




Abstract
Although systematic genomic studies have identified a broad spectrum of non-coding RNAs (ncRNAs) that are involved in breast cancer, our understanding of the epigenetic dysregulation of those ncRNAs remains limited. Here, we systematically analysed the epigenetic alterations of microRNAs (miRNAs) and long non-coding RNAs (lncRNAs) in two breast cancer subtypes (luminal and basal). Widespread epigenetic alterations of miRNAs and lncRNAs were observed in both cancer subtypes. In contrast to protein-coding genes, the majority of epigenetically dysregulated ncRNAs were shared between subtypes, but a subset of transcriptomic and corresponding epigenetic changes occurred in a subtype-specific manner. In addition, our findings suggested that various types of epi-modifications might synergistically modulate ncRNA transcription. Our observations further highlighted the complementary dysregulation of epi-modifications, particularly of miRNA members within the same family, which produced the same directed alterations as a result of diverse epi-modifications. Functional enrichment analysis revealed that epigenetically dysregulated ncRNAs were significantly involved in several hallmarks of cancers. Finally, our analysis of epigenetic modification-mediated miRNA regulatory networks revealed that cancer progression was associated with specific miRNA-gene modules in two subtypes. This study enhances understanding of the aberrant epigenetic patterns of ncRNA expression and provides new insights into the functions of ncRNAs in breast cancer subtypes.
Introduction
Breast cancer is a genetically heterogeneous disease with substantial variations at the molecular level, including both genetic and epigenetic aberrations [1–3]. Over the past decade, knowledge of cancer has been broadened through the use of molecular profiling technologies. Breast cancers have been classified into three molecular subtypes on the basis of the level of mRNA expression of specific genes: luminal (A and B), HER2-positive and basal (A and B). Different breast cancer subtypes are thought to arise through different ‘evolutionary paths’ reflecting distinct patterns of mutated cancer genes, gene/microRNA (miRNA) expression or epigenetic regulation [4]. Although numerous genetic mutations have been linked to different breast cancer subtypes, accumulating evidence suggests that abnormal alterations of epigenetic modifications are also hallmarks of human cancers [5]. In a previous study, we have compared changes in six types of histone modification and DNA methylation between two cell lines, representing two different breast cancer subtypes (luminal and basal) [6]. We used integrative analysis of genetic and epigenetic data to reveal distinct patterns of oncogenic pathway activation in different cancer subtypes. The study of epigenetics in breast cancer has made substantial progress, and these data hold significant implication in cancer diagnosis and the identification of potential molecular therapeutic targets.
It is now understood that most of the mammalian genome is transcribed, producing large amounts of non-coding RNAs (ncRNAs) [7]. The two main types of ncRNA are miRNAs and long non-coding RNAs (lncRNAs). MiRNAs are a recently discovered and well-characterized class of ncRNAs that are frequently dysregulated in different breast cancer subtypes [8–10]. Bhattacharyya et al. have examined miRNA expression profiles over a large set of breast cancer tumour samples to identify breast cancer subtypes [11]. Their experimental results have demonstrated that miRNAs carry a unique signature that can distinguish cancer subtypes and even reveal new cancer subtypes [12, 13]. The subtype-specific expression of ncRNAs has been shown to play key roles in the regulation of subtype-specific pathways; for example, miR-17, miR-19a, miR-25 and miR-200b show high miRNA regulatory activity in the triple-negative, basal-like cancer subtype, whereas miR-22 and miR-24 do so in the HER2 subtype [14]. Furthermore, pathway analysis has associated miR-17, miR-19a and miR-200b with leukocyte transendothelial migration. In contrast, the molecular basis of the functions of many lncRNAs is just emerging, although substantial evidence indicates that lncRNAs play intricate roles in the regulation of a wide variety of biological processes [15, 16]. Aberrant expression of lncRNAs, such as HOTAIR [17] and MIR31HG [18], has been reported in prevalent cancer types, including breast cancer. As described above, these results suggest that ncRNAs show distinct expression patterns in different cancer subtypes and regulate distinct downstream pathways.
Although ncRNAs have been demonstrated to participate in the modulation of gene expression, the epigenetic regulation of ncRNAs remains poorly understood in different breast cancer subtypes. We have previously reported widely aberrant methylation in ncRNA promoters in breast cancer [19] and have determined that this abnormal methylation is more frequent than that of protein-coding genes. Moreover, ncRNA expression is strictly regulated by histone modification, as shown by Saito et al. [20]. In addition, Scott et al. have found that treatment with histone deacetylase inhibitor leads to a rapid change in miRNA expression [21]. In lncRNAs, in contrast to miRNAs, the study of epigenetic regulation has just begun. Zheng et al. have identified a new lncRNA, designated SRHC, that is capable of inhibiting cancer proliferation and is down-regulated in tumours, at least partly by DNA methylation [22]. Sati et al. have performed genome-wide analysis of DNA methylation and histone modifications across the promoters of all known lncRNA genes in different cell and tissue types [23]. Their results suggest that lncRNAs have histone marks associated with active transcription, similar to protein-coding genes, although they differ in repressive marks such as DNA methylation and H3K9me3 histone modification. Although these reports have described epigenetic regulation in specific miRNAs and lncRNAs in cancer, the upstream epigenetic regulatory mechanisms that control breast cancer subtype-specific ncRNA expression remain unexplored.
