




Abstract
The recent years have witnessed a rapid increase in the number of scientific articles in biomedical domain. These literature are mostly available and readily accessible in electronic format. The domain knowledge hidden in them is critical for biomedical research and applications, which makes biomedical literature mining (BLM) techniques highly demanding. Numerous efforts have been made on this topic from both biomedical informatics (BMI) and computer science (CS) communities. The BMI community focuses more on the concrete application problems and thus prefer more interpretable and descriptive methods, while the CS community chases more on superior performance and generalization ability, thus more sophisticated and universal models are developed. The goal of this paper is to provide a review of the recent advances in BLM from both communities and inspire new research directions.
1 Introduction
Due to the rapid development of biomedical research, there is a large number of biomedical literature available online in electronic format. For example, COVID-19 was first detected in December 2019 in Wuhan, China, and then it led to an outbreak in China in January 2020, then in March 2020 it became a global pandemic. According to LitCOVID [1], there has already been more than 1000 research articles about COVID-19 until March 2020. In reality, it is almost impossible for the readers to keep up with all the articles they are interested in. This makes automatic knowledge extraction and mining from biomedical literature highly demanding.
Biomedical literature mining (BLM) refers to the field of developing text mining and natural language processing (NLP) techniques for automatic knowledge extraction and mining from biomedical literature. Comparing with other biomedical texts such as clinical notes, biomedical literature has the following characteristics: (1) they are more easily accessible thanks to the publicly available database MEDLINE and free search engine PubMed; (2) they tend to use professional language and have diverse ways of expressing the same concept; and (3) they can be lengthy with diverse contents about the new biomedical knowledge. As a research field, BLM integrates NLP, biomedical informatics (BMI) and data mining. BLM techniques have been successfully applied in applications including biomedical literature retrieval, biomedical question/answering, clinical decision support, etc.
In the past decade, researchers from both BMI and computer science (CS) communities have made great efforts on BLM. In general, the BMI community tends to focus more on specific problems, while the CS community works more on developing new algorithms. Because of the popularity and importance of BLM, there have been some surveys about the early efforts on related topics [2–8].
SOTA studies for different tasks discussed in this paper
| Task | Authors | Dataset | Method | F-score |
|---|---|---|---|---|
| Biomedical NER | Zhao et al. [22] | BC5CDR / NCBI Disease | BiLSTM–CNN–CRF-based multi-task learning | 0.8763 / 0.8745 |
| Biomedical NEN | Zhao et al. [22] | BC5CDR / NCBI Disease | BiLSTM–CNN–CRF-based multi-task learning | 0.8917 / 0.8823 |
| Relevant topic recognition | Jiang et al. [23] | MGI database and Mouse GXD | Random forest classifier | 0.923 |
| Biomedical literature indexing | Dai et al. [24] | PMC Open Access Subset | Attention-based CNN | 0.7021 |
| PPIs extraction | Hsieh et al. [25] | AIMed and BioInfer | RNNs | 0.769 / 0.872 |
| Genotype-phenotype associations extraction | Xing et al. [26] | TAIR database | Representation learning | 0.6683 |
| Drug-Drug interactions extraction | Zhang et al. [27] | DDI 2013 extraction corpus | Hierarchical RNNs | 0.729 |
| Pathway extraction | Poon et al. [28] | GENIA event extraction dataset | Distant supervision method | 0.371 |
| Hypothesis generation | Sang et al. [29] | SemKG | Graph-based link prediction | 0.892 |
| Task | Authors | Dataset | Method | F-score |
|---|---|---|---|---|
| Biomedical NER | Zhao et al. [22] | BC5CDR / NCBI Disease | BiLSTM–CNN–CRF-based multi-task learning | 0.8763 / 0.8745 |
| Biomedical NEN | Zhao et al. [22] | BC5CDR / NCBI Disease | BiLSTM–CNN–CRF-based multi-task learning | 0.8917 / 0.8823 |
| Relevant topic recognition | Jiang et al. [23] | MGI database and Mouse GXD | Random forest classifier | 0.923 |
| Biomedical literature indexing | Dai et al. [24] | PMC Open Access Subset | Attention-based CNN | 0.7021 |
| PPIs extraction | Hsieh et al. [25] | AIMed and BioInfer | RNNs | 0.769 / 0.872 |
| Genotype-phenotype associations extraction | Xing et al. [26] | TAIR database | Representation learning | 0.6683 |
| Drug-Drug interactions extraction | Zhang et al. [27] | DDI 2013 extraction corpus | Hierarchical RNNs | 0.729 |
| Pathway extraction | Poon et al. [28] | GENIA event extraction dataset | Distant supervision method | 0.371 |
| Hypothesis generation | Sang et al. [29] | SemKG | Graph-based link prediction | 0.892 |
SOTA studies for different tasks discussed in this paper
| Task | Authors | Dataset | Method | F-score |
|---|---|---|---|---|
| Biomedical NER | Zhao et al. [22] | BC5CDR / NCBI Disease | BiLSTM–CNN–CRF-based multi-task learning | 0.8763 / 0.8745 |
| Biomedical NEN | Zhao et al. [22] | BC5CDR / NCBI Disease | BiLSTM–CNN–CRF-based multi-task learning | 0.8917 / 0.8823 |
| Relevant topic recognition | Jiang et al. [23] | MGI database and Mouse GXD | Random forest classifier | 0.923 |
| Biomedical literature indexing | Dai et al. [24] | PMC Open Access Subset | Attention-based CNN | 0.7021 |
| PPIs extraction | Hsieh et al. [25] | AIMed and BioInfer | RNNs | 0.769 / 0.872 |
| Genotype-phenotype associations extraction | Xing et al. [26] | TAIR database | Representation learning | 0.6683 |
| Drug-Drug interactions extraction | Zhang et al. [27] | DDI 2013 extraction corpus | Hierarchical RNNs | 0.729 |
| Pathway extraction | Poon et al. [28] | GENIA event extraction dataset | Distant supervision method | 0.371 |
| Hypothesis generation | Sang et al. [29] | SemKG | Graph-based link prediction | 0.892 |
| Task | Authors | Dataset | Method | F-score |
|---|---|---|---|---|
| Biomedical NER | Zhao et al. [22] | BC5CDR / NCBI Disease | BiLSTM–CNN–CRF-based multi-task learning | 0.8763 / 0.8745 |
| Biomedical NEN | Zhao et al. [22] | BC5CDR / NCBI Disease | BiLSTM–CNN–CRF-based multi-task learning | 0.8917 / 0.8823 |
| Relevant topic recognition | Jiang et al. [23] | MGI database and Mouse GXD | Random forest classifier | 0.923 |
| Biomedical literature indexing | Dai et al. [24] | PMC Open Access Subset | Attention-based CNN | 0.7021 |
| PPIs extraction | Hsieh et al. [25] | AIMed and BioInfer | RNNs | 0.769 / 0.872 |
| Genotype-phenotype associations extraction | Xing et al. [26] | TAIR database | Representation learning | 0.6683 |
| Drug-Drug interactions extraction | Zhang et al. [27] | DDI 2013 extraction corpus | Hierarchical RNNs | 0.729 |
| Pathway extraction | Poon et al. [28] | GENIA event extraction dataset | Distant supervision method | 0.371 |
| Hypothesis generation | Sang et al. [29] | SemKG | Graph-based link prediction | 0.892 |
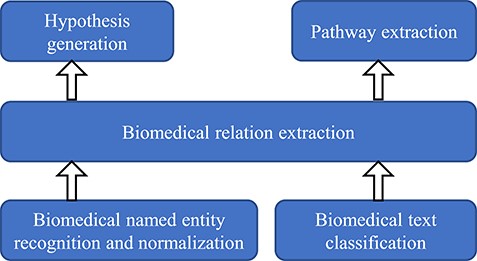
The hierarchy of different tasks of BLM.
Recently, deep learning technologies [9] have been developing rapidly and they have demonstrated strong potentials in various disciplines including NLP [10]. Deep learning models such as long short-term memory (LSTM) [11], convolutional neural network (CNN) [12] and bidirectional encoder representations from transformers (BERT) [13] have been firmly established as the state-of-the-art (SOTA) approaches in NLP tasks such as named entity recognition (NER) [14] and relation extraction (RE) [15, 16]. Numerous efforts have since continued to make advances in BLM with deep learning models (e.g. [17–21]).
This paper aims to survey the recent advances in BLM with special focus on various deep learning techniques. The PRISMA diagram of the literature surveyed in this paper is shown in Figure 2. We organize this survey into five different sections: biomedical NER and normalization, biomedical text classification, RE, pathway extraction and hypothesis generation. These topics have been widely studied in BLM in recent years. In particular, biomedical NER and normalization are the most basic tasks for extracting meaningful and interesting entities from biomedical articles, whose relationships can be identified through RE. Biomedical text classification is crucial for tasks like categorizing and indexing biomedical articles. Pathway extraction can merge connected relations and generate pathways by integrating them. New potential biomedical discovery can be discovered through hypothesis generation from biomedical literature. Among these tasks, biomedical NER and normalization along with text classification are the basis of the other tasks. They are necessary steps for enabling the implementation of other downstream tasks including RE. Pathway extraction and hypothesis generation are usually conducted on top of RE. Figure 1 illustrates the hierarchical relationships among these different tasks. Table 1 summarizes the SOTA performances achieved for these tasks together with their corresponding models.
