




Abstract
An increasing number of functional studies shows that long noncoding RNAs (lncRNAs) are involved in many aspects of cellular physiology and fulfills a wide variety of regulatory roles at almost every stage of gene expression. A major feature of lncRNAs is the highly folded modular domains in transcripts. With improved modeling and definition, it is now feasible to explore and gain novel insights into the structural–functional relationship of lncRNAs and their association with complex human diseases. In this study, we utilized an automatic computational pipeline to scan lncRNA architecture at the genome-wide scale and to obtain a landscape of functional domains. An accurate alignment algorithm was performed to identify 40 triple pairs between single-nucleotide polymorphisms (SNPs), lncRNAs and diseases. In order to detect the potential contribution of a lncRNA’s modular character, we estimated and evaluated structural rearrangements, which were derived from disease-associated SNPs. In addition, we focused on annotating and comparing the global and local heterogeneity of the wild-type and mutant lncRNAs. Assessing lncRNA architecture has yielded how variations in structured regions impact the molecular mechanisms of lncRNAs and how SNPs disturb binding and recruiting ability. These observations are the first glimpse of the ‘lncRNA structurome’ and make it possible to robustly explore and assemble intricate space conformation and their stress variation. This result also successfully demonstrates that lncRNA transcripts contain a complex structural landscape and highlights the proposed contribution of lncRNA structure in controlling RNA functions and disease mechanisms.
Introduction
Long noncoding RNAs (lncRNAs) are major regulatory factors that are found extensively in eukaryotic cells. They are classified as functional RNAs and are necessary participants in gene expression, regulation and protein synthesis [1, 2]. LncRNAs realize their own functions in various ways, including transcriptional regulation, translational modification and interaction with other biological molecules [3, 4]. A growing number of studies have demonstrated that dysregulation or aberrant expression of lncRNAs might be associated with human diseases such as cancer, cardiovascular disease and neuropathic disorders [5–8]. According to previous research, the evolutionary conservation of RNA secondary structure is a prominent molecular feature of lncRNAs and this secondary structure may modulate lncRNA function [9]. A series of examples show that RNA secondary structural changes mediated by single-nucleotide polymorphisms (SNPs) have the potentiality to cause human diseases. Halvorsen et al. [10] found that the SNPs of genes FTL and RB1 could alter the structure of the 5′and 3′ untranslated regions and lead to hyperferritinemia cataract syndrome and some other diseases [11]. Gong and their colleagues [12] illustrated that SNP rs920778 could impact on the structure of HOTAIR and result in gastric cancer. Ramírezbello et al. [13] revealed SNP rs11655237 in LINC000673 contribute to the susceptibility of pancreatic cancer by varying the flanking structure of miR-1231 binding site and causing molecular regulation disorder. The diversity of molecular mechanisms, in conjunction with emerging structural insights, has fueled considerable excitement and enthusiasm for research examining the structural biology of lncRNAs. Moreover, exploring the relationship between a disease phenotype and RNA molecular structure is of the utmost importance landscape.
The most common type of genetic variations, SNPs, is highly abundant and is widely distributed in the population; therefore, they have become vital research subjects. Recent genome-wide association studies identified a large number of SNPs that are associated with common human diseases [14, 15]. However, the complex mechanisms of how SNPs affect human diseases need to be explored further [16]. Genome variants, including SNPs and single-nucleotide variants (SNVs), might appear in seemingly arbitrary regions of a gene or other RNAs, thus they can contribute to change molecular function and structure. The disturbance of lncRNAs at a nucleotide level could play an important role in gene expression, translation and splicing of mRNAs, enhancing the inherent stability of RNAs and altering interactive protein function. These changes could increase the susceptibility to complex human diseases [17, 18]. According to the hypothesis SNPs have an effect on RNA’s molecular-level variation, it is more effective to interpret the occurrence and development mechanisms of diseases and offer a reference for clinical research.
Genome-wide association studies aim to predict the relationship of genetic variations and clinical phenotypes but do not give insights into the mechanisms of disease [10]. Based on recent research, several regulatory RNAs were found to undergo structural rearrangements that were specific to SNP-induced conformational changes [19]. These elements are called ‘RiboSNitchs’, a kind of SNPs or SNVs that alter RNA structure. The binding of RNAs and proteins can be altered by RiboSNitchs. Additionally, they can also influence the interactive affinity between biological molecules [20]. Studies have elucidated that RiboSNitchs could function as switches that turn genes on and off, which could result in a disease phenotype. Therefore, it is of utmost importance to identify and evaluate how RNA structural modifications are mediated by SNPs. To evaluate how RNA secondary structures and molecular functions are influenced by SNPs, researchers generally combined data from genome variants and RNA structure.