In this study, which builds on the results of our recent studies, we carried out a series of integrative analyses to compare the alterations in six types of histone modifications (H3K4me1, H3K4me3, H3K27ac, H3K36me3, H3K9me3 and H3K27me3) and DNA methylation in the promoters of miRNAs and lncRNAs between two cell lines representing different breast cancer subtypes (luminal and basal). We found that alterations in epigenetic modifications in the two breast cancer subtypes affected not only protein coding genes but also many ncRNAs. Moreover, we found a combinatory effect of different epigenetic modifications on ncRNA expression in both cancer subtypes. Further, through integrative analysis of genetic and epigenetic data, we identified miRNA-gene modules associated with the survival of patients in each subtype. This study presents the aberrant epigenetic patterns of ncRNAs in two cancer subtypes, providing a valuable resource for the investigation of epigenetic regulation in breast cancer.
Materials and methods
Annotation of ncRNA gene promoters
The annotation information of miRNAs was obtained from miRBase V21 [24], a searchable database of published miRNA sequences and annotations. The majority of transcriptional start sites (TSSs) of miRNA genes were obtained from a previous study [25] that had integrated three different data sources and has identified 1286 miRNA gene TSSs. Because these data sources were based on miRBase V18, the TSSs of newly added miRNAs in V21 were defined as the start sites of primary miRNA transcripts. In addition, we obtained the miRNA family and cluster information from miRBase V21. A miRNA cluster is defined as a group of miRNAs located with a genomic distance of <10 kb.
The lncRNA gene annotation was collected from GENCODE V18 [26]. In total, 13 467 lncRNAs were analysed in our study. In this study, similar to previous studies [19, 27], the genomic regions 2 kb up- and downstream of miRNA or lncRNA TSSs were identified as promoters.
Genome-wide epigenetic modification and expression data in breast cancer subtypes
The genome-wide epigenetic modifications data were the same as those used in one of our previous studies [6], wherein the processing of raw data and identification of epigenetically dysregulated genomic regions were described. In total, we compared six types of histone modifications (H3K4me1, H3K4me3, H3K27ac, H3K36me3, H3K9me3 and H3K27me3) and DNA methylation between two cell lines (MCF-7 and HCC1954) representing two breast cancer subtypes (luminal and basal). As a control, the human mammary epithelial cell (HMEC) line was used. Differential histone modification regions (DHMRs) and differentially methylated cytosines (DMCs) identified in the previous work were directly used in the present study.
The miRNA expression data of hundreds of breast tumour samples were obtained from The Cancer Genome Atlas (TCGA) program (http://cancergenome.nih.gov/), including 89 luminal subtypes, 46 basal subtypes and 87 normal controls (Supplementary Table S1). Separate expression profiles of luminal and basal subtypes were extracted and combined into one single profile by using customized Perl scripts. In total, the expression levels of 1046 miRNAs were profiled. The RNA-seq data for three cell lines were downloaded from public resources, as described previously [6]. LncRNA expression was qualified by using the Cufflinks program [28], which assembles mapped reads with lncRNA annotation from GENCODE [26]. Furthermore, the lncRNA expression data of TCGA breast cancer samples were collected from MiTranscriptome [29], which has computationally analysed RNA-seq data derived from >1000 samples. In the present study, we mapped these lncRNAs to the GENCODE lncRNAs, and then an expression profile of 3164 lncRNAs was retained for each subtype. There were 431 luminal subtype, 141 basal subtype and 117 normal samples (Supplementary Table S1). In addition, we obtained the gene expression profiles of 433 luminal subtype and 140 basal subtype patients from TCGA (Supplementary Table S1). Finally, the survival data for the patients corresponding to these samples were also downloaded from the TCGA database.
Collection of breast cancer-associated ncRNAs and miRNA-gene regulations
Breast cancer-related miRNAs were retrieved from the following five databases: HMDD [30], miR2Disease [31], PhenomiR [32], SM2miR [33] and miREnvironment [34]. MiRNAs that were reported in at least one of these databases were considered in our research (Supplementary Table S2). Breast cancer-associated lncRNAs were obtained from the LncRNADisease database [35] and manually selected from the published literature [36] (Supplementary Table S2).
Experimentally validated miRNA-gene regulation was compiled from four different public databases: DIANA-TarBase v7.0 [37], miRTarbase [38], miRecords [39] and miR2Disease [31]. Targets from DIANA-TarBase v7.0 and miRTarbase were retrieved from the published literature on text mining techniques. miRecords and miR2Disease manually curate targets from selected publications. Gene targets without an Entrez ID were filtered out, thus resulting in 35 011 unique miRNA–gene target pairs among 273 miRNAs and 10 852 genes.
Identification of epigenetically dysregulated ncRNAs
Promoter regions of ncRNAs were searched for DHMRs or DMCs. MiRNAs and lncRNAs whose promoters overlap at least one DHMR or DMC were deemed ‘epigenetically dysregulated miRNAs or lncRNAs’. NcRNA promoters harbouring more than one differential epigenetic modification region that changed in different directions were filtered out in the subsequent analysis.