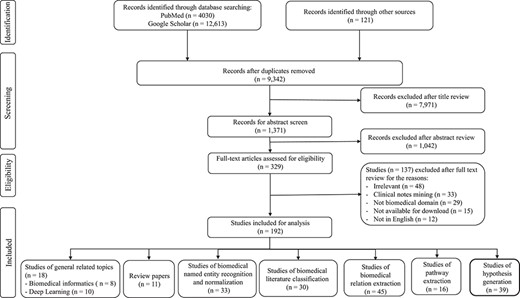
PRISMA flow diagram: recent advances in BLM.
2 Biomedical NER and normalization
In order to structualize the unstructured texts in biomedical literature to facilitate further analysis, a fundamental task is to accurately identify the various biomedical entities that are of interests to the readers, such as chemical ingredients, genes, proteins, medications, diseases, symptoms, etc. This is referred to as the NER problem.
Effective NER has been widely studied in general NLP [30–35], so does biomedical NER [36–46]. Biomedical NER recognizes the entities in texts as predefined categories (e.g. diseases, chemicals, genes, etc.), and it is the basis for many downstream analytical tasks such as enabling the search engines to index, organize and link biomedical documents, mining entity relations from the biomedical literature, etc.
Building a high-performance (e.g. measured by precision and recall) biomedical NER system is quite challenging due to the limited availability of high-quality labeled data, as well as the linguistic variation of texts such as the use of abbreviations, non-standardized names (e.g. Zomig, Zomigon and Zolmitriptan actually refer to the same medication) and lengthy descriptions. This makes biomedical named entity normalization (NEN) also a critical task.
2.1 Task definition
Technically, the goal of biomedical NER is to find the boundaries of mentions of biomedical entities from the text. Biomedical NEN is to map obtained biomedical named entities into a controlled vocabulary. On one hand, NEN can be considered as a follow-up task of NER because normalization is typically conducted on the NER outputs. On the other hand, both NER and NEN can be regarded as sequence labeling problems. Figure 3 shows an example of the task of biomedical NER and NEN, in which the input is a sentence ‘Takotsubo syndrome secondary to Zolmitriptan’, which contains a disease name ‘Takotsubo syndrome’ and a chemical name ‘Zolmitriptan’. The output is B-I-O (Begin-Inside-Outside) tag for each word in this sentence and the entity ID for each biomedical entity.
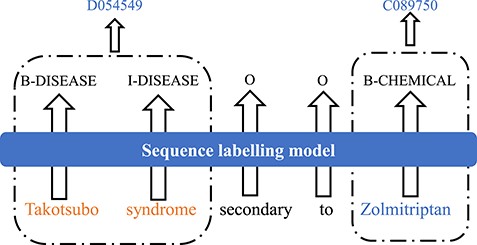
An example of the task of biomedical NER and normalization.
On this topic, Alshaikhdeeb et al. [6] provided an early review on biomedical NER, where they mainly focused on traditional methods. In this paper, we will focus on more recent studies.
2.2 Methods of biomedical NER
Conventional biomedical NER methods can be roughly categorized into three classes: dictionary-based approaches, semantic approaches and statistical approaches. Dictionaries-based approaches [39, 41] use term matching strategy to find the same entities in text that also appear in dictionaries, thus the method is difficult to generalize to recognize the entities outside the vocabulary. Semantic approaches require rich domain-knowledge to construct rules or patterns for identifying the named entities (e.g. [47–50]). Statistical approaches treat NER as a classification problem and train statistical models (e.g. decision trees or SVMs [37, 51], or Markov models-based sequence tagging methods such as HMM and CRFs [38, 40, 42]) to achieve the goal.
In recent years, deep learning models (e.g. [13, 52]) have been firmly established as the SOTA approaches in sequence modeling. Numerous efforts have since been devoted to applying those deep learning techniques to NER as they can be trained in an end-to-end way without additional feature engineering.
Figure 4 demonstrates a typical neural network model for NER, which consists of the following layers:
Character level embedding which represents each character within every word as a vector.
A CNN which extracts features encoding the morphological and lexical information observed in the characters within each word. The final output of this CNN would be a vector representation of each word, which is typically combined with another word embedding vector pre-trained on a large external biomedical text corpus such as PubMed abstracts. (As an alternative, Sahu and Anand [44] also exploited the power of recurrent neural network (RNN) to obtain the morphological and shape features of words in character level word embedding and used it as a feature concatenating with the word embedding for recognition.)
Word-level bidirectional long short-term memory (BiLSTM) layer to model the long-range dependency structure in medical texts. A BiLSTM [53] computes two separate latent embedding vectors for every word in a sequence that capture both the forward and backward semantic dependencies of the word sequence. The two vectors will be concatenated.
A decoder layer that transforms the BiLSTM representation by an affine transformation.
A conditional random field (CRF) layer that calculates the word sequence likelihood.
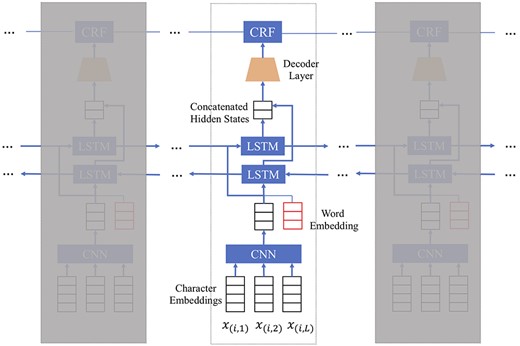
A neural network model for biomedical NER tasks, where character level embeddings are combined as one vector and then concatenated to form the pre-trained word embedding.
One specific model that is getting popular in NLP recently is the BERT model [13], wherein the major building block is transformer [54]. Transformer applies the attention mechanism to learn contextual relations between words in sentences. It is composed of two components including an encoder that encodes textual input and a decoder that predict labels for a particular task. BERT is a new paradigm of Transformer which pre-trains deep bidirectional representations from unlabeled text by considering both left and right context in all layers of attentions. There are two steps in BERT: pre-training and fine-tuning. During pre-training, the model is trained on large amount of unlabeled texts over different pre-training tasks like predicting masked tokens in text and predicting next sentence. For fine-tuning, the BERT model is first initialized with the pre-trained parameters, and all of the parameters are fine-tuned using labeled data from the downstream tasks. Unlike the traditional left-to-right language modeling objective, BERT is pre-trained on two tasks: predicting randomly masked tokens and predicting whether two sentences follow each other. This setting is very different from previous language modeling studies which encode text sequence either from left to right or in a bidirectional order. The BERT framework is shown in Figure 5. BERT broke several records for how well computational models can handle language modeling tasks. Using BERT, an NER model can be trained by feeding the output vector of each token into a classification layer that predict the NER label. Beltagy et al. [55] released a pre-trained contextualized embedding model for scientific text based on BERT, named SciBERT. SciBERT achieved SOTA biomedical NER performance on both the BC5CDR [56] and NCBI-disease [57] data sets, which are used for benchmarking biomedical NER. BioBERT [17] trained a BERT model on biomedical texts sourced from PubMed article abstracts and full texts. They found that BioBERT can improve performance on several biomedical NLP tasks including biomedical NER. Peng et al. [58] introduced a biomedical language understanding evaluation benchmark and evaluated several baselines. They found that the BERT model pre-trained on PubMed abstracts and MIMIC-III clinical notes achieved best results on biomedical NER.
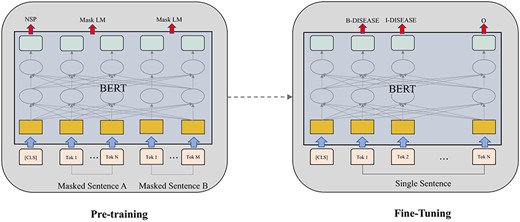
Overall pre-training and fine-tuning procedures for BERT, which uses the same same multi-layer architecture (except the output layer) for pre-training and fine-tuning.
2.3 Methods of biomedical NEN
Biomedical NEN another crucial task for BLM. It has been highlighted as a subtask in different NLP related evaluation or competition series such as SemEval [59] and BioCreative [60]. A variety of approaches have been proposed on this topic [61–65]. Most of them assumed that the named entities are already identified and focused on developing techniques for normalization afterwards. For example, Kang et al. [62] applied a rule-based NLP technique to improve disease normalization in biomedical text. Leaman et al. [64] developed a system called DNorm, which performs disease name normalization with a pairwise learning to rank approach based on CRF. Lee et al. [65] leveraged a dictionary-lookup method for medical NEN. In all these approaches, the biomedical NER and normalization were treated as two separate processes, and the accuracy of biomedical NER directly affects the normalization performance.
