matrix, whose elements belong to the set of known associations remained unchanged, is completed via two iteration steps based on the Uzawa algorithm [91] and the linearized Bregman iteration [92]. It also expands similarity calculation methods by considering the microorganism-inhabited organs and measuring gene-based disease similarity differently.
Experiment and comparison
In this section, we selected one or two prediction methods from each category to conduct comparative experiments. Candidates were chosen after comprehensive consideration including the aspects of similarity, the characteristics of their algorithms, code availability and reproducibility. In total, five typical methods including KATZHMDA, BRWMDA, NGRHMDA, LRLSHMDA and NMFMDA were experimentally compared. These methods adopt a common similarity calculation method, the GIP kernel similarity (some of them integrated with symptom-based disease similarity). Their parameters were set to default values according to the primary literature for optimal prediction performance.
Assessment methods
Owing to the single data source (i.e. HMDAD [29]), we selected two of the widely used evaluation methods, the global leave-one-out cross validation (LOOCV) and 5-fold cross validation (CV) [93], to obtain a performance comparison. In the meantime, the local LOOCV was also selected to assess the prediction performance for reference. We took turns leaving out one known MDA (i.e. set value of the corresponding entity in the association matrix to be 0) as the test sample in LOOCV. Conducting LOOCV on a small dataset is not time-consuming, and the result from LOOCV is stable and non-random. The global LOOCV differs from the local LOOCV because they have different scales of candidate samples that determine the ranking range of a predicted score. The global LOOCV indicates that the predicted score of a test sample ranks among all candidate samples that are not yet verified, whereas the score is compared with those of candidate samples connecting the tested disease in the local LOOCV.
By taking 5-fold CV for 100 times, we also obtained a stable result. The known MDAs are randomly divided into 5 folds, and each fold is drawn out in turn as the test sample set while the remaining folds are considered as the training sample set.
The receiver operating characteristic (ROC) curve reflects that the relationship between sensitivity and 1-specificity changes along with the varying cut-off threshold. The area under curve (AUC) of the ROC has been widely used as an evaluation metric to measure the performances of MDA prediction methods at this stage [94, 95]. We primarily compared prediction methods by their AUCs of the global and local LOOCV. As for 100 times of 5-fold CVs, AUCs were averaged as the final metric.
We additionally assessed the influences of using different similarity data on prediction performances. Six combinations of processed similarity data were tried with selected methods and judged by AUCs of the global and local LOOCV.
The AUCs and ROC curves of three types of CVs are drawn in Figure 4. Figure 5 shows the numbers of predicted MDAs ranking in different top portions with global LOOCV. Based on this, we visualize the overlaps of MDAs predicted by each method in the form of Venn diagrams which are placed in Figure 6. Table 3 shows the results of a predictive ability comparison via using different similarity data. Then, we discuss the prediction performances of different methods and give brief comments.

Three types of ROC curves of five representative methods.
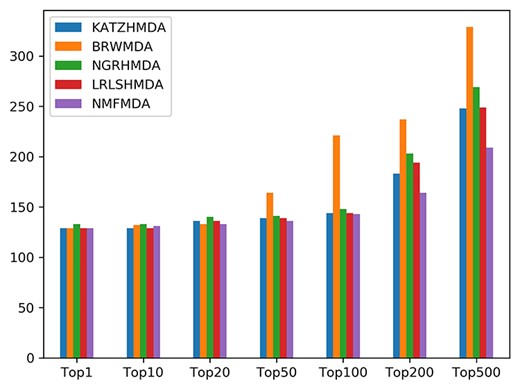
The numbers of correctly predicted MDAs from five representative methods with the global LOOCV.