In this paper, we utilized an automated algorithm, sliding windows model, based on the minimum free energy theory, to scan lncRNA sequences derived from the whole genome to detect potential functional structural domains [21]. Using a short sequence alignment algorithm, the exact location of SNPs relative to lncRNAs was accomplished. Furthermore, systemic analysis was implemented to determine the global and local conformation of lncRNAs. Through quantifying the evaluation of wild-type (WT) and mutant lncRNA secondary structures, we obtained and compared structural feature attribute differences between them. More importantly, we estimated the significance of lncRNA structural variation generated by SNPs and analyzed the disturbance of the binding affinity of RNA binding proteins (RBPs) and transcription factors (TFs). Finally, we revealed that RNA structural variation can be associated with human disease states, further demonstrating the interdependency between lncRNA secondary structure and disease phenotypes.
Methods
Overview and processing of data
All lncRNA sequences from the human genome were downloaded from the GENCODE human genome annotation databases in Version 26 (v26) [22], involving a comprehensive genome annotation of lncRNAs and the mature sequences that was recruited from GRCh38. We identified 15787 mature lncRNA genes and 27720 alternative isoform sequences. Sequences longer than 200 bp in length remained in our study.
The dbGap database, which contains human disease-related risk-associated SNPs, was utilized as a primary source of genotype–phenotype association [23]. We obtained 31 classes in total from 2770 SNPs related to complex diseases or characteristics, including WT and mutant genotypes. After attempting a series of subsequence lengths, we selected an optimized and suitable length of the flanking sequence (25 bp) related to SNPs to get the exact location on lncRNAs. The flanking upstream and downstream of each identified sequence was collected from the dbSNP database, which provides reliable data of SNP flanking sequence [24]. In total, 2770 SNP sites were used to carry out the following study.
Identifying lncRNA functional domains via a sliding windows model
We defined high-folded fragments of lncRNA as functional domains and revised an auto-pipeline based on the sliding windows model with a foundation of lower minimum energy to scan genomics and map domains of lncRNA [25]. Empirical parameters were selected (150 bp window with 10 bp steps) to search the full-length lncRNAs, because the length of sequence expected to form a structural RNA is roughly 150 bp.
We calculated the |$z- score$| for all the window fragments in the lncRNA transcripts. Then, we set a strict threshold |$\overline{\alpha}-\sigma$| (the deviation of average Gibbs free energy and SD) to define the potential structural domains. Structuralized RNA is expected to have a lower minimal free energy of fragments compared with a randomized sequence. Thermodynamic |$z- score$| was used to measure and assess the stability of the native sequences and the structural extent of local region of lncRNAs. The lower the value of energy, the more stable the lncRNA conformation. Based on this principle, we used the score as a numeric value to evaluate whether the fragments form modular region or not. We retained the sequence fragments with the score less than the threshold and merged the overlapping windows into candidate functional domains of one lncRNA. Those functional domains constituted lncRNA modular scaffold structures.
Repositioning SNPs in lncRNA transcripts
We detected variations in lncRNA sequences based on the reposition of SNPs. Progress was measured by a short-read alignment strategy. Bowtie 2, a memory-efficient and an ultrafast tool, was utilized to align the flanking sequence of a SNP to the relatively long genomes (lncRNA transcripts) [27]. For phase 1, human mature lncRNAs from GENCODE were chosen as reference sequences. The preprocessed lncRNA sequences were used to build a comparison library in Bowtie 2. Then, we adjusted the parameters and used the Bowtie 2 aligner to ensure exact match between the upstream and downstream flanking fragments of a SNP and a lncRNA sequence. Ultimately, we performed an end-to-end read alignment, searching for alignments involving all of the sequence characteristics to obtain the matched pairs. The output was comprised of the start/stop location, the positive/negative chain of SNPs relative to lncRNAs, WT and mutant alleles and matched fragments.
Quantifying variations of lncRNA structural domains
Quantifying global variation of lncRNA structures
PSMAlign score was applied to appraise the global disturbance of WT and mutant lncRNAs [28]. PSMAlign is a heuristics strategy that is defined as a progressive stem-matching algorithm. It employed the full-length structure profile of two types of lncRNAs to align the stems, pseudo-knots and the nested components. This progress was composed of pattern finding, feature identifying, conformational comparing and significance assessing. We detected the optimal secondary structure of WT and mutant lncRNAs via RNAFold software and used PSMAlign to quantify global structural variations between a pair of WT and mutant lncRNA sequences. The numeric values ranged from zero to thousands, where the higher the score was, the greater the difference between two structure pairs.