2.4 Methods of joint modeling biomedical NER and normalization
Recently there has been research on joint modeling of biomedical NER and normalization because of their inter-dependency. For example, semi-CRF has been used for joint entity recognition and disambiguation [66], where Viterbi decoding is used for assigning part-of-speech tags and normalizing non-standard tokens simultaneously. Semi-Markov models are also used for joint disease entity recognition and normalization. Leaman and Lu [67] leverage a joint scoring function for both tasks. Their model uses exact inference with dynamic programming to discourage non-local features. Leaman et al. [68] developed a high-performance chemical named entity recognizer and normalizer by combining two independent machine learning models in an ensemble. Lou et al. [69] proposed a transition-based model to jointly perform disease NER and normalization, which casts the output construction process into an incremental state transition process. Zhao et al. [22] further proposed a deep neural multi-task learning framework with explicit feedback strategies to jointly model biomedical NER and normalization. This method incorporates both the feedback strategies from the low-level task (biomedical NER) to the high-level task (biomedical NEN) and vice versa, which makes it possible to convert task hierarchy into the parallel mode while maintaining mutual supports between tasks.
2.5 Challenges
There are many challenges on accurate biomedical NER and NEN. A large number of synonyms and alternative expressions of the same entity leads to the explosion of word vocabulary. Moreover, many entities involve long sequences of tokens, which makes it more difficult to detect the boundaries exactly. These entities may also be mentioned by abbreviations, sometimes in non-standard ways. Polysemy or ambiguity could be a potential issue as well. For example, proteins (normally class GENE) are also chemical components and depending on the context. They occasionally should be classified as class CHEMICAL; tokens that are sometimes of class SPECIES can be part of a longer entity of class DISEASE referring to the specialization of disease on specific species.
Nested entities are common in biomedical text, where different biological entities of interest are often composed of one another. For example, the GENIA corpus (http://www.geniaproject.org/) is labeled with entity types such as protein and DNA, wherein roughly 17% of entities are embedded within another entity. However, the current biomedical NER research typically just focus on the outermost entities.
Biomedical NEN also remains a challenging problem. Even though in some cases normalization can be considered as a database look-up, usually no exact match can be found between the recognized entity in the text and the reference entity set. The main reason is that biomedical terms have many variations, which can be categorized as three main types. The first is syntactic variations, where the identified entity contains relatively small character differences with its canonical form present in the reference set, such as different capitalization, reordering of words, typos, or errors (e.g. ‘FOXP2’ and ‘FOX-P2’). The second is the different forms of the same biomedical term such as synonyms and abbreviations. The third is semantic variations, where the recognized entity does not exist in the reference set even when taking external knowledge bases to get synonyms of the recognized biomedical entity.
3 Biomedical literature classification
The problem of text classification has been widely studied in NLP and it has been applied in diverse domains. In this section, we will review the text classification research in BLM.
3.1 Task definition
There are two typical biomedical article classification tasks, relevant topic recognition and biomedical literature indexing. Relevant topic recognition determines if a biomedical publication is related to a given topic. For example, one task of BioCreative III (https://biocreative.bioinformatics.udel.edu/tasks/biocreative-iii/) is to determine if a biomedical publication is related to PPI (protein-protein interaction) [70, 71]. Participants were asked to extract DDIs from biomedical articles and classify them into four predefined classes in the DDI extraction task of SemEval 2013 [72].
Biomedical literature indexing is another important biomedical text classification problem. It assigns a set of terms (e.g. MeSH (Medical Subject Headings) terms) to each specific biomedical article to denote concepts that are discussed in the article. For example, Jimeno et al. [73] exploited the MeSH indexing of MEDLINE papers to generate a data set for word sense disambiguation.
3.2 Methods
Conventional studies on relevant topic recognition utilized classical machine learning models, such as supervised machine learning models, ranking models and ontology matching models, to achieve the goal. For example, Donaldson et al. [74] adopted an support vector machine (SVM) trained with the words in MEDLINE abstracts to identify the abstracts related to PPIs. Polavarapu et al. [75] also applied the SVM to categorize biomedical literature into topics including epidemiology, cancer, congenital disabilities. Dobrokhotov et al. [76] used a probabilistic latent categoriser (PLC) with Kullback-Leibler (KL) divergence to re-rank documents returned from PubMed search to curate information in the Swiss-Prot database [77]. Dollah et al. [78] proposed to classify a collection of biomedical abstracts with an ontology alignment algorithm.
The problem of assigning MeSH terms to biomedical articles is essentially a multi-label classification problem, which treats each MeSH term as a binary classification task [79–81]. Zhang et al. [82] provided a literature review on multi-label classification, and many models therein have been applied in this context, such as k-nearest neighbor (KNN) [83], Naive Bayes [84], SVM [85], learning to rank approaches [79, 86–88], etc. There are also studies emphasizing more on feature engineering instead of the classifier. For example, MeSHLabeler [87] tackled the MeSH indexing problem by integrating multiple types of evidence generated from BOW representation in the framework of learning to rank. Peng et al. [89] proposed a DeepMeSH model that incorporates the deep semantic information to generate large-scale MeSH indexing.
3.3 Recent advances with deep learning
Recent advances in deep neural networks have been established as SOTA models for biomedical text classification. While conventional supervised machine learning models require manual feature engineering, deep learning models can directly take raw text inputs and work in an end-to-end way.
Many models on MeSH indexing has been proposed with deep learning methods [24, 90–95]. These studies typically comprises two modules: 1) a neural network to produce a likelihood scores for each MeSH term and 2) a classifier to determine if the term relevant or irrelevant. Different neural network architectures, including multi-layer feed-forward neural networks [90, 93], convolution neural networks (CNN) [91], RNNs, pre-trained deep neural language models like BERT and ELMo [96, 97], and attention-based model [24, 94, 95], have been adopted. It is worth mentioning that the FullMeSH model, which trained an attention-based CNN for each section has achieved SOTA performance on infrequent MeSH headings [24].
3.4 Challenges
Although biomedical text classification is a classical topic, there are still challenges that have not been completely resolved yet.
Large label space. There are more than 29,000 MeSH terms for indexing biomedical articles, which makes efficient multi-label learning in such a large space difficult.
Label relationship. The relations among labels (MeSH terms) are complicated. Effective exploration of such relationships in the learning process is challenging.
Label bias. Due to the large label space, it is difficult for the ground truth labels (MeSH terms) provided by domain experts to be precise on the training data set. This could potentially impact the quality of the learned classifiers. Creating an accurate unbiased training data set is another challenge.
4 Biomedical RE
4.1 Task definition
RE for BLM refers to the detection and classification of relation mentions among different biomedical concepts within the main texts. The goal of RE is to detect the occurrences of pre-specified types of relationships between entity pairs. Comparing with the types of biomedical entities, the types of entity relations are more diverse. Figure 6 presents an example of the task of biomedical entity RE, where the input is a set of sentences, and the output is the set of identified relations.

An example of biomedical RE. There are three biomedical entities co-occurred in this text piece. The biomedical RE process is to assemble them and determine if a specific relation holds for each pair.
There have been many existing studies on biomedical relation extraction. Template/rule-based methods use patterns (usually in the form of regular expressions) generated by domain experts to extract relations and involved concepts from text [98]. Automatic template construction methods create relationship templates automatically by checking the text patterns surrounding the concept pairs [99–101]. Statistical methods identify these relationships by looking for concepts that frequently co-occur [102]. NLP-based methods perform sentence parsing to decompose the text into a structure from which relationships can be readily extracted [103]. Shahab [7] provided a survey on existing techniques for extracting different types of biomedical relations.
4.2 Methods for different relation extraction tasks
According to the concrete types, we categorize the biomedical relation extraction research into 4 different classes: protein–protein interactions, genotype–phenotype relations, chemical–protein interactions and drug–drug interactions.
4.2.1 Protein–protein interactions (PPIs)
PPIs are indispensable for understanding the complex disease mechanisms and designing appropriate treatments. As we mentioned above, existing PPI extraction methods can be either rule/template based [104–107] or automatic [108, 109]. Simple rules, such as co-occurrence, have been used in the early efforts of PPI extraction. These methods assume that two proteins are likely to interact with each other if they co-occur in the same sentence/abstract. One potential issue of these approaches is that their false positive rates tend to be high. Later studies used manually specified rules, which can achieve a much lower false-positive rate, but often suffer from a low recall rate [104, 110]. Recently, machine learning approaches have been leveraged in automatic PPI extraction. By learning the language rules from annotated texts, machine learning techniques can perform better than rule-based methods in terms of both decreasing the false-positive rate and increasing the coverage. For example, Huang et al. [108] developed a dynamic programming algorithm that extracts patterns from sentences with part-of-speech labels from part-of-speech taggers. Kim et al. [109] developed a kernel-based approach for learning gene and protein-protein interaction patterns. Chowdhary et al. [111] developed a Bayesian network-based approach for extracting PPI triplets from unstructured text. Yu et al. [25] proposed to exploit the grammatical relationship within each PPI triplet extracted by NLP techniques and construct shortest path based features to build a classifier for PPI extraction.
4.2.2 Genotype–phenotype associations (GPA)
Identification of GPA from biomedical literature plays a central role in precision medicine. There have been existing studies on on this topic. Regarding the specie type, most of these research focuses on the associations between human genes and phenotypes [112, 113]. Regarding the entity type, these research usually focus on specific phenotypes such as disease and gene associations [114, 115]. According to the extraction methodology, there were pattern-based [116] or learning-based approaches [26] approaches as well.