Classification of ncRNAs on the basis of epigenetic alterations
To investigate the effects of epigenetic alterations on transcription, we grouped the ncRNAs into three groups on the basis of epigenetic alterations. The increased active or decreased repressive epigenetic modifications might increase the expression of ncRNAs, whereas decreased active or increased repressive epigenetic modifications might decrease the expression. Thus, if epigenetic alterations for ncRNAs were all increased active or decreased repressive modifications, we classified those ncRNAs in the ‘Act’ group. In contrast, if epigenetic alterations for ncRNAs were all either decreased active or increased repressive modifications, we classified those ncRNAs in the ‘Rep’ group. The other epigenetically altered ncRNAs with mixed active and repressive modifications were classified in the ‘Ind’ group because we could not infer transcriptional alterations from mixed epigenetic alterations.
Functional analysis of epigenetically dysregulated ncRNAs
A list of Gene Ontology (GO) terms associated with cancer hallmarks was obtained from Plaisier et al. [40]. To investigate the functional significance of epi-modifications, we focused on ncRNAs that showed both differential epi-modifications and a corresponding distinct expression change as compared with the normal samples. The dysregulated miRNA-gene network was constructed for luminal and basal subtypes only using the cancer samples. In each subtype, the correlations between epigenetically dysregulated miRNAs and their targets were assessed by Pearson correlation using paired miRNA and gene expression data from the TCGA project. Only negatively correlated miRNA–gene pairs in each subtype were used for further analysis.
MiRNA functions were interrogated via their targets in biological processes associated with hallmarks of cancer. Specifically, for each epigenetically dysregulated miRNA, enrichment of a target gene set in cancer hallmark-associated GO terms was assessed by computation of a hypergeometric P-value with the Benjamini–Hochberg correction (false discovery rate, FDR). Biological processes that the epigenetically dysregulated lncRNAs might participate in were investigated through their co-expressed genes among TCGA samples. Potential co-expressed relationships between lncRNAs and candidate genes were assessed on the basis of Pearson correlation among TCGA samples of each breast cancer subtype. Co-expressed genes (P-adjusted < 0.05) for each epigenetically dysregulated lncRNA were grouped into one set in each subtype, and functional enrichment was performed as described for miRNAs.
Identification of epigenetic-mediated miRNA-gene prognostic biomarkers
Statistical analysis and data visualization
Statistical analyses in this study were conducted by using R statistical software [42]. P values or FDRs < 0.05 were regarded to be statistically significant. Heatmaps were presented by using the pheatmap program. Visualization of epi-modifications data was performed by using the Integrative Genomics Viewer tool [43].
Results
NcRNAs are differentially expressed in breast cancer subtypes
To determine whether ncRNAs are significantly differentially expressed between breast tumours and normal samples, we analysed their expression in breast tumours and normal breast tissue by using available next-generation sequencing data. We found that 511 (48.8%) miRNAs were differentially expressed and that 3580 (26.6%) lncRNAs exhibited at least a 2-fold change in breast tumours compared with normal breast tissue (Figure 1A, B). Furthermore, 79 and 157 miRNAs showed subtype-specific aberrant expression in luminal and basal cancer subtypes, respectively. Expression analysis revealed subtype-specific differential expression of 1224 lncRNAs in luminal and 511 lncRNAs in basal cancer subtype (Figure 1C, D). These results indicate that, in addition to the considerable fraction of ncRNAs that exhibit abnormal expression in breast tumours, a subset of ncRNA are differentially expressed in a subtype-specific manner.
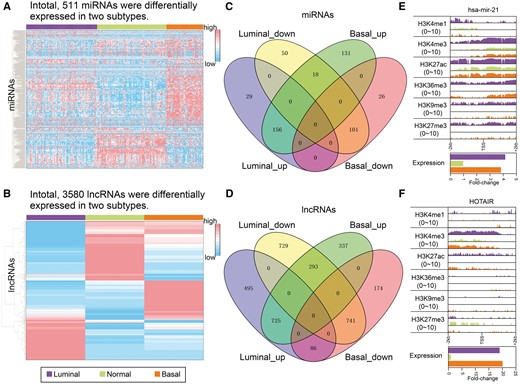
ncRNAs showed transcriptomic and epigenomic changes in breast cancer subtypes. (A) Heatmap of expression profiles of miRNAs demonstrating differential expression in breast cancer subtypes compared with normal breast tissues (Student’s t-test, adjusted P ≤ 0.05). (B) Heatmap of lncRNA expression profile in luminal (MCF-7) and basal (HCC1954) subtype cell lines and a normal breast cell line (HMEC). Expression of the presented lncRNAs showed changes of at least 2-fold in the breast cancer subtypes compared with the normal sample. (C) Overlap of differentially expressed miRNAs in the luminal and basal subtypes. (D) Overlap of differentially expressed lncRNAs in the luminal and basal subtypes. (E) Visualization of histone modifications within promoter regions (2 kb up- and downstream of TSSs, upper panel) and average relative expression abundance (lower panel) of mir-21. Expression abundance is shown relative to that detected in normal samples. (F) Visualization of histone modifications within promoter region (upper panel) and relative expression abundance (lower panel) of the lncRNA HOTAIR.