Quantifying local variations of lncRNA structure
To determine local changes of structural WT and mutant lncRNAs, we utilized RNAsmc score, a smartly evaluative criterion, which is an R package developed by our group and is an open download from https://mirrors.tuna.tsinghua.edu.cn/CRAN/. The detailed instructions are also uploaded in the website. RNA secondary structure can be divided into five conformational units including stem loop (S), bulge loop (B), interior loop (I), hairpin loop (H) and multibranched loop (M). We counted the location and the number of the five substructures and calculated the variation of the two types of lncRNA architectures. According to the calculative criteria, the range of RNAsmc score is (0, 10]. Comparison of two local conformations could get a score of 10 if they contained identical subunits. Otherwise, if the RNAsmc score was lower than the sum of five subunits (sum is 10), we consider that a disturbance in local structure existed between WT and mutant lncRNAs.
Evaluating the structural change of the SNP flanking region in lncRNA transcripts
To assess the impact of SNPs on flanking regions of lncRNA transcripts, we generated an allele-specific sequence by compiling an empirical 30 bp sequence fragment (15 bp upstream and 15 bp downstream of the SNP site) of both WT and mutant alleles [19]. The paired-probability changes of two corresponding regions were predicted, which represented the alterative region of lncRNA secondary structure. In order to show the structural change in brief, we used Mfold and VARNA to draw the local structure of fragments [29, 30].
Evaluating the variation of molecular combinative ability mediated by lncRNA structural disturbance
To reveal the biological functions of lncRNA generated by structural changes, we analyzed molecular mechanisms of binding through predicting and analyzing biomolecular interactions of TF, RBP and microRNAs with lncRNA. Using the catRAPID online analysis tool we calculated the targeted interactions of lncRNAs and proteins and searched for targeted binding sites and interactive strength scores of WT and mutant lncRNAs within a protein [31]. In addition, we predicted the targeted interactions between lncRNAs and miRNAs via RegRNA [32]. The combined ability and confidence of lncRNAs and other molecules were realized and the functional annotation of structural heterogeneity was completed by searching databases and retrieving papers. The NPInter 3.0 database gave concrete evidence that lncRNAs interact with other molecules [33]. Annotation information of disease-associated lncRNAs were confirmed by experiments that were extracted from the MNDR2.0 database [34].
Results
Identification of the whole genomic lncRNA functional domains
Our study looked for lncRNA functional domains within the whole genome using a computational method. Nucleotide sequences of lncRNA transcripts from reference chromosomes in GENCODE (v26) were preprocessed and used to predict structural domains. The functional domains of 27720 lncRNAs were spliced differentially by a sliding windows model (Supplementary Table S1). According to the genomic location, we classified lncRNAs into five classes (Figure 1A): long, intervening noncoding RNA (lincRNA, 45%) that can be found in intergenic regions and it can take up a large proportion of the whole genome; antisense lncRNA (antisense, 36%); sense overlapping lncRNA (sense overlapping, 1%); sense intronic lncRNA (sense intronic, 3%); and unknown lncRNA (others, 15%).
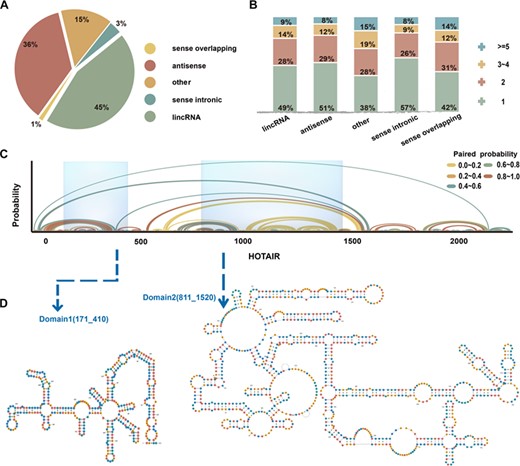
The landscape of lncRNA functional domains was derived from the human genome using a sliding-windows pipeline. HOTAIR is shown as an example. (A) The proportion of the five groups of lncRNA in 27 720 alternative isoforms. (B) Distribution of domains in five categories. (C) Base-pair probabilities of HOTAIR. Arches represent base pairing and are color-coded according to probability. (D) Modular scaffold structure and genome location in HOTAIR transcripts.
The automated sliding window algorithm made it possible for us to identify functional domains of lncRNAs. Figure 1B shows the overview of lncRNA domains from the whole genome separated into five categories. It demonstrates that lncRNAs contain highly folded regions, which might act as functional fragments (domains). In Figure 1C, we show an example that depicts base-pair probabilities, distribution of domains and the folding space of lncRNA HOTAIR. This result illustrates that HOTAIR is characterized by two domains present in a complex spatial architecture. Previous research demonstrated these modular scaffold structures of HOTAIR recruit two distinct repressive complexes, PRC2 and LSD1 [4, 35]. We also perform the visualization of complex domains of some other lncRNAs, and another example of TPT1_AS1 is shown in Supplementary Figure S1. These structured RNA domains provide insights into regulatory regions and action models of lncRNA structural biology.