4.2.3 Chemical–protein interactions (CPI)
CPI identifies the interactions among chemical compounds and proteins in human body, which is a fundamental task in drug discovery and development. Because of the large number of chemical compounds and genes, it is exhaustive for domain experts to identify them from literature. This makes automatic extraction methods attractive. In particular, Zhu et al. [117] proposed a probabilistic model called mixture aspect model (MAM) to mine implicit CPIs in the text based on compound-target co-occurrence patterns. Wariko et al. [118] utilized a linguistic pattern-aware dependency tree kernel to extract CPIs, and their method obtains an F-score of 36.54% on the BioCreative challenge VI [119]. Lung et al. [120] constructed CPI pairs and triplets and exploited sophisticated features by analyzing the sentence structures. They achieved an F-score of 56.71% in the same challenge.
4.2.4 Drug–drug interactions (DDI)
DDI identification is an essential task in post-market drug safety surveillance, or pharmacovigilance. In general, the problem of DDI detection can be regarded as a binary classification problem. Existing methods for DDI extraction include co-occurrence-based, rule-based and machine learning approaches [121]. Co-occurrence-based methods establish a relationship between two drugs based on their co-occurrences [122]. Linguistic rule-based approaches combine shallow parsing and syntactic simplification with pattern matching [123]. In particular, complex sentences are broke down into clauses from which trigger words or subject-predicate-object patterns can be used to recognize the relations among them. With the better availability manually annotated corpora, methods based on machine learning, especially deep neural networks have become popular in DDI relation extraction tasks as well. One can refer to Zhang et al. [21] for a recent survey.
4.3 Biomedical knowledge base curation
Biomedical relation extraction can support the curation of biomedical knowledge bases, which comprise biomedical entities and relations (e.g. gene A inhibits gene B and gene C is involved in disease G) and are natural assemblies of biomedical NER and relation extraction. On this topic, Ren et al. [124] developed the iTextMine system including an automated workflow to run multiple text-mining tools on a large text corpus for knowledge base curation. Singhal et al. [115] proposed a machine learning approach to curate biomedical knowledge base via extracting disease-gene-variant triplets from biomedical literature.
4.4 Recent advances with deep learning
Relation extraction is essentially a classification problem which can be solved by classical supervised machine learning techniques. These methods take handcraft features as inputs, such as surface, lexical, syntactic features, or features derived from existing ontologies. The use of kernels based on dependency trees has also been explored [125]. The construction of useful handcrafted features is difficult and time-consuming. More recently, a few studies have investigated the use of deep neural networks, such as CNNs (see Figure 7) and RNNs (see Figure 8), for relation extraction, which are detailed below.
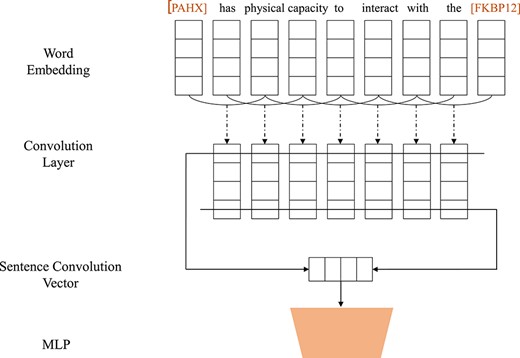
CNN-based framework for relation extraction.
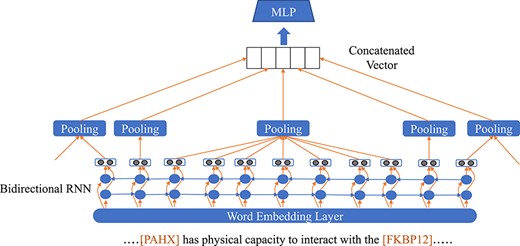
RNN-based framework for relation extraction.
The CNN architecture, illustrated in Figure 7, consists of four main layers, similar to the one used in text classification. For better encoding the information of input sentence, the model usually uses CNN layers to capture n-gram level features. The embedding layer converts each word into an embedding vector via a lookup table. The convolutional layer with rectified linear unit (ReLU) activation transforms the embeddings into feature maps by sliding filters over the word tokens. The pooling layer reduces the dimensionality of the feature map vectors by selecting the highest, lowest, or mean feature values. The multi-layer perceptron (MLP) layer outputs the probability of each relation. Under this framework, Liu et al. [126] proposed a method for DDI extraction.
RNNs model text by exploring the long and short term dependencies in word sequences. It can extract lexical and sentence level features without any complicated NLP preprocessing procedures such as parsing. RNNs can directly represent essential linguistic structures, i.e. word sequences and constituent/dependency trees. On this topic, Hsieh et al. [25] proposed a novel approach based on RNNs to capture the long-term relationships among words in order to identify PPIs. Cross-validation results demonstrate that it outperforms existing methods in the two largest corpora, BioInfer [127] and AIMed [128], with relative improvements of 10% and 18%, respectively on these two datasets [25].
Comparing with previous research that focused on extracting binary relations from single sentences, there are recent studies on more general setting of extracting n-ary relations that span multiple sentences. For example, Peng et al. [129] explored a general framework for cross-sentence n-ary relation extraction based on graph LSTM. The graph structure makes it easy to incorporate rich linguistic characteristics. Experiments on biomedical corpus showed that extraction beyond the sentence boundary exploited much more knowledge, and encoding such rich linguistic knowledge can lead to consistent performance gain.
Another thing we want to emphasize there is that the previously discussed BERT model has also been demonstrated to be highly successful for relation extraction. In particular, Beltag et al. [55] used 1.14M papers randomly picked from Semantic Scholar to fine-tune BERT and built SciBERT. The corpus includes 18% CS papers and 82% biomedical papers. SciBERT obtained results comparable to SOTA models in relation extraction. Lee et al. proposed BioBERT [17], which is a pre-trained language representation model in biomedical domain. The overall process for pre-training and fine-tuning BioBERT is illustrated in Figure 9. First, BioBERT is initialized with pre-trained BERT on general domain corpora. Then, BioBERT is pre-trained on biomedical texts (e.g. PubMed articles). BioBERT was further fine-tuned on several biomedical corpora for BLM tasks. BioBERT only requires a limited number of task-specific parameters but outperforms the SOTA models in biomedical relation extraction by 3.49 F1 score. Both SciBERT and BioBERT share the same basic BERT model architecture shown in Figure 5.
Summary of notable papers in this review
| Author | Dataset | Architecture | Highlight |
|---|---|---|---|
| Medical NER and normalization | |||
| Carreras et al. (2003) [30] | CoNLL-2003 | AdaBoost classifier | Named Entity Extraction system for the CoNLL-2003 |
| Leaman and Gonzalez (2008)[38] | BioCreative 2 GM | CRF | Executable survey of advances in biomedical NER |
| Finkel and Manning (2009) [32] | GENIA and JLPBA | Discriminative Constituency Parsing | using a discriminative constituency parser |
| Hettne et al. (2009)[39] | Corpora for Chemical Entity Recognition | Rule-based method | Rule-based term filtering for small molecules and drugs in text |
| Chowdhury and Lavelli (2010) [42] | Arizona Disease Corpus | CRF | Use a feature set specifically tailored for disease names |
| Abacha and Zweigenbaum (2011)[43] | i2b2 corpus | Hybrid approach | Hybrid approach based on both machine learning and domain knowledge |
| Rocktäschel et al. (2012) [41] | SCAI corpus | CRF | Combining a CRF with a dictionary |
| Leaman et al. (2013) [64] | NCBI disease corpus | Pairwise learning to rank | Use BANNER [38] to locate disease mentions |
| Leaman et al. (2015) [68] | CHEMDNER dataset | CRF | Ensemble two independent models for chemical NER and normalization |
| Leaman and Lu (2016) [67] | NCBI Disease corpus and BioCreative 5 CDR corpus | Semi-Markov model | The first ML model for joint modelling recognition and normalization |
| Lee et al. (2016) [65] | BioCreative V CDR corpus and DNER corpus | CRF | Ensemble CRF models for disease recognition/normalization |
| Sahu et al. (2016) [44] | NCBI disease corpus | RNN | Use CNN and RNN to get character-based embedded features |
| Chen et al. (2017) [45] | BioCreative II gene mention and JNLPBA 2004 corpus | RNN | BLSTM-CRF model with attention for medical NER |
| Lou et al. (2017) [69] | BC5CDR corpus and NCBI disease corpus | Transition-based model | Cast the output construction process into an incremental state transition process |
| Zhao et al. (2019) [22] | BC5CDR corpus and NCBI disease corpus | Deep multi-task learning | Convert hierarchical tasks into parallel multi-task mode |
| Biomedical text classification | |||
| Donaldson et al. (2003) [74] | PubMed abstract | SVM | locate protein-protein interaction data in literature |
| Rios et al. (2015) [91] | MEDLINE articles | CNN | a CNN-based model to conduct a multi-label classification |
| Peng et al. (2019) [97] | Ten Benchmarking Datasets | BERT and ELMo | Compassion between BERT and ELMo on 10 datasets |
| Singh et al. (2018) [95] | PubMed abstract | GRU | a neural sequence-to-sequence (seq2seq) model for structured multi-label classification |
| Yan et al. (2018) [176] | PubMed abstract | CNN-based hybrid model | can adaptively deal with features of documents that have sequence relationships |