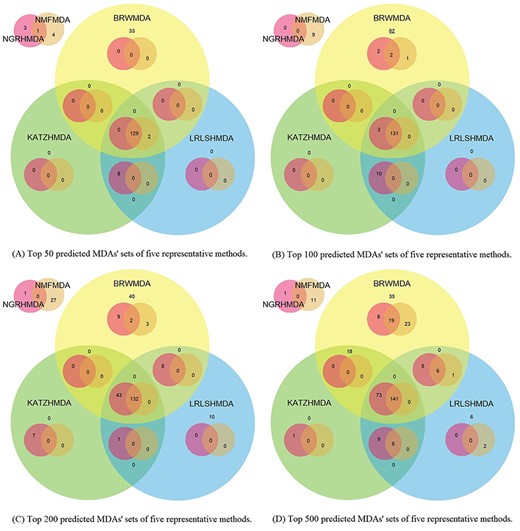
Four overlapping relationships among top 50 (a), top 100 (b), top 200 (c) and top 500 (d) ranked MDAs of five representative methods. The top left Venn diagram of each sub-figure shows the overlapping relationships of high-score MDAs which are just predicted by NGRHMDA and NMFMDA but not by the other three methods. The main Venn diagram of each sub-figure shows the overlapping relationships of high-score MDAs predicted by five methods together.
Experimental analysis
From the results of the prediction performances, the random walk methods represented by BRWMDA achieved the best performance among the selected methods in terms of all three validations. The good results are caused by the logistic function and the SNF method, which bring reliable transition matrices for bi-walk [68]. The performances of random walk methods highly depend on the property of the walking network. A well-integrated and informative network leads the walker to a proper destination. The SNF processing captures the vital information and integrates the complementary information from diverse networks by an iterative update based on message-passing [96]. According to Table 3, fusing the Spearman correlation coefficient similarity and the symptom-based disease similarity by the SNF method outperformed all other combinations (AUC = 0.9527), even is better than the original results. It indicates that both the SNF method and the Spearman correlation coefficient similarity generally fit this method, since the iterative fusion approach and the monotonic relationship assessment of two profiles may play an important role in constructing a more reasonable similarity network for walkers [97].
According to the ROC curves in Figure 4, NMFMDA failed to achieve a superior result. However, the matrix factorization methods are common alternatives in other field such as drug–target interaction prediction [32]. Although the MDA matrix factorization is less intuitive as the image decomposition where NMF learns the combination of components, it indeed captures latent properties from both sides. The dissatisfactory prediction result could be partly explained by the unbalanced distribution of associations. A significant fraction of associations are clustered with several common diseases. Hoyer et al. who improve the NMF algorithm by incorporating sparseness constraints provide us with a solution [98]. In our case, the unbalanced distribution (disease-specific centralized distribution of MDAs) simulates a ‘rare’ phenomenon [98]. To overcome this defect, the basis vectors W containing latent properties of diseases should be sparse [98]. Due to this phenomenon, NMFMDA performed better under 5-fold CV when the centralized distribution was alleviated in contrast to LOOCV. Table 3 illustrates that the effect of the GIP kernel similarity far exceeded other similarities. By observing different similarity distributions, the values computed by the GIP kernel similarity were found densely located in the neighborhoods of 0 and 1, having a higher distinguishability, while those computed by the Spearman correlation coefficient similarity mainly ranged from 0 to 0.5. This binary centralized distribution resulting from the GIP kernel similarity may generate sparser basis vectors W in terms of the deduction of the graph regularization term [56, 74, 98]. As a result, matrix factorization methods are not the optimal approach.
Among path-based methods, the representative method, KATZHMDA, did not perform as well as others. It could be explained that the sparse data cause rare paths linking with isolated nodes and therefore these nodes are unable to be identified precisely. It also indicates that path-based methods are more intuitive methods based on the association network. While random walk methods focus on the possible locations that a walker may arrive at, path-based methods pay more attention to the details of all paths. Hence, a convincing roadmap deserves to be assured. However, path across homogeneous networks measured with computational association-based similarities are still not reliable enough due to the computational dimension-reduction deviation during the similarity calculation. The results derived from a series of experiments on different similarity data in Table 3 are supportive evidence for our arguments. When adopting path-based methods, we should concern about new similarity sources and measurements to construct more efficient homogeneous paths like what we tested on extra similarity data.
Relatively, BLMs cover an extensive range of methods based on diverse ideas. From the prediction results of the two selected BLMs, they presented a satisfactory solution to the MDA prediction by taking advantage of disease and microbe similarity information separately. In addition, the further two-step diffusion in NGRHMDA and the modified graph Laplacian regularization term in LRLSHMDA also contribute to the reliable prediction performance. Notably, BLMs are not entirely independent for their processes share mutual associations, but neither of the models participates in the operation of each other until the mergence. We recommend that this approach could be considered as the prime candidate owes to its fast speed and weak dependence on conditional hyperparameters and similarity data as shown in Table 3.