Mapping SNPs in lncRNA transcripts
We downloaded a total of 2770 SNPs, which associated with 31 types of diseases from dbGap. SNPs were mapped accurately on lncRNA transcripts using the short-read alignment algorithm. A total of 36 SNPs located on 29 lncRNAs were associated with 18 diseases. We obtained 40 kinds of correspondences between SNPs, lncRNAs and diseases. (Figure 2, Supplementary Table S2).
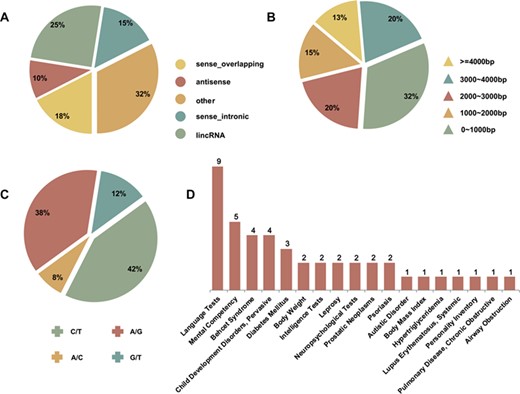
The quantitative annotations of secondary structure variation related lncRNAs, SNPs and diseases. (A) Five categories of disease-associated lncRNAs. (B) The full-length structure characteristic of disease-associated lncRNAs. (C) The distribution of disease-associated SNPs. (D) The number distribution of lncRNA–SNP pairs in diverse human diseases.
Figure 2A illustrates the classification ratios of lncRNAs, which includes SNPs. It shows that the proportion of sense-intronic and sense-overlapping lncRNAs increased, while antisense lncRNA and lincRNA decreased. This observed phenomenon indicates that the sense-strand sequences might undergo allelic mutations. Figure 2B reveals the proportional distribution of the sequence length of 29 lncRNAs, which have an exact SNP location. Four kinds of SNP alleles were involved in our study, A/G, C/T, G/T and A/C (Figure 2C). Among those alleles, the C/T allele had the highest frequency at about 42%. SNPs in the lncRNA transcripts were associated with 18 complex diseases or phenotypes, including diabetes mellitus, prostatic neoplasms and psoriasis (Figure 2D).
In our study, two corresponding lncRNA-disease associations, Prostatic Neoplasms-CCAT2 and Prostatic Neoplasms-PRNCR1, were confirmed in the MNDR2.0 database, which is the ncRNA-disease association’s repository. Correlations among the databases have been updated by integrating experimental and predicting associations from manual literature curation. These results suggest our study is a feasible way to detect functional lncRNA-disease associations by repositioning and associating SNPs. The obtained relative relationships provide a theoretical foundation to give insights into lncRNA structural variations mediated by SNPs and it may allow for further identification of the etiology of disease and the ability to give precise diagnoses at the molecular level.
Detecting variations of lncRNA functional domains mediated by SNPs
To reveal potential molecular mechanisms driven by lncRNA functional domains and to evaluate structural changes influenced by the effects of a mutation in a lncRNA transcript, we analyzed variations in lncRNA functional domains in detail. We evaluated changes in the location and span of lncRNA domains using the Jaccard index to assess the standard of variant extent. Figures 3A and B illustrate the quantitative changes of domains between WT and mutant lncRNAs. We discovered that all domains were changed more or less, with 72.5% of the domains changing significantly (Jaccard index < 0.7 and Wilcoxon rank–sum test, P < 0.05, Supplementary Table S3). Figure 3C and D depict the variations between WT and mutant domains: blue represents the WT lncRNA domain distribution, red represents the mutant lncRNA domain distribution and the khaki histogram indicates the diversity of nucleic acid-pairing probability between two sequence states. This result illustrates that SNPs can affect the folding of the sequence and the configuration of functional domains.
In 40 items of SNPs, lncRNAs and diseases, we summarized the relationship between the functional domains of WT and mutant lncRNAs and the location of SNPs. Four conditions are included: (I) The four items of WT lncRNA domains contain a SNP, but the corresponding mutant domains do not cover the site, (II) four lncRNA domains that change from distancing SNP sites to covering those sites, (III) 19 items of the WT and mutant lncRNAs contain a SNP (Figure 3C and D) and (IV) the remaining 13 items that do not cover the span of domains are turned (Supplementary Table S3). These results suggest that the location of a SNP on a lncRNA transcript is diverse and non-regular. SNPs may change the structural features of its adjacent regions or alter the structural attributes of sequences a greater distance away. In general, SNPs will affect the folded state of lncRNA sequences and will have an effect on spatial structures.