Chromatin modifications play a crucial role in transcriptional regulation, and subsets of genes are subject to epigenetic changes in breast cancer subtypes [6]. We postulated that aberrant chromatin modifications might also regulate numerous ncRNAs in breast cancer. MiR-21 has been shown to participate in process in various cancers, including breast cancer [44]. Overexpression of mir-21 has been observed in both luminal and basal subtypes (Figure 1E). Inspection of epigenetic modifications within the promoter region of mir-21 revealed incremental signals of H3K4me1, H3K4me3, H3K27ac, H3K36me3, H3K9me3 and H3K27me3 in the luminal subtype. The strongest changes were exhibited by H3K4me3 and H3K27ac, which may alter transcription (Figure 1E). However, only H3K36me3 showed an increased signal in the basal subtype compared with the normal sample. Another breast cancer-associated miRNA, mir-125b-1, displayed down-regulated expression in both luminal and basal subtypes (Supplementary Figure S1A). In the luminal subtype, apart from decreased H3K4me1/2/3 signals, mir-125b-1 showed decreased signals of H3K36me3. However, mir-125b-1 showed no alterations in H3K36me3. In addition to miRNAs, the lncRNA HOTAIR, which has been proven to promote cancer metastasis [45], showed significantly higher transcriptional abundance in both luminal and basal subtypes (Figure 1F). Consistently with these transcriptional changes, we determined that HOTAIR exhibited epigenetic changes within the promoter regions, wherein we observed increased H3K4me1, H3K4me3 and H3K27ac signals and a decreased H3K27me3 signal. Recent studies have shown that apoptosis is regulated by GAS5, a lncRNA aberrantly expressed in breast cancer [46, 47]. We observed GAS5 down-regulation in the luminal subtype and up-regulation in the basal subtype (Supplementary Figure S1B). Investigating the epigenetic abundances within the GAS5 promoter region in the luminal subtype revealed remarkably lower H3K4me1 signal and slightly decreased H3K27me3, in addition to slightly increased H3K27ac signal. In the basal subtype, we observed significantly higher abundance of the H3K36me3 signal, which might lead to the overexpression of GAS5.
Collectively, transcriptomic and epigenomic inspection in breast cancer subtypes revealed the abnormal expression and chromatin modification of numerous ncRNAs, including oncogenic miRNAs and lncRNAs. Furthermore, a subset of transcriptomic and corresponding epigenetic changes was observed to be subtype specific, thus indicating that dysregulation of ncRNAs in breast cancer subtypes might be under subtype-specific epigenetic control.
Aberrant epigenetic alterations are associated with ncRNAs in breast cancer subtypes
The above analyses indicated that ncRNA expression is regulated by epigenetic modifications in luminal and basal breast cancer subtypes. Next, we systematically analysed the DHMRs and DMCs in the context of ncRNA promoters. Annotation of 1286 miRNA TSSs was taken from a study by Chen et al., comprising 793 intragenic and 491 intergenic miRNAs. Interestingly, these intragenic and intergenic miRNAs had an analogous distribution in terms of distances between the TSSs and the corresponding start sites of the miRNA body (K-S test, P = 0.74, Supplementary Figure S2A). Similarly to observations for protein-coding genes, miRNAs and lncRNAs may also be subject to epigenetic control at the chromatin level, through modifications including H3K4me3 and H3K27ac (Supplementary Figure S2B–E).
Next, we dissected genome-wide epigenetic alterations within ncRNA promoter regions. Promoters overlapping with DHMRs or DMCs were identified, including 1150 (60.4%) miRNAs and 7178 (53.3%) lncRNAs in luminal subtype, and 1013 (53.2%) miRNAs and 6373 (47.3%) lncRNAs in basal subtype (Figure 2A and Supplementary Table S3). Notably, miRNAs exhibited a much higher aberrant frequency than did protein-coding genes in both subtypes (Figure 2A). In contrast to protein-coding genes, the majority of epigenetically deregulated ncRNAs were identified in both luminal and basal subtypes (Supplementary Figure S3A, C), thus suggesting that most of the epigenetically regulated ncRNAs participate in biological pathways underlying both subtypes. In addition, large set of these miRNAs were associated with breast cancer. Notably, 252 (21.9%) and 218 (21.5%) breast cancer-related miRNAs were identified in the luminal and basal subtypes, respectively (Figure 2B). Among these, 168 miRNAs were shared by both subtypes, and these miRNAs constituted most of the breast cancer-related miRNAs in either subtype (66.7% in luminal and 77.1% in basal) (Supplementary Figure S3A). Furthermore, most of the high-confidence breast cancer-related miRNAs were also among the shared miRNAs (Figure 2B). CpG islands (CGIs) have been shown to play an important role in the regulation of gene transcription [48], thus leading us to probe for CGIs in epigenetically dysregulated ncRNA promoters. Our results revealed that over half of the epigenetically altered miRNA promoters harboured at least one CGI in both luminal and basal subtypes (Supplementary Figure S3B), and this fraction was over one-third in the case of lncRNAs (Supplementary Figure S3D). This observation suggests that epigengetic marks may play roles in transcriptional regulation, and abnormalities in these marks are likely to contribute to the molecular mechanisms underlying breast cancer development.