| Relation extraction | |||
| Blaschke et al. (1999) [104] | Medline abstracts | Rule-based method | automatic detection of protein-protein interactions |
| Giuliano et al. (2006) [177] | AImed corpus and LLL Challenge | Kernel method | Use shallow linguistic information only |
| Chowdhary et al. (2009) [111] | Mannuly labeled PubMed abstracts | Bayesian network | Extract PPI triplets from medical literature |
| Chowdhury et al. (2011) [178] | DDIExtraction 2011 | Ensemble method | Combine two different machine-learning approaches to extract DDI |
| He et al. (2013) [179] | DDIExtraction 2011 | Stacked generalization | Combine the feature-based, graph and tree kernels |
| Bui et al. (2014) [180] | DDI-2011 and DDI-2013 | SVM | Very traditional RE method for extracting drug-drug interactions |
| Collier et al. (2015) [112] | BMC collection | Search and retrieve | A novel technique for flexibly capturing diverse phenotypes |
| Pathway extraction | |||
| Craven and Kumlien (1999) [134] | Yeast Protein Database | ML method | Map information from text sources into structured representations |
| Ng and Wong (1999) [181] | GENBANK and MEDLINE | IR and IE | Automatic pathway discovery |
| Yao et al. (2004) [182] | 12 papers and 116 abstracts | user-involved extraction | Designed with appropriate level of users’ involvement on information extraction |
| Kemper et al. (2010) [135] | MEDLINE | Text mining | Integrate a pathway visualizer, text mining systems and annotation tools as PathText |
| Miwa et al. (2013) [136] | MEDLINE | IR based method | Integrating and ranking the evidence for biochemical pathways to extend PathText |
| Poon et al. (2014) [28] | Pathway Interaction Database and PubMed abstracts | Distant supervision method | The first attempt to formulate the distant supervision problem for pathway extraction |
| Hypothesis generation | |||
| Shang et al. (2014) [158] | MMB and SemMedDB | Reflective Random Indexing and PSI | Investigate the ability of LBD methods to identify side effects of drugs |
| Hristovski et al. (2016) [159] | 44,250,865 sentences from MEDLINE | LBD based method | Use LBD to explain adverse drug effects |
| Rastegar et al. (2015) [183] | SemMedDB | Random Forest | Propose a method to prioritize the hypotheses generated by LBDs |
| Zhao et al. (2018) [100] | TCM abstracts | Factor graph | Use textual and structural knowledge to infer hypothesis |
| Author | Dataset | Architecture | Highlight |
|---|---|---|---|
| Medical NER and normalization | |||
| Carreras et al. (2003) [30] | CoNLL-2003 | AdaBoost classifier | Named Entity Extraction system for the CoNLL-2003 |
| Leaman and Gonzalez (2008)[38] | BioCreative 2 GM | CRF | Executable survey of advances in biomedical NER |
| Finkel and Manning (2009) [32] | GENIA and JLPBA | Discriminative Constituency Parsing | using a discriminative constituency parser |
| Hettne et al. (2009)[39] | Corpora for Chemical Entity Recognition | Rule-based method | Rule-based term filtering for small molecules and drugs in text |
| Chowdhury and Lavelli (2010) [42] | Arizona Disease Corpus | CRF | Use a feature set specifically tailored for disease names |
| Abacha and Zweigenbaum (2011)[43] | i2b2 corpus | Hybrid approach | Hybrid approach based on both machine learning and domain knowledge |
| Rocktäschel et al. (2012) [41] | SCAI corpus | CRF | Combining a CRF with a dictionary |
| Leaman et al. (2013) [64] | NCBI disease corpus | Pairwise learning to rank | Use BANNER [38] to locate disease mentions |
| Leaman et al. (2015) [68] | CHEMDNER dataset | CRF | Ensemble two independent models for chemical NER and normalization |
| Leaman and Lu (2016) [67] | NCBI Disease corpus and BioCreative 5 CDR corpus | Semi-Markov model | The first ML model for joint modelling recognition and normalization |
| Lee et al. (2016) [65] | BioCreative V CDR corpus and DNER corpus | CRF | Ensemble CRF models for disease recognition/normalization |
| Sahu et al. (2016) [44] | NCBI disease corpus | RNN | Use CNN and RNN to get character-based embedded features |
| Chen et al. (2017) [45] | BioCreative II gene mention and JNLPBA 2004 corpus | RNN | BLSTM-CRF model with attention for medical NER |
| Lou et al. (2017) [69] | BC5CDR corpus and NCBI disease corpus | Transition-based model | Cast the output construction process into an incremental state transition process |
| Zhao et al. (2019) [22] | BC5CDR corpus and NCBI disease corpus | Deep multi-task learning | Convert hierarchical tasks into parallel multi-task mode |
| Biomedical text classification | |||
| Donaldson et al. (2003) [74] | PubMed abstract | SVM | locate protein-protein interaction data in literature |
| Rios et al. (2015) [91] | MEDLINE articles | CNN | a CNN-based model to conduct a multi-label classification |
| Peng et al. (2019) [97] | Ten Benchmarking Datasets | BERT and ELMo | Compassion between BERT and ELMo on 10 datasets |
| Singh et al. (2018) [95] | PubMed abstract | GRU | a neural sequence-to-sequence (seq2seq) model for structured multi-label classification |
| Yan et al. (2018) [176] | PubMed abstract | CNN-based hybrid model | can adaptively deal with features of documents that have sequence relationships |
| Relation extraction | |||
| Blaschke et al. (1999) [104] | Medline abstracts | Rule-based method | automatic detection of protein-protein interactions |
| Giuliano et al. (2006) [177] | AImed corpus and LLL Challenge | Kernel method | Use shallow linguistic information only |
| Chowdhary et al. (2009) [111] | Mannuly labeled PubMed abstracts | Bayesian network | Extract PPI triplets from medical literature |
| Chowdhury et al. (2011) [178] | DDIExtraction 2011 | Ensemble method | Combine two different machine-learning approaches to extract DDI |
| He et al. (2013) [179] | DDIExtraction 2011 | Stacked generalization | Combine the feature-based, graph and tree kernels |
| Bui et al. (2014) [180] | DDI-2011 and DDI-2013 | SVM | Very traditional RE method for extracting drug-drug interactions |
| Collier et al. (2015) [112] | BMC collection | Search and retrieve | A novel technique for flexibly capturing diverse phenotypes |
| Pathway extraction | |||
| Craven and Kumlien (1999) [134] | Yeast Protein Database | ML method | Map information from text sources into structured representations |
| Ng and Wong (1999) [181] | GENBANK and MEDLINE | IR and IE | Automatic pathway discovery |
| Yao et al. (2004) [182] | 12 papers and 116 abstracts | user-involved extraction | Designed with appropriate level of users’ involvement on information extraction |
| Kemper et al. (2010) [135] | MEDLINE | Text mining | Integrate a pathway visualizer, text mining systems and annotation tools as PathText |
| Miwa et al. (2013) [136] | MEDLINE | IR based method | Integrating and ranking the evidence for biochemical pathways to extend PathText |
| Poon et al. (2014) [28] | Pathway Interaction Database and PubMed abstracts | Distant supervision method | The first attempt to formulate the distant supervision problem for pathway extraction |
| Hypothesis generation | |||
| Shang et al. (2014) [158] | MMB and SemMedDB | Reflective Random Indexing and PSI | Investigate the ability of LBD methods to identify side effects of drugs |
| Hristovski et al. (2016) [159] | 44,250,865 sentences from MEDLINE | LBD based method | Use LBD to explain adverse drug effects |
| Rastegar et al. (2015) [183] | SemMedDB | Random Forest | Propose a method to prioritize the hypotheses generated by LBDs |
| Zhao et al. (2018) [100] | TCM abstracts | Factor graph | Use textual and structural knowledge to infer hypothesis |
Summary of notable papers in this review
| Author | Dataset | Architecture | Highlight |
|---|---|---|---|
| Medical NER and normalization | |||
| Carreras et al. (2003) [30] | CoNLL-2003 | AdaBoost classifier | Named Entity Extraction system for the CoNLL-2003 |
| Leaman and Gonzalez (2008)[38] | BioCreative 2 GM | CRF | Executable survey of advances in biomedical NER |
| Finkel and Manning (2009) [32] | GENIA and JLPBA | Discriminative Constituency Parsing | using a discriminative constituency parser |
| Hettne et al. (2009)[39] | Corpora for Chemical Entity Recognition | Rule-based method | Rule-based term filtering for small molecules and drugs in text |
| Chowdhury and Lavelli (2010) [42] | Arizona Disease Corpus | CRF | Use a feature set specifically tailored for disease names |
| Abacha and Zweigenbaum (2011)[43] | i2b2 corpus | Hybrid approach | Hybrid approach based on both machine learning and domain knowledge |
| Rocktäschel et al. (2012) [41] | SCAI corpus | CRF | Combining a CRF with a dictionary |
| Leaman et al. (2013) [64] | NCBI disease corpus | Pairwise learning to rank | Use BANNER [38] to locate disease mentions |
| Leaman et al. (2015) [68] | CHEMDNER dataset | CRF | Ensemble two independent models for chemical NER and normalization |
| Leaman and Lu (2016) [67] | NCBI Disease corpus and BioCreative 5 CDR corpus | Semi-Markov model | The first ML model for joint modelling recognition and normalization |
| Lee et al. (2016) [65] | BioCreative V CDR corpus and DNER corpus | CRF | Ensemble CRF models for disease recognition/normalization |
| Sahu et al. (2016) [44] | NCBI disease corpus | RNN | Use CNN and RNN to get character-based embedded features |