Global structural heterogeneity analysis of lncRNAs mediated by SNPs
We defined the global variations of lncRNAs for those structural transformations emerging distinctly and extensively in the transcript (Figure 3C). The secondary structure heterogeneity of global transcripts induced by SNPs was quantified using PSMAlign, a tool for evaluating secondary structure differences. The threshold of distinction was set, and when a PSMAlign score was higher than 10, we considered that the spatial conformation of a lncRNA transcript was changed by a SNP (In total, about 55% lncRNA transcripts secondary structure changed; Figure 4A, Supplementary Table S4). This result indicates that spatial folding of some lncRNAs can be changed by SNPs and that these changes can have an influence on global structure.
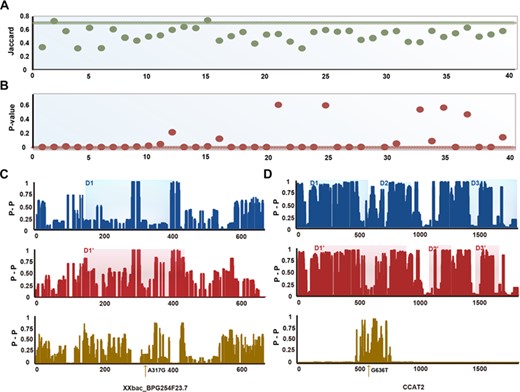
The quantitative variations ofWT and mutant lncRNA domains. (A, B) The Jaccard index (A) and Wilcoxon rank–sum test P-value (B) of WT and mutant-lncRNA domains. (C) The pairing probability and the difference of WT and mutant lncRNA XXbac_BPG254F23.7 and functional domains. (D) The pairing probability and the difference of WT and mutant lncRNA CCAT2 and functional domains.
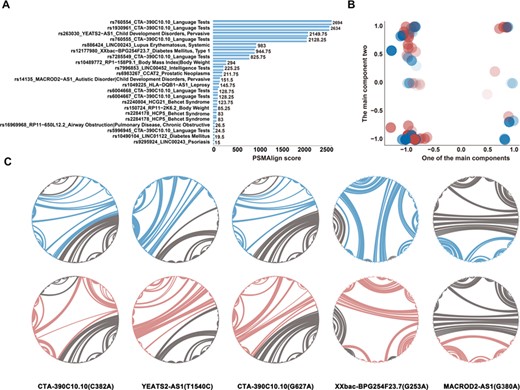
Global secondary structural variation of lncRNAs mediated by SNPs. (A) The global PSMAlign score of WT and mutant lncRNAs. (B) The structural principle component of disease-associated lncRNAs. The paired blue and red bubbles with same concentrations represent wild and mutant lncRNAs, respectively. (C) The circular structural diagram of five lncRNAs which have a significant diversity in global level. The arched line represents the base pairing between two nucleotides. The color of blue and red represents wild and mutant type, respectively.
We identified spatial structural transformations mediated by SNPs in lncRNAs. Figure 4B shows the principal component plot based on extracting the base-pairing probability of WT and mutant lncRNAs on the basis of the secondary structures. The structural properties of the WT and mutant lncRNAs are represented by blue and red bubbles, respectively, which have the same potency in color. This principal component analysis diagram allows for visualization of the structural heterogeneity of all lncRNAs. There is a significant variety of architecture between WT and mutant lncRNAs. Figure 4C depicts five instances of lncRNA structural variation where the sequence is represented by a circular structure plot and the relationship of paired bases is indicated by an arched curve. All grey lines show the spatial conformation of WT and mutant lncRNAs and the blue or red lines display structural differences between each other. In this research, we identified 18 disease states, including mental competency and diabetes mellitus, which are associated with SNPs in lncRNAs. Most importantly, we demonstrated that global structural variation is induced by SNPs in some lncRNAs. Changes in lncRNA structure may give rise to the development of diseases.
Local structural heterogeneity analysis of lncRNAs mediated by SNPs
Among the 40 triple pairs (SNPs, lncRNAs and diseases) in our study, SNPs in a couple of lncRNAs resulted in global and local structural changes. An example of this was changes in XXbac-BPG254F23.7 (Figure 3C). A number of SNPs resulted in centralized and local variations in the structure of lncRNAs. We developed a toolkit, RNAsmc, which estimates local structural heterogeneity. The sub-units of lncRNA secondary structure were evaluated quantitatively by comparing the change in location and the amount of five impacts: stem loop (S), bulge loop (B), interior loop (I), hairpin loop (H) and multibranched loop (M). By analyzing the local structural variation of WT and mutant lncRNA, we found that 65% of transcripts were altered significantly at the local level (Supplementary Table S5). In order to explore the local effects mediated by SNPs, we obtained the flanking-sequence fragments (15 bp upstream and 15 bp downstream of the mutated site) in XXbac-BPG254F23.7. Figure 5A illustrates the paired probability of the WT and mutant lncRNA flanking region. Many of the sites surrounding the SNPs have been disturbed (Figure 5B). Figure 5C depicts the concrete secondary structure of the WT and mutant lncRNA flanking region, showing that the local region around a SNP site is structurally altered. Local variations of lncRNAs may alter their function and could be an underlying molecular mechanism that leads to diseases.