Results of different similarity data applied to typical MDA prediction methods. M: microbe; D: disease; GIP kernel: the GIP kernel similarity; Spearman: the Spearman correlation coefficient similarity; Ave(X + sym): the average of the X similarity and the symptom-based disease similarity and SNF(X + sym): the SNF fusion of the X similarity and the symptom-based disease similarity. We underline the original results and highlight the best result of each method in bold and the global best result in italics
| AUC of global/local LOOCV | M: GIP kernel D: GIP kernel | M: Spearman D: Spearman | M: GIP kernel D: Ave (GIP kernel + sym) | M: GIP kernel D: Ave (Spearman + sym) | M: GIP kernel D: SNF (GIP kernel + sym) | M: GIP kernel D: SNF (Spearman + sym) | Average AUC |
|---|---|---|---|---|---|---|---|
| KATZHMDA | 0.8382/0.6806 | 0.8769/0.5632 | 0.8644/0.7 | 0.876/0.5576 | 0.8786/0.7688 | 0.8632 /0.5262 | 0.8662/0.6327 |
| BRWMDA | 0.9293/0.8595 | 0.9406/0.8895 | 0.922/0.8552 | 0.9431/0.8861 | 0.9397/0.8614 | 0.9527/0.8894 | 0.9379/0.8735 |
| NGRHMDA | 0.9111/0.7951 | 0.8930/0.5967 | 0.9117/0.7964 | 0.8943/0.5917 | 0.9025/0.7889 | 0.8931/0.6020 | 0.901/0.6951 |
| LRLSHMDA | 0.8909/0.7657 | 0.8610/0.6429 | 0.8930/0.7612 | 0.8429/0.5244 | 0.8854/0.7367 | 0.832/0.5222 | 0.8675/0.6589 |
| NMFMDA | 0.8470/0.6973 | 0.6167/0.4817 | 0.8568/0.6949 | 0.6433/ 0.4766 | 0.8510/0.7012 | 0.6963/0.4836 | 0.7519/0.5892 |
| Average AUC | 0.8833/0.7596 | 0.8376/0.6348 | 0.8896/ 0.7615 | 0.8399/0.6073 | 0.8914/0.7714 | 0.8475/0.6047 | 0.8649/0.6899 |
| AUC of global/local LOOCV | M: GIP kernel D: GIP kernel | M: Spearman D: Spearman | M: GIP kernel D: Ave (GIP kernel + sym) | M: GIP kernel D: Ave (Spearman + sym) | M: GIP kernel D: SNF (GIP kernel + sym) | M: GIP kernel D: SNF (Spearman + sym) | Average AUC |
|---|---|---|---|---|---|---|---|
| KATZHMDA | 0.8382/0.6806 | 0.8769/0.5632 | 0.8644/0.7 | 0.876/0.5576 | 0.8786/0.7688 | 0.8632 /0.5262 | 0.8662/0.6327 |
| BRWMDA | 0.9293/0.8595 | 0.9406/0.8895 | 0.922/0.8552 | 0.9431/0.8861 | 0.9397/0.8614 | 0.9527/0.8894 | 0.9379/0.8735 |
| NGRHMDA | 0.9111/0.7951 | 0.8930/0.5967 | 0.9117/0.7964 | 0.8943/0.5917 | 0.9025/0.7889 | 0.8931/0.6020 | 0.901/0.6951 |
| LRLSHMDA | 0.8909/0.7657 | 0.8610/0.6429 | 0.8930/0.7612 | 0.8429/0.5244 | 0.8854/0.7367 | 0.832/0.5222 | 0.8675/0.6589 |
| NMFMDA | 0.8470/0.6973 | 0.6167/0.4817 | 0.8568/0.6949 | 0.6433/ 0.4766 | 0.8510/0.7012 | 0.6963/0.4836 | 0.7519/0.5892 |
| Average AUC | 0.8833/0.7596 | 0.8376/0.6348 | 0.8896/ 0.7615 | 0.8399/0.6073 | 0.8914/0.7714 | 0.8475/0.6047 | 0.8649/0.6899 |
Results of different similarity data applied to typical MDA prediction methods. M: microbe; D: disease; GIP kernel: the GIP kernel similarity; Spearman: the Spearman correlation coefficient similarity; Ave(X + sym): the average of the X similarity and the symptom-based disease similarity and SNF(X + sym): the SNF fusion of the X similarity and the symptom-based disease similarity. We underline the original results and highlight the best result of each method in bold and the global best result in italics
| AUC of global/local LOOCV | M: GIP kernel D: GIP kernel | M: Spearman D: Spearman | M: GIP kernel D: Ave (GIP kernel + sym) | M: GIP kernel D: Ave (Spearman + sym) | M: GIP kernel D: SNF (GIP kernel + sym) | M: GIP kernel D: SNF (Spearman + sym) | Average AUC |