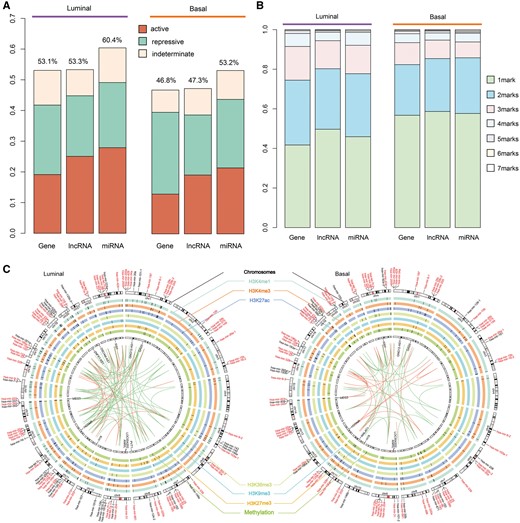
The majority of ncRNAs were epigenetically dysregulated in breast cancer subtypes. (A) The proportion of genes, lncRNAs and miRNAs that were epigenetically dysregulated in each breast cancer subtype. (B) The proportion of genes, lncRNAs and miRNAs with different numbers of aberrant epigenetic modifications. (C) The circles show epigenetically dysregulated miRNAs and lncRNAs in each subtype; only breast cancer-associated miRNAs and lncRNAs with high confidence TSSs are indicated. Red indicates the miRNAs common to both subtypes and black indicates subtype-specific miRNAs. Each circle presents a type of epigenetic modification; epigenetically dysregulated miRNAs and lncRNAs are marked in the corresponding locations. miRNAs of the same family are linked by lines.
Combined effects of epigenetic alterations on ncRNA expression in breast cancer subtypes
To gain further insight into how epigenetic alterations are organized in the promoter areas of ncRNAs, we examined the patterns of aberrant epigenetic marks. Both miRNAs and lncRNAs showed patterns similar to those of protein-coding genes (Figure 2B and Supplementary Figure S4A). MiRNA promoters with one type of epi-modification contributed a large fraction (45.9% and 57.7% in luminal and basal subtype, respectively) of epigenetically altered promoters in both subtypes (Figure 2B). The remaining promoters were regulated by combinations of two or more epi-modifications, which contribute to the high-order reprogramming of chromatin modifications in breast tumours. Despite the similar global distributions of epi-modification alterations within miRNA promoters in the luminal and basal subtypes, noticeably different patterns were observed. For singular epi-modifications, different types of modifications contributed to various extents in these subtypes. Specifically, many miRNA promoters exhibited epigenetic alterations in H3K4me1 and H3K27ac in the luminal subtype, whereas the basal subtype displayed predominantly H3K27ac and DNA methylation alterations (Supplementary Figure S4). Bivalent alterations also made a substantial contribution because co-occupancy of two epi-modifications constituted 31.8% and 28.1% of the epigenetically regulated miRNA promoters in the luminal and basal subtypes, respectively. In the luminal subtype, H3K4me1/H3K27ac and H3K4me3/H3K27ac were the most frequently observed bivalent alterations, whereas in the basal subtype, H3K27ac/DNA methylation and H3K4me1/H3K27ac were most frequently observed (Supplementary Figure S4). As for lncRNAs, virtually the same overall patterns were observed in both subtypes (Supplementary Figure S5).
Combinations of these epigenetic marks form an ‘epigenetic code’ that regulates the expression status of transcriptional units [49–51]. The frequent occupancy and remodelling of epigenetic marks in the promoter regions of ncRNAs led us to question whether the epigenetic alterations might control ncRNA expression, as previously observed for protein-coding genes. First, ncRNAs were classed into three clusters (active, repressive and indeterminate; details in Methods) according to the regulatory direction of the epigenetic changes within their promoter regions. We found that epigenetic modifications regulated most genes in the same direction (Figure 2A). Next, we investigated expression changes separately among the previously defined ‘active’, ‘repressive’ and ‘indeterminate’ classes of ncRNAs. Overall, ‘active’ miRNAs have a relatively higher level of expression changes than ‘repressive’ miRNAs in both subtypes (Figure 3A). We also observed marked differences in expression between these two classes for lncRNAs (Figure 3B). In addition, ‘indeterminate’ miRNAs and lncRNAs exhibited relatively intermediate expression shifts in both the luminal and basal subtypes, a result that might be attributed to antagonistic action between epigenetic modifications changing in different directions. To further investigate the association between patterns in epi-modification changes and ncRNA expression, ncRNAs were categorized into four groups according to the number of different types of epi-modification alterations that were co-located within promoter areas. We next observed that the effect of epi-modification alterations on transcriptions became more apparent as the number of types of altered epi-modifications increased, particularly for lncRNAs in both subtypes (Figure 3C, D). For example, lncRNAs with both H3K4me3 and H3K27ac were corresponded to higher expression changes that those with either H3K4me3 or H3K27ac in both subtypes (Supplementary Figure S6). Although it was not as apparent as for lncRNAs, a similar relationship between the numbers of altered epi-modifications and expression changes was observed for miRNAs (Supplementary Figure S7).
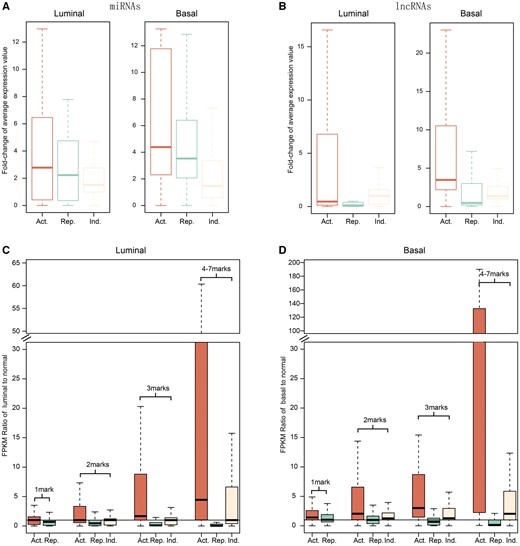
Alterations in epi-modifications affect ncRNA expression in both subtypes. (A) and (B) Fold change distributions of epigenetically dysregulated miRNAs and lncRNAs in luminal and basal breast cancer subtypes. (C) and (D) Altered expression of lncRNAs with different chromatin pattern categories. The x-axis indicates lncRNAs with different numbers and types of epi-alterations, and the y-axis represents the expression fold changes.