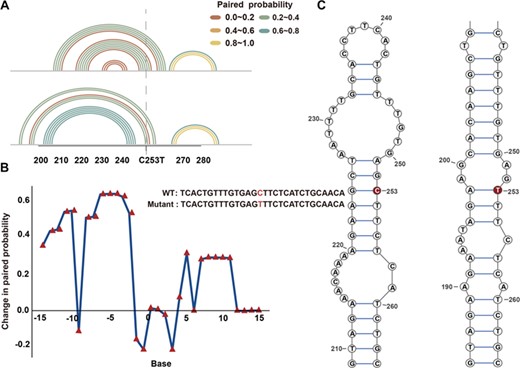
Local secondary structure of lncRNA XXbac-BPG254F23.7. (A) The paired probability of the flanking sequence in wild and mutant XXbac-BPG254F23.7. (B) The paired-probability difference of wild and mutant XXbac-BPG254F23.7. (C) The secondary structure of wild and mutant XXbac-BPG254F23.7 around the flanking region. The bases in red represents SNPs on XXbac-BPG254F23.7.
Disease risk analysis induced by RNA conformational variation
To address the hypothesis that SNPs-induced structural rearrangement of RNA transcripts leads to disease, we mapped all known disease-associated SNPs to lncRNAs. We also evaluated global or local changes in the conformations of WT and mutant lncRNAs. Analyzing the interaction of lncRNA with other molecules is an effective way to understand the regulatory mechanism of lncRNA. Utilizing the methods described above, we predicted the relationship of disease-associated SNPs to diseases from a structural perceptive.
By seeking the interactive information of lncRNAs from databases (LncDisease and MNDR2.0 database), we determined the relationship of four lncRNAs and 16 TFs. Among those interactions, our work shows that 25% of the combinative regions between three WT and mutant lncRNAs and four TFs increased (CCAT2-MYC, CCAT2-CDC25A, AC003084.2-CDX2, MACROD2-AS1-E2F4); however, others failed to transform.
For the purpose of this study, we were particularly interested in identifying disease-associated SNPs, like rs14135 in MACROD2-AS1, which had a significant effect on lncRNA structure. Previously, the relationship between MACROD2-AS1 and autism was verified [36]. The structural conservation of lncRNAs is low. There are two domains in WT MACROD2-AS1, D1:110–300 bp and D2:320–640 bp, which are mutated to D1:60–260 bp and D2:410–650 bp, respectively, in the mutant MACROD2-AS1 (Figure 6A). As shown in Figure 6A, the location of these domains in the WT and mutant MACROD2-AS1 produced an apparent structural change. We predicted the interactive regions of the WT and mutant MACROD2-AS1 and E2F4 using catRAPID (the innermost and outermost blue bar). Blue and red shadows represent the WT and mutant interactive region. Therefore, we can infer that rs14135 alters the structure of lncRNA, which could affect its interaction with proteins and could lead to the occurrence of autism.
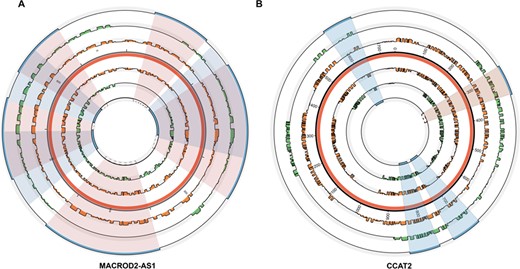
The paired probability, distribution of domains and interactive regions of wild and mutant secondary structure of MACROD2-AS1 and CCAT2. (A) The situation of lncRNA MACROD2-AS1. (B) The situation of lncRNA CCAT2. The red interlayer demonstrates the overall length of each lncRNA. The inner and outer orange bars refer to paired probability of wild and mutant lncRNAs. The green bar in the inner and outer layer shows the domains of wild and mutant lncRNAs and the corresponding innermost and outermost blue bars represent the interactive region of wild and mutant lncRNAs with TF. The shadow section in blue represents the coherent interactive regions of wild and mutant lncRNAs and the incarnadine represents incongruous regions of them. The region displayed by broken line shows the interactive region appears in mutant lncRNA rather than wild lncRNA.