|---|---|---|---|---|---|---|---|
| KATZHMDA | 0.8382/0.6806 | 0.8769/0.5632 | 0.8644/0.7 | 0.876/0.5576 | 0.8786/0.7688 | 0.8632 /0.5262 | 0.8662/0.6327 |
| BRWMDA | 0.9293/0.8595 | 0.9406/0.8895 | 0.922/0.8552 | 0.9431/0.8861 | 0.9397/0.8614 | 0.9527/0.8894 | 0.9379/0.8735 |
| NGRHMDA | 0.9111/0.7951 | 0.8930/0.5967 | 0.9117/0.7964 | 0.8943/0.5917 | 0.9025/0.7889 | 0.8931/0.6020 | 0.901/0.6951 |
| LRLSHMDA | 0.8909/0.7657 | 0.8610/0.6429 | 0.8930/0.7612 | 0.8429/0.5244 | 0.8854/0.7367 | 0.832/0.5222 | 0.8675/0.6589 |
| NMFMDA | 0.8470/0.6973 | 0.6167/0.4817 | 0.8568/0.6949 | 0.6433/ 0.4766 | 0.8510/0.7012 | 0.6963/0.4836 | 0.7519/0.5892 |
| Average AUC | 0.8833/0.7596 | 0.8376/0.6348 | 0.8896/ 0.7615 | 0.8399/0.6073 | 0.8914/0.7714 | 0.8475/0.6047 | 0.8649/0.6899 |
| AUC of global/local LOOCV | M: GIP kernel D: GIP kernel | M: Spearman D: Spearman | M: GIP kernel D: Ave (GIP kernel + sym) | M: GIP kernel D: Ave (Spearman + sym) | M: GIP kernel D: SNF (GIP kernel + sym) | M: GIP kernel D: SNF (Spearman + sym) | Average AUC |
|---|---|---|---|---|---|---|---|
| KATZHMDA | 0.8382/0.6806 | 0.8769/0.5632 | 0.8644/0.7 | 0.876/0.5576 | 0.8786/0.7688 | 0.8632 /0.5262 | 0.8662/0.6327 |
| BRWMDA | 0.9293/0.8595 | 0.9406/0.8895 | 0.922/0.8552 | 0.9431/0.8861 | 0.9397/0.8614 | 0.9527/0.8894 | 0.9379/0.8735 |
| NGRHMDA | 0.9111/0.7951 | 0.8930/0.5967 | 0.9117/0.7964 | 0.8943/0.5917 | 0.9025/0.7889 | 0.8931/0.6020 | 0.901/0.6951 |
| LRLSHMDA | 0.8909/0.7657 | 0.8610/0.6429 | 0.8930/0.7612 | 0.8429/0.5244 | 0.8854/0.7367 | 0.832/0.5222 | 0.8675/0.6589 |
| NMFMDA | 0.8470/0.6973 | 0.6167/0.4817 | 0.8568/0.6949 | 0.6433/ 0.4766 | 0.8510/0.7012 | 0.6963/0.4836 | 0.7519/0.5892 |
| Average AUC | 0.8833/0.7596 | 0.8376/0.6348 | 0.8896/ 0.7615 | 0.8399/0.6073 | 0.8914/0.7714 | 0.8475/0.6047 | 0.8649/0.6899 |
In conclusion, each computational method has its own advantages, applicability and disadvantages. An informative similarity network integrated effectively with prior knowledge could generally improve the prediction results for most methods. Compared with the GIP kernel similarity, the Spearman correlation coefficient similarity fits the methods better based on topological networks (the random walk and path-based methods). Considering the specific predictive capability and the characteristic of each method, it is possible to conduct a combination prediction on the basis of results as shown in Figure 6. Although we analyzed the experimental results by category, we would like to give supplementary explanations on prior experiments: (i) hindered by the unbalanced dataset, we were more focused on comparing computational methods mathematically and horizontally and ignored their biomedical authenticity judged by the empirical validation. This will be discussed in Subsection ‘Biomedical interpretation for computational discovery’. (ii) The prediction accuracy of top-ranked associations is a valuable metric to evaluate the applicability and practical significance of a method. (iii) As shown in Figure 6, the overlapping top-ranked associations predicted by several methods are the highest confidence alternatives for the biomedical validation.