| Chen et al. (2017) [45] | BioCreative II gene mention and JNLPBA 2004 corpus | RNN | BLSTM-CRF model with attention for medical NER |
| Lou et al. (2017) [69] | BC5CDR corpus and NCBI disease corpus | Transition-based model | Cast the output construction process into an incremental state transition process |
| Zhao et al. (2019) [22] | BC5CDR corpus and NCBI disease corpus | Deep multi-task learning | Convert hierarchical tasks into parallel multi-task mode |
| Biomedical text classification | |||
| Donaldson et al. (2003) [74] | PubMed abstract | SVM | locate protein-protein interaction data in literature |
| Rios et al. (2015) [91] | MEDLINE articles | CNN | a CNN-based model to conduct a multi-label classification |
| Peng et al. (2019) [97] | Ten Benchmarking Datasets | BERT and ELMo | Compassion between BERT and ELMo on 10 datasets |
| Singh et al. (2018) [95] | PubMed abstract | GRU | a neural sequence-to-sequence (seq2seq) model for structured multi-label classification |
| Yan et al. (2018) [176] | PubMed abstract | CNN-based hybrid model | can adaptively deal with features of documents that have sequence relationships |
| Relation extraction | |||
| Blaschke et al. (1999) [104] | Medline abstracts | Rule-based method | automatic detection of protein-protein interactions |
| Giuliano et al. (2006) [177] | AImed corpus and LLL Challenge | Kernel method | Use shallow linguistic information only |
| Chowdhary et al. (2009) [111] | Mannuly labeled PubMed abstracts | Bayesian network | Extract PPI triplets from medical literature |
| Chowdhury et al. (2011) [178] | DDIExtraction 2011 | Ensemble method | Combine two different machine-learning approaches to extract DDI |
| He et al. (2013) [179] | DDIExtraction 2011 | Stacked generalization | Combine the feature-based, graph and tree kernels |
| Bui et al. (2014) [180] | DDI-2011 and DDI-2013 | SVM | Very traditional RE method for extracting drug-drug interactions |
| Collier et al. (2015) [112] | BMC collection | Search and retrieve | A novel technique for flexibly capturing diverse phenotypes |
| Pathway extraction | |||
| Craven and Kumlien (1999) [134] | Yeast Protein Database | ML method | Map information from text sources into structured representations |
| Ng and Wong (1999) [181] | GENBANK and MEDLINE | IR and IE | Automatic pathway discovery |
| Yao et al. (2004) [182] | 12 papers and 116 abstracts | user-involved extraction | Designed with appropriate level of users’ involvement on information extraction |
| Kemper et al. (2010) [135] | MEDLINE | Text mining | Integrate a pathway visualizer, text mining systems and annotation tools as PathText |
| Miwa et al. (2013) [136] | MEDLINE | IR based method | Integrating and ranking the evidence for biochemical pathways to extend PathText |
| Poon et al. (2014) [28] | Pathway Interaction Database and PubMed abstracts | Distant supervision method | The first attempt to formulate the distant supervision problem for pathway extraction |
| Hypothesis generation | |||
| Shang et al. (2014) [158] | MMB and SemMedDB | Reflective Random Indexing and PSI | Investigate the ability of LBD methods to identify side effects of drugs |
| Hristovski et al. (2016) [159] | 44,250,865 sentences from MEDLINE | LBD based method | Use LBD to explain adverse drug effects |
| Rastegar et al. (2015) [183] | SemMedDB | Random Forest | Propose a method to prioritize the hypotheses generated by LBDs |
| Zhao et al. (2018) [100] | TCM abstracts | Factor graph | Use textual and structural knowledge to infer hypothesis |
| Author | Dataset | Architecture | Highlight |
|---|---|---|---|
| Medical NER and normalization | |||
| Carreras et al. (2003) [30] | CoNLL-2003 | AdaBoost classifier | Named Entity Extraction system for the CoNLL-2003 |
| Leaman and Gonzalez (2008)[38] | BioCreative 2 GM | CRF | Executable survey of advances in biomedical NER |
| Finkel and Manning (2009) [32] | GENIA and JLPBA | Discriminative Constituency Parsing | using a discriminative constituency parser |
| Hettne et al. (2009)[39] | Corpora for Chemical Entity Recognition | Rule-based method | Rule-based term filtering for small molecules and drugs in text |
| Chowdhury and Lavelli (2010) [42] | Arizona Disease Corpus | CRF | Use a feature set specifically tailored for disease names |
| Abacha and Zweigenbaum (2011)[43] | i2b2 corpus | Hybrid approach | Hybrid approach based on both machine learning and domain knowledge |
| Rocktäschel et al. (2012) [41] | SCAI corpus | CRF | Combining a CRF with a dictionary |
| Leaman et al. (2013) [64] | NCBI disease corpus | Pairwise learning to rank | Use BANNER [38] to locate disease mentions |
| Leaman et al. (2015) [68] | CHEMDNER dataset | CRF | Ensemble two independent models for chemical NER and normalization |
| Leaman and Lu (2016) [67] | NCBI Disease corpus and BioCreative 5 CDR corpus | Semi-Markov model | The first ML model for joint modelling recognition and normalization |
| Lee et al. (2016) [65] | BioCreative V CDR corpus and DNER corpus | CRF | Ensemble CRF models for disease recognition/normalization |
| Sahu et al. (2016) [44] | NCBI disease corpus | RNN | Use CNN and RNN to get character-based embedded features |
| Chen et al. (2017) [45] | BioCreative II gene mention and JNLPBA 2004 corpus | RNN | BLSTM-CRF model with attention for medical NER |
| Lou et al. (2017) [69] | BC5CDR corpus and NCBI disease corpus | Transition-based model | Cast the output construction process into an incremental state transition process |
| Zhao et al. (2019) [22] | BC5CDR corpus and NCBI disease corpus | Deep multi-task learning | Convert hierarchical tasks into parallel multi-task mode |
| Biomedical text classification | |||
| Donaldson et al. (2003) [74] | PubMed abstract | SVM | locate protein-protein interaction data in literature |
| Rios et al. (2015) [91] | MEDLINE articles | CNN | a CNN-based model to conduct a multi-label classification |
| Peng et al. (2019) [97] | Ten Benchmarking Datasets | BERT and ELMo | Compassion between BERT and ELMo on 10 datasets |
| Singh et al. (2018) [95] | PubMed abstract | GRU | a neural sequence-to-sequence (seq2seq) model for structured multi-label classification |
| Yan et al. (2018) [176] | PubMed abstract | CNN-based hybrid model | can adaptively deal with features of documents that have sequence relationships |
| Relation extraction | |||
| Blaschke et al. (1999) [104] | Medline abstracts | Rule-based method | automatic detection of protein-protein interactions |
| Giuliano et al. (2006) [177] | AImed corpus and LLL Challenge | Kernel method | Use shallow linguistic information only |
| Chowdhary et al. (2009) [111] | Mannuly labeled PubMed abstracts | Bayesian network | Extract PPI triplets from medical literature |
| Chowdhury et al. (2011) [178] | DDIExtraction 2011 | Ensemble method | Combine two different machine-learning approaches to extract DDI |
| He et al. (2013) [179] | DDIExtraction 2011 | Stacked generalization | Combine the feature-based, graph and tree kernels |
| Bui et al. (2014) [180] | DDI-2011 and DDI-2013 | SVM | Very traditional RE method for extracting drug-drug interactions |
| Collier et al. (2015) [112] | BMC collection | Search and retrieve | A novel technique for flexibly capturing diverse phenotypes |
| Pathway extraction | |||
| Craven and Kumlien (1999) [134] | Yeast Protein Database | ML method | Map information from text sources into structured representations |
| Ng and Wong (1999) [181] | GENBANK and MEDLINE | IR and IE | Automatic pathway discovery |
| Yao et al. (2004) [182] | 12 papers and 116 abstracts | user-involved extraction | Designed with appropriate level of users’ involvement on information extraction |
| Kemper et al. (2010) [135] | MEDLINE | Text mining | Integrate a pathway visualizer, text mining systems and annotation tools as PathText |
| Miwa et al. (2013) [136] | MEDLINE | IR based method | Integrating and ranking the evidence for biochemical pathways to extend PathText |
| Poon et al. (2014) [28] | Pathway Interaction Database and PubMed abstracts | Distant supervision method | The first attempt to formulate the distant supervision problem for pathway extraction |
| Hypothesis generation | |||
| Shang et al. (2014) [158] | MMB and SemMedDB | Reflective Random Indexing and PSI | Investigate the ability of LBD methods to identify side effects of drugs |
| Hristovski et al. (2016) [159] | 44,250,865 sentences from MEDLINE | LBD based method | Use LBD to explain adverse drug effects |
| Rastegar et al. (2015) [183] | SemMedDB | Random Forest | Propose a method to prioritize the hypotheses generated by LBDs |
| Zhao et al. (2018) [100] | TCM abstracts | Factor graph | Use textual and structural knowledge to infer hypothesis |
![BioBERT framework proposed by Lee et al. [17].](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/bib/22/3/10.1093_bib_bbaa057/1/m_bbaa057f9.jpeg?Expires=1749128841&Signature=asHzokqniS2HQ0bBBK-~TJpdyL4f8C12BR1ZykKhzuIPxnImkrzaIWgigDRPk6cRuxWyiXCmpMH9-4rPxJ5murbuAfvhfn1rmM1YJ7eTxNY2r0rmPfLVOPFfFT8eC89efZ8tmyX1sMLhZnk2XiZBw-X2WEGf~LXrtjMmVx42fNyae12oqBwWKWJhL9qIonEP1B-2WYdpXq518JIED2hPsxH1KspjkuUgi0Yo0lybrcgbtz4eLeyhc2lco4-UOJz10gXeLf1igJxREwcrGymeNIjXOL~pL8ZU6J~ZclekLWV0hVp-614xKVD8sMLkx8s6W6v5vlNYulchY~9npWrzeg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
BioBERT framework proposed by Lee et al. [17].