Additionally, we examined the relationship of CCAT2, rs6983267 and prostatic neoplasm [37]. It was previously demonstrated that the genomic locus of CCAT2 is highly conserved and harbors rs6983267, which was shown to be associated with the risk of developing prostatic neoplasm. Instead of meta-analysis, we interpreted the regulatory mechanism at the structural level. We observed that the WT and mutant CCAT2 functional domains changed (Figure 6B). The interactive regions of two CCAT2 sequences and MYC were calculated using catRAPID. Compared with the WT CCAT2, a new interactive fragment appears: 281–352 bp in the mutant CCAT2. Therefore, we can infer that SNPs have an important influence on the structure of CCAT2.
As an alternative to TF interaction, we further analyze the effects of miRNA targets, under the situation of WT and mutant lncRNAs, by using RegRNA. We find different SNP alleles could affect both the amount of miRNA binding sites and the changes of interactive regions. For example, the WT RP11-508 N22.12 has only one target (hsa-miR-1304-3p: 4700–4721 bp); however, the alternative rs2474596 allele exposes four targets (hsa-miR-1304-3p: 4700–4721 bp; hsa-miR-3661: 5728–5757 bp; hsa-miR-4459:2105–2126 bp and hsa-miR-4459: 2136–2158 bp). As a case of altering the interactive region of miRNA targets, rs7996853 alternative allele in LINC00452 cold reduce the seed region of hsa-miR-296-3p (1868–1891 bp). We speculate that increasing or deleting of miRNA binding targets mediated by SNP could have an impact on human diseases.
Discussion
Our analysis of the effects of disease-associated genetic variation on lncRNAs reveals the extent to which specific SNPs affect the lncRNA structure and give rise to diseases. Structural changes were assessed by examining functional domains and local and global variation of WT and mutant lncRNAs. This is mainly based on two considerations. Firstly, in the field of RNA-folding study, there are still some differences between global optimization and local optimization. These two viewpoints are both supported by some reliable examples. Then, in our previous study, we found that the global and local analysis did not always show consistent results for different lncRNAs, which might reflect the variation of lncRNA nucleic acid sequence composition [35]. In order to explore the proximal regulatory nature of lncRNAs, we observed the variations effect both at global and local levels. A couple of criterions can quantify lncRNA structural changes. However, in this study, only small-scale variations are just attributed to global structure variations. Most of the significative SNPs have effects both on local context and the whole length of lncRNAs. LncRNA domains were obtained from the whole human genome by a sliding windows model. By quantifying the secondary structure at a local and a global level, we found that 55% of SNPs had an effect on global structure and 65% of SNPs had an influence on sub-structure. More importantly, our analysis revealed the extent to which SNPs affect RNA structure through disrupting molecular interactions, which can further have an effect on recruiting functional proteins. SNPs associated with certain disease states may become molecular markers for clinical diagnosis.
According to our study, the domains are defined as high-folded region and could function as structural scaffold to recruit biomolecules. High-structured domains interact with TFs, RBPs and other functional RNAs and then influence gene expression and regulation. Among 40 triple pairs of our results (SNPs, lncRNAs and diseases), the results inferred that all domains have structural diversity more or less, about 72.5% domains transform significantly. But Jaccard index could measure the diversity of WT and mutant lncRNA domains at the level of sequence scope only. In order to further consider the structural changes of lncRNA mediated by SNP from the base interaction level, we introduced a global and local structural variation evaluation system. In our study, some lncRNA domains have no significant variations, such as XXbac-BPG254F23.7 (Jaccard index = 0.41, P = 1), but SNP in the lncRNA induces obvious disturbance in both global and local structural level. The result illustrated rs12177980 (A253G) in XXbac-BPG254F23.7 leads to the greatly changed pattern of base pairing and then influence the spatial conformation and function of lncRNA. However, the sequence range of these domains has no variation (150–420 in WT lncRNA; 140–410 in mutant lncRNA). In addition, some individuals (such as XXbac-BPG181B23.7-Behcet Syndrome) have no significant changes in global and local structural level, as well as domains. We inferred that lncRNAs might actualize their function through regulating expression level rather than just using structural interaction with other molecules.
The lncRNAs are involved in a wide range of cellular processes, including gene expression, protein translation and protein stability. Previously, a few studies found that lncRNAs play an important role in the development of diseases. For example, mutations in the lncRNA XIST can lead to X-chromosome inactivation. HOTAIR is associated with a variety of cancers, including breast, colorectal, nasopharyngeal and hepatocellular cancers [38, 39]. The regulatory mechanisms of a large number of lncRNAs are unknown. Structural conservation is much stronger than sequence conservation [40]. Studying the structures of lncRNAs and the interaction between their structure and sequence are keys to elucidating lncRNA function. In this paper, we analyzed the extent to which specific SNPs affect lncRNA structure. The study of lncRNAs’ functional domains has laid the foundation for study of the complex regulatory mechanisms of lncRNAs.