Future work
According to previous studies and our observations, we make some suggestions in perspectives of the data, methods and formulations in order to achieve higher precision, better generalization ability and wider applicability of the MDA prediction in the future.
Enriching data sources
Collecting data from different sources is the top priority which could be divided into two parts. On the one hand, original association data are still sparse and unevenly distributed. So firstly, rigorously verified prediction results could be added to the original association data. A webtool, EviMass, has been developed and conduced to solve this issue [99]. Reported literature will return when users query with an MDA or even association networks. It provides a channel for biologists to test their hypotheses by mining biomedical evidence. Secondly, searching recent related literature for a novel association data is necessary. For example, Yan et al. extended the MDA dataset as HMDAD-SUP [89]. Furthermore, the MDA prediction prefers more independent datasets for training and testing models. For example, Janssens et al. constructed the Disbiome database which continuously organizes experimental cases linking the microorganism to disease from PubMed publications [100]. Recently, an insightful review meticulously outlined existing knowledge bases of human MDAs and discussed current challenges in the process of constructing them [101]. Readers can obtain relevant information of natural language processing, text mining, terminology unification and data consolidation to have an overall grasp on a new MDA-related knowledge base.
On the other hand, multi-source data need to be introduced to enhance the generalization performance and support innovative approaches. For example, other data for similarity, including GDA, symptom-based disease similarity and so on [36, 40, 46, 47, 53, 89], have been successfully applied to the MDA prediction. However, these data could also be used as microbe and disease features in feature-based learning models. Likewise, other microbe-related and disease-related information that could identify a microorganism or a disease should be considered in the MDA prediction. For example, PHI-base stores a lot of genetic information about pathogens and their various mutant phenotypes [102]. EuPathDB is a large-scale retrieval platform for eukaryotic microbes, serving to identify genes with an all-round search strategy [103]. The therapeutic drug list of a given disease can be retrieved in KEGG [104].
There is yet no certain microbe-disease non-association dataset to date. The label ‘0’ has two possible interpretations, unknown association or non-association. It affects the effectiveness of supervised learning methods. Although some technical solutions have already been proposed in ABHMDA (balanced sampling from bi-class sets) [87], BMCMDA (unobserved states with the probability of being non-associations) [88] and other studies (in silico screening) [105, 106], it is necessary to curate verified non-association entries manually.
Unifying taxonomy and terminology
Another measure for the enhancement of prediction reliability is to define the taxonomic level of each microorganism and perform the prediction at the same level [71]. Organisms could be classified and mapped with taxonomy (e.g. NCBI [107] and SILVA [21] taxonomy). Moreover, the introduction of taxonomy would facilitate a precise identification of microorganisms among microbiology databases, which aids in incorporating microbiome (e.g. microbial genome sequences [21, 22] and patient-derived microbial metagenome [22, 108], transcriptome and metabolome [24–26]) into the MDA prediction. Microbiomic data could be used to identify microorganisms and measure inter-microorganism similarities, whereas relevant work has been done with the aim at predicting virus-host associations. Ahlgren et al. measured inter-viruses dissimilarities utilizing genomic oligonucleotide frequencies and Liu et al. constructed the virus similarity network based on the prediction of virus-host associations, which provide examples of using microbiome in microbe-related association prediction [109, 110].