These observations suggested that various types of epi-modifications might work synergistically or antagonistically in modulating the transcription of ncRNAs that may play a crucial role in biological processes in breast cancer.
Complementary aberration patterns of miRNAs with identical seed sequences
Among the epigenetically alterated miRNAs, we found that the most miRNAs could be separated into miRNA families. Owing to their identical seed sequences, miRNAs in the same family may share common target genes. Thus, miRNA genes in the same miRNA families suggest shared functions and even transcriptional regulation [52]. However, whether miRNAs with identical seed sequences are subject to similar or complementary patterns of epi-modification changes remains unknown. To investigate this issue, we examined the epi-modification alteration profiles of epigenetically dysregulated miRNAs on the basis of miRNA family. ‘Epigenetically dysregulated miRNAs’ refers to miRNAs that show changes in both epi-modification and expression in each subtype. In the luminal subtype, epigenetically regulated miRNAs were grouped into 127 different families, 93 of which were overlapped with the basal subtype (Figure 4A, B). Next, we computed the proportion of miRNAs in the specific family that showed the same epigenetic dysregulations. However, we found that approximately 78.7% of members of common miRNA families showed distinct epi-modification aberrations between the two subtypes (Figure 4B). This result suggests that different members of miRNA families might work in combination to exert their functions in diverse cancer subtypes, and epigenetic regulation may affect these functions.
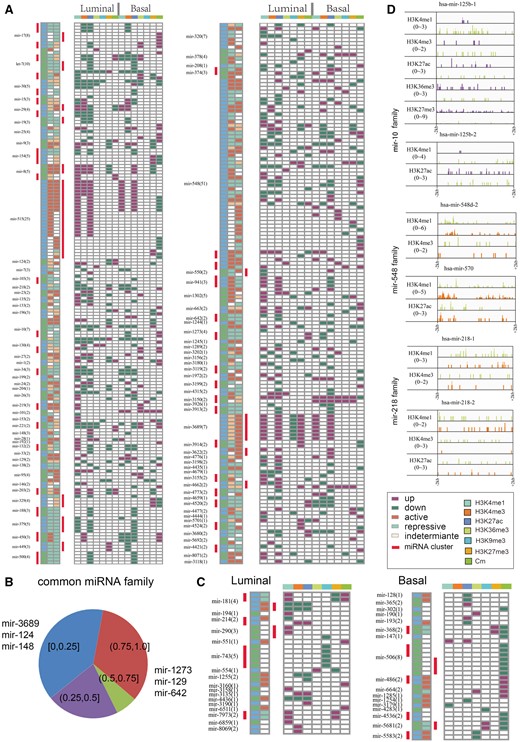
Complementary aberration pattern of miRNAs with identical seed sequences. (A) Common miRNAs that were epigenetically dysregulated in luminal and basal breast cancer subtypes. MiRNAs are ordered by miRNA family, and miRNA families are spaced by blue-green colour. miRNAs in the same clusters are marked by a red rectangle. Pink indicates increased epigenetic modifications and green indicates decreased epigenetic modifications. (B) The proportion of miRNA families with different number of members with the same epigenetic regulations. (C) Subtype-specific epigenetically dysregulated miRNA families. (D) Members of the same miRNA family with diverse alterations of epigenetic modifications.
Because some miRNA genes are located within a short distance of each other, thus forming miRNA clusters, and hence tend to be regulated by the same epi-modifications, we further analysed cluster distribution within these miRNA families. Intriguingly, for both subtypes, a subset of the miRNA families with accordant directions of epigenetic changes formed part of a single miRNA cluster. This observation suggests the efficiency of epigenetic regulation in modulating multiple miRNA genes in the same cluster, thus potentially affecting a broad range of targets. Furthermore, in both subtypes, almost half of the miRNA families with accordant directions of epigenetic changes had members that did not belong to the same cluster (Figure 4A, B). Apart from the most common miRNA family, we also observed families showing subtype-specific epigenetic regulations, 17 each in the luminal and basal subtypes (Figure 4C). Of particular interest was that miRNAs in these individual families were regulated by different epi-modifications in different subtypes. For example, mir-125b-1 and mir-125b-2 were grouped into the same family and were found to be down-regulated in the luminal subtype. However, these miRNAs were regulated by different epigenetic marks in this subtype. Specifically, decreased signals of H3K4me1, H3K4me3, H3K36me3, H3K27ac and increased H3K27me3 were observed within the promoter region of mir-125b-1 in luminal subtype, whereas mir-125b-2 showed decreased levels of H3K4me1 and H3K27ac (Figure 4D). In addition, we found that mir-125b-1 was also down-regulated in the basal subtype, which may have been the result of decreased H3K4me1, H3K4me3 and H3K36me3 signals as well as increased H3K27me3. Furthermore, some miRNA family members were regulated by disparate epi-modifications in the basal subtype. For instance, mir-548d-2 and mir-570, which are in the same family, were both up-regulated. but by different epigenetic marks. Notably, an increase in H3K4me3 and H3K4me1 signals was detected in the promoter region of mir-548d-2 compared with the normal sample (Figure 4D). However, increased H3K4me1 and H3K27ac signals were observed within the promoter region of mir-570 (Figure 4D). Analogously, mir-218-1 and mir-218-2 were epigenetically down-regulated by different epi-modifications in the basal subtype. Mir-218-1 exhibited a decrease in only H3K4me3 and H3K4me1 signals, whereas mir-218-2 exhibited a decrease in the H3K27ac signal (Figure 4D). These observations highlight the complementary dysregulation of epi-modifications, in which members of the same miRNA family converged in the same direction as a result of diverse epi-modifications. This complementary mode of aberrations further suggests that these miRNAs play important oncogenic roles in breast cancer.