The SNP rs6983267 in CCAT2 increases the risk of many cancer types, such as colon, ovarian and inflammatory breast cancer [41, 42]. CCAT2 plays a variety of molecular roles in cellular processes, but our study focuses on changes in CCAT2 domains induced by SNP and influence MYC binding. Furthermore, according to previous research, CCAT2 promotes cell growth and chromosomal instability in several tumors. MYC-regulated miR-17-5p also plays an important role in the development of cancer [43]. The mechanisms underlying these associations are unknown. It is possible that the SNP rs6983267 affects the domain responsible for interactions between CCAT2 and MYC, thus altering the expression of one or more downstream genes.
Understanding the molecular mechanisms of disease-associated lncRNAs is an important area of study and requires systematic probing of lncRNA functional domains. The functional elements of lncRNAs, such as XIST and HOTAIR, have been validated, and we expect other lncRNA domains will be useful for advancing the mechanistic understanding of lncRNAs [21]. With the rapid development of high-throughput sequencing of the ‘RNA structurome’, current algorithms will be optimized to recognize the flexible structures of RNA molecules. Ultimately, the long-term goal of this study is to create a catalog of lncRNA structures and their inferred interactions with sequence motifs to conduct future functional and mechanistic studies.
The first glance to investigate the disease risk SNP mechanism from the level of allosteric effect of lncRNA.
A sliding windows method was used to evaluate and compare the domain characteristics of genome-wide lncRNAs.
The global and local analysis algorithms to detect the differences of lncRNA structure under the situation of different SNP alleles.
Acknowledgements
The authors wish to thank all the members and the support from the Training Center for Students Innovation and Entrepreneurship Education, Harbin Medical University and the Institute of Biomedical Big Data, Wenzhou Medical University.
Funding
National Natural Science Foundation of China (grant nos 31501062 and 31801098), the Research Projects of Education Department of Heilongjiang Province (grant nos 1254G041 and 12511273), the Harbin Science and Technology Bureau project (grant nos RC2013QN004112), the Fundamental Research Funds for the Provincial Universities (grant nos 2017JCZX50) and the Talent Startup Fund Project of Wenzhou Medical University.
Hong Wang is an assistant professor at Harbin Medical University and cooperates with Wenzhou Medical University. Her research interests include complex disease bioinformatics and molecular genetics.
Xiaoyan Lu is a graduate student jointly trained by Harbin Medical University and Wenzhou Medical University. Her main research interests include RNA molecular genetics and structural bioinformatics.
Fukun Chen is an undergraduate student jointly trained by Harbin Medical University and Wenzhou Medical University. His main research interests include RNA structural bioinformatics and system biology.
Yu Ding is a graduate student jointly trained by Harbin Medical University and Wenzhou Medical University. His main research interests include RNA and protein structural biology.
Hewei Zheng is a graduate student jointly trained by Harbin Medical University and Wenzhou Medical University. His main research interests include disease genetics and structural bioinformatics.
Lianzong Wang is a graduate student jointly trained by Harbin Medical University and Wenzhou Medical University. His main research interests include lncRNA regulatory bioinformatics and system biology.
Guosi Zhang is a graduate student jointly trained by Harbin Medical University and Wenzhou Medical University. His main research interests include circRNA regulation and RNA structural bioinformatics.
Jiaxin Yang is a graduate student jointly trained by Harbin Medical University and Wenzhou Medical University. Her main research interests include microbiological genomics and computational biology.
Yu Bai is a graduate student jointly trained by Harbin Medical University and Wenzhou Medical University. Her main research interests are RNA molecular regulation and structural bioinformatics.
Jing Li is a graduate student jointly trained by Harbin Medical University and Wenzhou Medical University. Her main research interests include molecular network analysis and RNA system biology.
Jingqi Wu is a graduate student jointly trained by Harbin Medical University and Wenzhou Medical University. Her main research interests include disease molecular genetics and structural bioinformatics.
Meng Zhou is an associate professor at Wenzhou Medical University. His main research interests include bioinformatics and computational RNomics.
Liangde Xu is an associate professor and the deputy director of the Institute of Biomedical Big Data at Wenzhou Medical University. His main research interests include functional genomics and complex disease bioinformatics.
The College of Bioinformatics Science and Technology, of Harbin Medical University, is one of the largest computational biology research organizations in China, which has made valuable achievements in bioinformatics research of complex diseases.
The School of Ophthalmology & Optometry and Eye Hospital, School of Biomedical Engineering of Wenzhou Medical University, is one of the most famous ophthalmic medical organizations in China, which has achieved a high level of research in ophthalmic disease mechanism, biomedical big data and biomedical materials.
Reference
Author notes
These authors contribute equally to this work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}