Additionally, it is necessary to expand the current size of diseases and requires a basic terminology dictionary to regulate disease synonyms and classifications [101]. Because of complex aliases, extended description (e.g. ‘new-onset untreated’ rheumatoid arthritis) and ambiguity between symptoms and diseases [38], hierarchical classifications, such as Medical Dictionary for Regulatory Activities [111] and MeSH [112] are required. They could contribute in organizing disease terms with a structured standard and hence allow to retrieve standardized disease terms from different disease repositories in a consistent way.
Introducing deep learning methods
Machine learning is a powerful tool, and related algorithms have been widely applied to our issue, such as least squares, matrix factorization and completion. However, feature-based machine learning algorithms are trapped in the dilemma of lacking effective features and hence received little attention. Compared with machine learning, deep learning, which is regarded as a meaningful attempt, has not yet been introduced into MDA prediction. In response to the dilemma above, deep learning-based algorithms that target a complicated topological network and capture its node embeddings have been proposed in many studies [113–118].
Inspired by representation learning such as DeepWalk [115], refining characteristics of the topological structure of the MDA network by deep learning methods is an available way to obtain distinctive representations. For example, Masashi et al. extracted topological information from the drug molecular structure by encoding all atoms and chemical bonds in a variable dimensional space. They then converted these stochastic pre-encodings to final molecular embeddings in the form of vector representations by the graph neural network [119]. In our case, a microbe or disease node can be represented as the co-contribution of its neighbors and itself in the homogeneous network through graph neural networks, and then the end-to-end model is a preferred choice for handling the subsequent prediction.
Furthermore, Zitnik et al. proposed an approach for modeling a node encoder in the heterogeneous network constituted of multigraph convolutional networks [120]. When we introduce new interactive networks into the MDA prediction, the encoder integrating multi-graph information for a node would take effect. Although topography-based deep learning methods mentioned above, especially the graph neural network, are effective tools, these types of methods are unable to predict novel associations because the encoding dictionary (or one-hot codes) is pre-fixed. Hence, it is urgent to curate valid features capable of identifying a microorganism or a disease.
Fusing multi-source similarities in an effective manner is also an essential task that we can apply deep learning methods to. For example, Zeng et al. assembled 10 types of drug-drug networks and converted them to a common feature space that generates reconstructed drug features via multi-modal deep autoencoder [121]. The step of aggregating similarity features could be addressed by multi-input concatenation or summation layer.
Evaluating diversely
Comprehensively, evaluating prediction methods is also a critical part of the MDA prediction. In this article, we mainly focus on discussing non-feature-based prediction methods. These methods require a reconstruction of the association matrix via removing label ‘1’ when testing novel associations. Therefore, the association matrix changes by different test sampling modes. In order to evaluate the prediction ability comprehensively, more test sampling modes should be designed besides CVs. For example, to evaluate the capability of predicting associations of a new disease with known microbes (or vice versa), the corresponding profile should be excluded from the association matrix and recovered by a set of predicted scores. Note that supplementary similarities (i.e. based on data mentioned in Section ‘DATA’) are still available for predicting new diseases and microbes. Besides probabilistic outcome for a pair of microbe-disease, we could further determine a threshold to estimate whether they associate by maximizing the Matthews correlation coefficient [122] theoretically, which could serve as an evaluation metric other than the rank.
Reforming predictive tasks
The MDA prediction is a typical binary classification task but able to develop a much finer-grained prediction. Predicting drug–target binding affinities, for example, is a further work based on drug–target interaction prediction [123]. HMDAD records additional entries whether the quantity of microbial population is increased or decreased in the reported cases [29]. Furthermore, the Disbiome database provides the microbial population variation between control-derived and patient-derived groups [100]. Most of population indexes are given in the absolute quantitative value responded in a given unit (the unit is dependent on the used detection method) [100]. Such quantitative relationships could be used for predicting disease-induced microbial population variations, physiological disorders in response to microbial population dynamics, inter-microbe interactions and even synergisms in combinations of human microbiota in the future.