4.5 Challenges
There are several challenges for biomedical RE compared to general-domain RE tasks. The first is the non-standard expression variation of biomedical entities as we discussed in Section 2. Second, the general RE models typically extract binary relations from the text, such as ‘Founding-location(IBM, New York)’, but the relations involved in the medical literature could be unary, binary, or n-ary relations in which multiple entities are involved in a single relationship. Third, the availability of well-annotated biomedical relations is much less than general relations because of its requirements on domain expertise, which makes sufficient training of complex deep learning models challenging. Fourth, new findings keep appearing in biomedical domain. Developing models for identification of new unseen relations is a challenging problem as well.
5 Biological pathway extraction
Biological pathways are crucial for understanding the underlying mechanisms of complex diseases such as cancer. According to the definition on National Human Genome Research Institute (https://www.genome.gov/about-genomics/fact-sheets/Biological-Pathways-Fact-Sheet), ‘A biological pathway is a series of actions among molecules in a cell that leads to a certain product or a change in a cell. Pathways can also turn genes on and off, or spur a cell to move.’ Most of the pathway knowledge is contained in free text (such as biomedical literature) which needs huge human efforts to understand and interpret [130]. Therefore, it is highly demanding for developing computational approaches for extraction of biological pathways from biomedical literature in an automatic way.
5.1 Task definition
Biological pathways involve the interactions among heterogeneous entities such as genes, gene products and small molecules such as metabolites. Examples of interactions include transcriptional regulation (e.g. transcription factor binding for transcription initiation) and post-translational regulation (e.g. kinase phosphorylation for protein activity modulation). For simplicity, most existing studies focus on static pathways such as signaling transduction and gene regulation, rather than metabolic networks and dynamics. For example, ‘protein A is activated by protein B’ and ‘small molecule C can inhibit such a process’. This kind of information is critical to the understanding of disease mechanism and drug development.
Figure 10 shows an example cancer pathway extracted from biomedical literature. As shown in this example, pathways can form a graph where each node represents a gene or gene product, and each edge represents an interaction. The pathway extraction task is typically formulated as a classification problem in previous studies, i.e. classifying each extracted pairwise relation into one of the well-defined type of relations. The final pathway structure is obtained by merging these extracted relations.
![A pathways extraction from biomedical literature example from [28].](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/bib/22/3/10.1093_bib_bbaa057/1/m_bbaa057f10.jpeg?Expires=1749128841&Signature=UklqSn6IZ8pnVUG1yM6Isgd6d5Y4WDTndc~P8ChJimay23xkwA9INSu~N3fVGttlpRNXNu6PeNdF~z1lm~58r9uxmeLogK036UMSJJLEZAc4bHZ-6v2iVzESaJrNLdGNW8PmuZyhsPz49LEBR~a1tMoRspqC~2U3gMSEER0XXeEzH2JhPp-H55j800ieb3PcJfDmDNwY9jGykkv-sFAUmSrFHxuvEnDFua8aTB3gyatgjZJ7u3uh-7bfgsi5P0N0~OaOqZ8A9xLEdGn-8Y4lM2S6BN12LXhBoRNLH~sKTa3WDjeqmjrmrBALoSezxIYBUMvUM2JGco2RkUIt9BNyxA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
A pathways extraction from biomedical literature example from [28].
Examples of hypothesis generation by inferring unseen relations from biomedical articles.
5.2 Methods
Many existing studies on pathway extraction are rule-based systems (e.g. GeneWays system [131], Petri net [132], BioJAKE [133]), but extraction of hand-crafted rules are expensive and time-consuming. These approaches generally suffer from low recall due to the flexibility of textual expression. As an alternative, machine learning methods can perform effective and automatic rule engineering, but they require large scale annotated examples to achieve satisfactory performance, which still requires lots of human efforts. Craven and Kumlien [134] proposed a way to alleviate such problem with distant supervision from existing knowledge bases. Poon et al. [28] applied the distant supervision framework to extract pathway interactions from PubMed abstracts and showed that their proposed method is superior to the rule-based approach.
In addition, hybrid approaches which take advantage of both rule-based and machine learning approaches have also been actively investigated. For example, Kemper et al. [135] proposed a PathText system including a pathway visualizer, annotation tools, as well as a text mining system integrating syntactic analysis, NER, disambiguation of acronyms, real-time co-occurrence searches, etc. Through PathText, users can have a complete experience of visualized pathway extraction. Miwa et al. [136] extended the capabilities of PathText and developed PathText2, which combined multiple techniques from the various semantic search systems and introduced a new document ranking component. It also offered a new API supporting human computer interactions. Yao et al. [137] developed the PathwayFinder system for pathway extraction with user interactions. Specifically, it provides an interface through which any particular user can influence the result of NER and modify syntactic patterns, which makes the extracted pathway more robust and with better quality.
In addition to biological pathway, other structures such as protein interaction network and gene-disease-drug interaction network are also of paramount interests to biomedical researchers in the era of precision medicine. However, to the best of our knowledge, there is still no study yet on automatic extraction of such network structures directly from the literature, instead the current research typically extracts pairwise relations first and then ensemble them offline. We have reviewed the pairwise entity relation extraction methods in Section 2.4.

The example of ABC model for connecting fish oil and Raynaud’s disease.
5.3 Potential applications with deep learning
There is still no study to solve this pathway extraction task with deep learning techniques. The main reason is that there is no public available training data, which makes training supervised deep learning models challenging to achieve. If we have enough training data, several potential deep models are applicable for this task, such as RNN, CNN and Transformer.
5.4 Challenges
One major challenge of the computational pathway extraction systems is the insufficient involvement of users. More precisely, although some systems provide a way to interact with users, it is too ambitious to achieve a fully automated pathway extraction system without any user intervention because of the following reasons: 1) The diverse and complex expressions in the biomedical literature make it difficult to extract pathways accurately; 2) The low accuracy of the extracted results discourages the further utilization of the system; 3) Some necessary context information is missing, such as conditions of interactions; 4) The varied and continuously changing demands make it hard to adapt promptly. Moreover, many single sentences in scientific publications tend to have multiple biomedical entities involved, which makes the relation extraction procedure have minimal context as clues to use. Just like the example in Figure 10, the single sentence contains four entities, three event triggers and six roles.
6 Biomedical hypothesis generation
The increasing growth rate of the scientific literature makes it challenging for researchers to stay up-to-date with all relevant research in order to formulate novel research hypotheses in their specific disciplines. The generation of hypotheses, also known as literature-based discovery (LBD), attempts to make novel biomedical discoveries from the literature with computational approaches.
6.1 Task definition
Automated LBD, which uses published articles to discover new biomedical knowledge via generating new hypotheses, is a sub-field of BLM. The goal of hypothesis generation is to detect underlying relations that are not present in the text but instead are inferred by the presence of other explicit relations. In other words, it is a way to identify implicit connections by logically combining explicit facts scattered throughout different studies.
Specifically, hypothesis generation is usually referred to the process of connecting two pieces of knowledge previously regarded as unrelated [138]. For example, it may be known that disease A is caused by chemical B, and that drug C is known to reduce the amount of chemical B in the body. However, because the respective articles were published separately from one another (called ‘disjoint data’), the relationship between disease A and drug C may be unknown. Hypothesis generation aims to detect these implicit relations from biomedical articles. Figure 11 presents examples of hypothesis generation by inferring unseen relations.
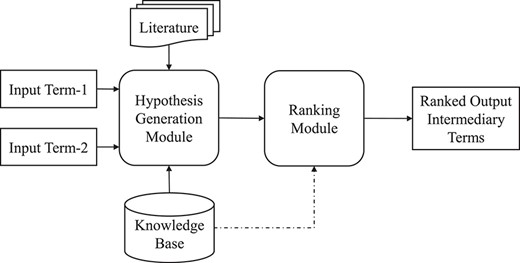
An overview schematic of a general end-to-end pipeline of LBD.
It is worthwhile to mention that biomedical hypothesis generation is different than relational extraction. Relation extraction focuses on extracting relationships between entities that have been explicitly identified in the text, while hypothesis generation attempts to reveal relationships that are unknown yet.
Thilakaratne et al. [8] surveyed the existing computational techniques used in the LBD process with a set of key milestones over a timeline of topics including LBD validation checks, major LBD tools, application areas, domains and generalizability of LBD methodologies. The survey did not mention applications with deep learning on LBD and challenges, which are covered in this paper.