Epigenetically mediated miRNA regulatory modules are associated with prognosis in breast cancer subtypes
Next, we further interrogated the roles of aberrant epi-modifications in breast cancer subtypes. Specifically, we focused on the ncRNAs that showed both differential epi-modifications and corresponding expression changes. Functional enrichment analysis was performed on these ncRNAs. It is well known that miRNAs perform their important functions via modifying the protein expression of their target genes [53]. Therefore, functional enrichment was performed on the set of target genes for each epigenetically dysregulated miRNA (see Methods). During their multi-step development, human tumours are driven by an organizing principle characterized by several hallmarks of cancer [54]. Accordingly, we investigated miRNA function in the context of cancer hallmarks. Among epigenetically dysregulated miRNAs, 14 and 12 miRNAs were particularly enriched in cancer hallmarks in the luminal and basal subtype, respectively, and 38 dysregulated miRNAs were shared by both subtypes (Supplementary Figure S7 and Table S4). Although these hallmarks represent notable cancer traits, they are acquired or lost alternately during the evolution to malignant cancer. Our results showed that most of the epigenetically dysregulated miRNAs were associated with four hallmarks (Supplementary Figure S8), including the ‘tumor promoting inflammation’, ‘tissue invasion and metastasis’, ‘limitless replicative potential’ and ‘evading immune detection’ hallmarks. Because the general large-scale functional annotation of lncRNAs has been based on the ‘guilt-by-association’ principle, we applied this principle to predict the probable functions of the dysregulated lncRNAs identified above. Functional analysis of epigenetically dysregulated lncRNAs also showed distinct hallmarks between luminal and basal subtypes (Supplementary Figure S9 and Table S5). These results indicated that some ncRNAs are regulated via epi-modification marks, which promote or impede tumour progressions, and that the variety of epigenetic dysregulation in different subtypes might lead to diverse characteristics of tumour cells.
Recent research on the biological significance and the promising prognostic value of miRNA-mediated regulatory networks in cancer prompted us to further investigate the prognostic value of these epigenetically regulated miRNAs in breast cancer subtypes. To interrogate the epigenetically mediated regulatory networks that may play a role during the development of breast cancer subtypes, we focused on miRNA-gene regulations in key breast cancer pathways (Figure 5A). To explore the prognostic values of these epigenetically regulated modules in each subtype, Cox regression analysis was performed to evaluate the significance of the association between the expression of each miRNA/gene and overall survival (see Methods). Consequently, our analysis revealed six bicliques in the luminal subtype and five bicliques in the basal subtype that were able to significantly distinguish patients in high-risk groups from those in low-risk groups in terms of overall survival (Supplementary Table S6). We found that some miRNAs and genes, such as miR-21-3p and miR-7-5p, CCND1 and PIK3R3, were associated with patient survival in both subtypes (Figure 5B). Present and previous findings show that miR-21 is significantly elevated in invasive breast carcinomas compared with healthy breast tissue. A previous analysis has also shown that changes in miR-21 levels might be important in the later and/or late phases of breast carcinogenesis rather than in the initial early phases [55]. In addition, the overexpression of miR-7-5p has been shown to inhibit cell proliferation and induce apoptosis [56]. These findings suggest that these miRNAs and genes may play key roles in both subtypes. Moreover, subtype-specific miRNA-gene modules were also identified (Figure 5C, D). A module consisting of five miRNAs and one gene (NCOA3) was observed to be associated with survival in patients with luminal, but not basal, cancer. We also found that NCOA3 was specifically epigenetically dysregulated in luminal subtype cases (Figure 5A). Evidence has shown that NCOA3 is an oestrogen receptor α (ERα) co-activator that is, amplified and overexpressed in a fraction of breast cancers [57]. Our observations might be helpful to identify those ERα-positive breast cancer patients who are candidates for adjuvant miRNA-based therapy. However, some basal-specific modules were also identified, such as the module comprising miR-100b-5p, FGFR3 and IGF1R (Figure 5D). IGF1R was observed to be epigenetically dysregulated specifically in the basal subtype and has been demonstrated to be a target of basal-specific drugs (Figure 5A). These results suggest that ncRNAs and protein-coding genes may cooperatively mediate pathway dysregulation during the development and progression of breast cancer.