The network analysis is a worthwhile strategy that further extends more refined prediction tasks methodologically for a better mechanistic inference. On the one hand, applying network analysis to the combined heterogeneous network is a popular trend along with the enrichment of microbe-centric and disease-centric networks. Microbe-centric networks (e.g. microbe co-occurrence [124, 125], microbe-gene [22], microbe-protein [44] and microbe-host [126]) and disease-centric networks (e.g. disease-gene [34, 127], disease-symptom [38] and disease-drug [128, 129]) could cooperate to construct a comprehensive relational database based on MDAs. This is a challenging task requiring the screening and consolidation of these massive data based on specialized knowledge. On the other hand, the network analysis has been more common in studying community-level microbiome-host relationships to explain pathogenesis [130]. For example, a literature-curated network, composed of the gut microbiome and host cells that metabolically interact annotated with small-molecule transport and macromolecule degradation events, has been constructed and serves to reveal microbial metabolism functionally for a specific disease [131]. Additionally, identifying enzyme-coding genes and their annotated enzymes from microbial metagenomics data derived from healthy individuals and diseased individuals has been used to compare topological differences based on metabolic networks [132]. These networks take enzymes as nodes, and there is an edge between two enzymes if they catalyze successive reactions. These studies indicate that networks constructed of a given population-derived microbiomic data truly help to analyze and infer disease mechanisms, which is worthy of consideration for predictive tasks ahead.
Developing related tools
There are only a few web-based platforms where researchers can perform a customized MDA prediction. For instance, MicroPattern is a web-based tool that divides microorganisms into different disease-related sets for reference and provides the function of similarity calculation [133]. Such platforms have integrated various state-of-the-art prediction methods, which are specialized for other fields (e.g. meta-PPISP [134], DINIES [135] and DIANA-microT [136]). However, we could not ignore other related works which explore microbe-disease and microbe-host relationships. For example, an open-source pipeline, MicroPro, can estimate abundance profiles of unknown microbial organisms based on unmapped reads from the metagenomic data and predict phenotypes using complete abundance profiles of the cases and the controls [137]. Furthermore, an online tool, NetCooperate, can quantify the ability of the nutritive support of a host for a parasitic or commensal organism and the complementarity of a pair of microorganisms based on their metabolic networks [138]. Such open web-based platforms and available software packages truly facilitate further studies of microorganism–human relationships from both methodological and biomedical perspectives.
Biomedical interpretation for computational discovery
A pair of high-confidence MDA could be interpreted from two biomedical perspectives as follows: (i) species-specific changes in microbial community composition are found within patients, but there is no evidence to suggest that it is a pathogen or even a diagnostic signature [139]. (ii) It is a pathogen [140]. The pathogenicity is not the intrinsic nature of microorganisms, and the host response to potential virulence factors varies [141]. More importantly, identifying the causality of microbe-outcome relationships is sometimes unrealistic [142]. Therefore, interpreting any computational discovery is a complex and tough work. When obtaining potential candidates, computational scientists usually seek for empirical literature to conduct case studies [72]. However, biologists can purposefully identify species-specific microbial biomarkers from the host response and link microorganism to disease with the novel computational discovery [143]. Based on this, biologists and bioinformaticians can work together and carry out a thorough research on the detailed mechanism, combining the subjects such as genetics, metabolism, toxicology and so on. For example, considering genetic and metabolic strategies of microbiota, biologists curated organ-specific and patient-specific microbial community-level metabolic networks and developed computational frameworks to study the biomedical interpretation of specific microbial impact on human health [131, 132]. Additionally, some biologists have modelled dynamic simulators based on immune responses and environment-driven microbial behaviors for the pathological interpretation. For example, Wendelsdorf et al. developed a gastrointestinal immune system simulator and applied it to find a treatment strategy for Brachyispira hyodysenteriae infection-induced dysentery [144]. The agent-based models have been established to study gastrointestinal cell-pathogen interaction mechanisms of Pseudomonas aeruginosa and Helicobacter pylori from the aspects of pathway regulation of immunity and virulence [145, 146]. The shift of computational discovery to biomedical discovery heavily relies on expert experiences. Therefore, computational scientists are encouraged to increase the accessibility of the MDA discovery to biomedical research community.