6.2 Problem settings
The core objective of hypothesis generation is to predict a possible relationship between two biomedical terms based on a text corpus [2, 139]. Different from a typical link prediction problem, which are usually either based on triangle closing models [140] or positive semi-definite graph kernels [141], hypothesis generation aims at providing a rationale and evidence in the form of connecting terms. There are two variations of the problem setting, closed discovery and open discovery. The former enables people to perform confirmatory analysis, while the latter is for scenarios that require more exploratory paradigms [142]. As an famous example, consider the queries ‘Is Fish oils and Raynaud’s Disease connected?’ versus ‘What are the therapeutic options for Raynaud’s Disease?’. The first question is a closed discovery problem, where the answer could either YES or NO. If the answer is Yes, the following step should be identifying the evidence that supports this claim. The second question is an open discovery problem. The answer needs to be obtained by exploring all concepts with Raynaud’s disease as potential therapeutic indication. Such questions usually have a ‘grounded’ biomedical concept on one side and a meta-type that defines the characteristics of the possible terms that can appear on the other side [143].
Biomedical hypothesis generation has led to many discoveries such as potential disease treatments [144–147], as well as understanding and discovering new health benefits of supplements [148]. It is particularly promising in a number of applications in pharmaceutical industry such as drug development [149–151], drug repurposing [144, 150, 152–157] and pharmacovigilance [152, 158–160], which are further highlighted below.
Drug discovery and development are expensive and time-consuming processes. Drug repurposing refers to the process of identification of disease targets as alternative potential indications of existing drugs. Successful repurposing can save lots of time and financial cost for drug development because it does not need to go through the initial in-silico and part of the in-vitro phases. For example, COVID-19 is now a global epidemic. Until March 2020 there have been nearly 300K confirmed cases with over 11K deaths 185 countries and areas (https://www.who.int/emergencies/diseases/novel-coronavirus-2019). There is an urgent need on the development of effective treatment for COVID-19. Complete de-novo drug discovery would be very time consuming in this case. Remdesivir, a drug that was originally developed for treating ebola, has demonstrated effectiveness of treating COVID-19 [161], so does hydroxychloroquine, whose original indication is malaria [162]. LBD can provide essential helps for the drug repurposing process. Andronis et al. [163] reviewed various LBD methods that are critical for the detection of hidden connections between biomedical entities and suggest that visualization techniques can help scientists perform tests. Tari et al. [153] used a declarative programming language, AnsProlog, to achieve the automated reasoning for the incomplete information of indirect relationships for drug indications. They also introduced several publicly available knowledge resources such as chemical structures, side effects and signaling pathways for identifying alternative drug indications [154].
Pharmacovigilance refers to ‘the pharmacological science relating to the collection, detection, assessment, monitoring and prevention of adverse effects with pharmaceutical products’ [164]. On this topic, Shang et al. [158] developed a scalable LBD method which uses distributional statistics to infer and apply discovery patterns to evaluate the plausibility of drug/adverse drug reaction pairs for pharmacovigilance. Hristovski et al. [159] presented a tool for providing pharmacological and pharmacogenomics explanations for known adverse drug effects through genes or proteins that link the drugs to the adverse effects. Mower et al. [160] extended this paradigm by evaluating machine learning classifiers when applied to high-dimensional representations of relationships extracted from the literature as a way to identify substantiated drug/adverse drug reaction pairs.
6.3 Methods
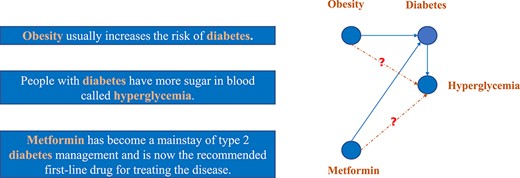
Most of the LBD systems are based on or derived from Swanson’s ABC co-occurrence model [139]. In such a model, explicit knowledge is encoded in the text in forms of ‘A implies B’ and ‘B implies C’ relations. Implicit knowledge can be discovered by drawing a ‘therefore A implies C’ conclusion. For instance, dietary fish oil is mentioned in articles with blood viscosity and vascular reactivity. These two terms are also mentioned in articles with Raynaud’s disease. Swanson proposed that it is reasonable that dietary fish oil and Raynaud’s disease might be associated. This result has been validated experimentally [165] (see Figure 12).
Various tools have been developed to use the ABC co-occurrence model for hypothesis generation [166]. For example, Swanson’s Arrowsmith tool has utilized the co-occurrence of biomedical terms in titles from MEDLINE abstracts to identify existing associations [167]. The user of the system needs to input a starting term and could get choices of appropriate intermediate terms provided by the system. The predicted target terms are ranked by counting the number of intermediate terms. Similar idea have been explored in other systems as well (e.g. CoPub [168] and FACTA+ [169]). These systems typically use the text in the abstract, not just the title. The BITOLA system made use of both the number of intermediate concepts and the number of publications that support these intermediate links as the score for ranking the association candidates [170]. These methods only considered local knowledge in terms of the intermediate terms that co-occur with the starting and the target terms. Recently Zhao et al. [100] discussed different situations of ABC co-occurrence and proposed a factor graph model, CausalTriad, which utilizes more holistic textual and structural knowledge to infer the causal hypothesis.
In addition to the above ABC co-occurrence based modeling paradigms, there were also other methodologies for LBD. For example, the rarity principle [171–173], which looks at infrequently co-occurring terms rather than frequently co-occurring ones. Bibliometric based systems used the citation information to find the linkage and target literature [173]. Sang et al. [29] further developed a biomedical knowledge graph-based LBD approach for drug discovery.
Figure 13 presents a typical end-to-end pipeline of LBD. This system takes a pair of medical terms (a medical term and meta information in the case of open discovery) as input. The task of the ‘Hypotheses Generation Module’ is to list a set of assumptions that relate two inputs by mediation, e.g. ‘Fish oils |$\rightarrow $| Beta-Thromboglobulin|$\rightarrow $| Raynaud Disease’. The ‘Ranking Module’ is then responsible for generating these postulates, which would be finally given to the end-user for further validations. The generated hypotheses can be ranked via selected tools and algorithms in the ranking module. Due to the cascading nature of these modules, it is assumed that the output quality of the generated module will affect the overall quality of the final result.
6.4 Potential applications with deep learning
Most of the studies in hypothesis generation are based on the ABC model. Deep learning models have rarely been used directly in this task potentially due to the high interpretability requirements of the LBD process. It can be envisioned that deep learning models training from large annotated biomedical text corpus should be able to achieve better numerical performance measured on the generated hypotheses. However, it is essential that those hypotheses are explainable. Therefore, effective deep learning interpretability mechanisms [174, 175] are needed.
6.5 Challenges
Despite the promises, challenges remain for biomedical literature based hypothesis generation. In particular, 1) the assumptions in certain methods (such as the ABC co-occurrence based approach) is too simple to capture the complexity of biomedical processes. Enrichment of these technologies with comprehensive biomedical context is important and challenging; 2) many existing LBD methodologies and systems are developed for research purpose. It is important to get them deployed in real application settings where those systems can really help with, such as basic science research, pharmaceutical research and development, as well as clinical care, so that the new discoveries can be prospectively evaluated; 3) the contents of the biomedical articles could be biased towards their specialized disciplines. Sometimes the discoveries from different articles could be contradictory. Obtaining reliable and convincing hypotheses in this scenario is challenging.
7 Conclusions
This paper surveyed the problems, methods and recent advances in BLM. Given the broadness of this area, we picked five critical tasks: biomedical NER and normalization, text classification, relation extraction, biological pathway extraction and hypothesis generation. Methods from both the CS and BMI communities along with their typical application scenarios are reviewed. We also emphasized the recent advances on these topics, especially the potential of deep learning models. At the end of each section we pointed out the challenges and future research directions.
Biomedical literature mining (BLM) is an important area for both computer science and biomedical informatics.
The topics covered under the umbrella of BLM are broad and diverse, such as biomedical named entity recognition and normalization, biomedical text classification, relation extraction, biological pathway extraction and hypothesis generation.
Many techniques based on conventional machine learning algorithms have been proposed for BLM in the last decade. Recently, deep learning models have achieved the state-of-theart performance in many BLM tasks.
Due to the complexity of biomedical systems, there are still many challenges faced by different BLM algorithms.
Funding
This research was supported by National Science Foundation IIS-1750326 (SZ, CS, FW) and NIH Intramural Research, National Library of Medicine (ZL).
Sendong Zhao, PhD, is a postdoctoral associate in the Division of Health Informatics, Department of Healthcare Policy and Research at Weill Cornell Medicine at Cornell University, New York, NY, USA
Chang Su, PhD, is a postdoctoral associate in the Division of Health Informatics, Department of Healthcare Policy and Research at Weill Cornell Medicine at Cornell University, New York, NY, USA
Zhiyong Lu, PhD, is the Deputy Director for Literature Search, National Center for Biotechnology Information (NCBI) at National Library of Medicine, National Institute of Health, Bethesda, MD, USA
Fei Wang, PhD, is an Assistant Professor in the Division of Health Informatics, Department of Healthcare Policy and Research at Weill Cornell Medicine at Cornell University, New York, NY
References

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}