Conclusion
The MDA prediction is critical in revealing relationships between human diseases and microorganisms. In this study, a comprehensive overview of the MDA prediction has been given. Firstly, we introduced multi-source data applied for the MDA prediction and their purposes, respectively. Secondly, we described several similarity calculation methods that are widely used in the MDA prediction. We then classified computational prediction methods and give detailed descriptions of them. Meanwhile, we conducted a comparative assessment of similarity calculation methods and computational prediction methods and then analyzed their prediction performances. Finally, we offered a series of recommendations on enhancing the prediction performance and discussed top tasks in the future.
The development of sequencing technologies lays the basis for conducting a detection in microorganism population abundance [147]. High-throughput sequencing technologies and advanced omics technologies allow diversified means to detect the changes in patient-derived microbial composition [148]. However, these data are still deficient with the problems of information loss, sparsity, small-scale data, unbalanced distribution, lack of a unified taxonomic standard and ambiguity in disease terms at this stage. The problems of small-scale data and unregulated use of terms need the most attention. Enriching data, introducing the taxonomy and the terminology dictionary and performing fine-grained predictions are effective approaches to alleviate these problems. Each method proposed its unique strategy by adopting, integrating, improving and inventing algorithms adapted to our issue. Besides the methodology, we should also guide future efforts to a practical and broad direction like the instances that we talked over in future work. Microbe-related association prediction is a rising research field and has broad prospects of development and application because its interdisciplinary perspective relates to fields including medication [149], genome [150], pathogenesis [151] and phenotype [152]. It is to be expected that more and more advanced computational methods as well as comprehensive datasets will be developed in the future.
The microbe-disease association (MDA) prediction is an in silico pre-screening instrument for the clinical trials of pathogenic mechanisms related to microorganisms. Various raw data used for predicting MDAs and computational similarity pre-processes methods are summarized.
Computational MDA prediction methods based on diverse strategies and algorithms, to our knowledge, are classified and elaborated. A series of experiments with different combinations of prediction methods and similarity data are performed. An analysis of the results and possible improvement based on their nature are discussed.
Considering the small-scale dataset and the lack of feature data, data enrichment and regularization are the prime tasks that could be ameliorated in many ways. Deep learning technologies can help address it by learning latent topological information based on the MDA network structure, and more machine learning models are encouraged to be proposed and adopted as the data are expanded and regularized.
Concerning further work after the MDA prediction, quantitative records of the microbial population variation in experimental cases enable the models to perform fine-grained prediction tasks, and the network analysis could be applied to the inference of microbiological pathogenesis with annotated networks of biological events in the future.
Funding
NSFC-Zhejiang Joint Fund for the Integration of Industrialization and Informatization under grant No. U1909208, National Natural Science Foundation of China (grant nos. 61772552, 61962050); 111 Project (grant no. B18059); Hunan Provincial Science and Technology Program (grant no. 2018WK4001).
Conflict of Interest
None declared.
Zhongqi Wen is a graduate student in the Hunan Provincial Key Lab of Bioinformatics, School of Computer Science and Engineering at Central South University, Hunan, China. His research interests include microbe-related and drug toxicity prediction.
Cheng Yan is currently a postdoctoral in the School of Computer Science and Engineering, Central South University, Changsha, Hunan, China. His current research interests include bioinformatics and machine learning.
Guihua Duan is an Associate Professor in the School of Computer Science and Engineering, Central South University. Her research interests include bioinformatics, network security and computer network.
Suning Li is a Research Assistant in the Hunan Provincial Key Lab of Bioinformatics, School of Computer Science and Engineering, Central South University, Changsha, Hunan, China.
Fang-Xiang Wu is a Professor in the College of Engineering and the Department of Computer Sciences, University of Saskatchewan, Saskatoon, Canada. His research interests include artificial intelligence and computational biology.
Jianxin Wang is a Professor in the Hunan Provincial Key Lab of Bioinformatics, School of Computer Science and Engineering at Central South University, Hunan, China. His research interests include computational genomics and proteomics.
References
Author notes
Zhongqi Wen and Cheng Yan contributed equally to this work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}