![Epigenetically dysregulated ncRNAs regulate extensive pathways and are associated with cancer progression. (A) The core part of this pathway was obtained from SnapShot from the journal Cancer cell. The digits on the top and bottom of each gene mark the type of epi-alteration in luminal and basal subtypes, respectively. Red colour indicates up-regulation, and green represents down-regulation of epigenetic modifications. The subtype-specific drug targets were obtained from a previous study (see details in [6]) and are also noted in the figure. The miRNA and lncRNA regulators were added and the digits on the top and bottom of each miRNA or lncRNA mark the type of epi-alteration in luminal and basal subtypes. The miRNA-gene links show miRNA-gene regulations and the differentially expressed lncRNAs that were linked to genes via coexpression. (B) miRNA-gene modules associated with patient survival in luminal and basal breast cancer subtypes. (C) miRNA-gene modules associated with the patient survival in luminal subtype patients only. (D) miRNA-gene modules associated with patient survival in basal subtype only.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/bib/19/1/10.1093_bib_bbw099/4/m_bbw099f5.jpeg?Expires=1749227972&Signature=vcRdU-lXyiwVVAeKSx-Yel~IgrbIdnMV1IYqCMpfZAD0IPAn5ADojnH7DFaojTvQk~JhzxUZK3Ayi5UySzihA68izGsFFEMntvjdY~djQqc-OmiSiCgD4lMKwrNNveP560Ta580fnhjyDyzBbSXTIWWPoEOapWab4jZfZDxWyhBbovouudVF0H3VPzhS5rmeG03LdcqS0Adt0G1lxsEm~H~iIpSXTN6FdpbszE9QwDVMA8y-dZZ1wiGMaLcoH4SacVedZ1Y5UCNiRZg9aTkQPaQIT8mdc72SF8PTCDjbmFvHi2nhe53wSQ-2qzQmpHsKGrJcA4AvSGmtNNufSO210g__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Epigenetically dysregulated ncRNAs regulate extensive pathways and are associated with cancer progression. (A) The core part of this pathway was obtained from SnapShot from the journal Cancer cell. The digits on the top and bottom of each gene mark the type of epi-alteration in luminal and basal subtypes, respectively. Red colour indicates up-regulation, and green represents down-regulation of epigenetic modifications. The subtype-specific drug targets were obtained from a previous study (see details in [6]) and are also noted in the figure. The miRNA and lncRNA regulators were added and the digits on the top and bottom of each miRNA or lncRNA mark the type of epi-alteration in luminal and basal subtypes. The miRNA-gene links show miRNA-gene regulations and the differentially expressed lncRNAs that were linked to genes via coexpression. (B) miRNA-gene modules associated with patient survival in luminal and basal breast cancer subtypes. (C) miRNA-gene modules associated with the patient survival in luminal subtype patients only. (D) miRNA-gene modules associated with patient survival in basal subtype only.
Collectively, our observations suggest a crucial role of epigenetically regulated regulatory networks in the development of breast cancer subtypes. The extracted miRNA-gene modules that can discriminate high-risk from low-risk patients may lead to a promising clinical applications for the prognostic and surgical management of breast cancer.
Discussion
In this study, we characterized changes in epi-modifications of ncRNAs in luminal and basal breast cancer subtypes. Our study revealed that a large number of ncRNA promoters were epigenetically regulated. Unlike protein-coding genes, the majority of epigenetically deregulated ncRNAs (approximately 70%) were shared by both luminal and basal subtypes. However, a subset of transcriptomic and corresponding epigenetic changes did show a subtype specificity, consistently with the results obtained for protein-coding genes. In addition, various types of epi-modifications might synergistically modulate ncRNA. Functional enrichment analysis indicated that epigenetically dysregulated ncRNAs were significantly involved in the hallmarks of cancers. In addition, we collected the breast cancer-related miRNAs in some databases and observed that about 70% of epigenetically dysregulated miRNAs were reported in more than one database, and with the increase of reporting rates, the percent also increased (Supplementary Figure S10). This observation provides novel evidence of the mechanisms behind ncRNA dysregulation in breast cancer subtypes. Moreover, this study contributes to the understanding of how methylation and histone modifications influence ncRNA expression and suggests novel miRNAs and lncRNAs that may be important in breast cancer subtypes.
Although members of the same miRNA family often reside on different chromosomes, they have probably arisen from gene duplication events [58], and their regulatory regions might be conserved among family members. Previous evidence has shown that miRNA family members share transcription factors [59]. Our observations also highlight the complementary dysregulation of epi-modifications, in which miRNA members within the same family converge to the same direction as a result of diverse epi-modifications. Hence, diverse epigenetic control elements are another mechanism for regulation of the expression of miRNA family members.
Tumorigenesis is a complex and dynamic biological process that involves multiple steps of genetic and epigenetic alterations. However, understanding of this intricate network of coding genes and ncRNAs in cancer biology remains at an early stage. In this study, we found that lncRNAs and miRNAs regulate key components of breast cancer-associated pathway (Figure 5). We observed that the majority of ncRNAs show consistent expression patterns in both subtypes, thus suggesting some molecular mechanisms are shared by these two subtypes. However, the common ncRNAs were altered by different types of epigenetic regulations in a subtype-specific manner. For example, miR-200b-3p was up-regulated in both subtypes, but through analysis of its epigenetic regulation, we observed elevated H3K4me1, H3K4me3, H3K27ac and decreased H3K27me3 in the luminal subtype but only elevated H3K27ac and decreased H3K27me3 in the basal subtype. Investigation of these ncRNAs might provide new approaches for the subtype-specific treatment of breast cancer. Together, these findings indicate that aberrant epigenetic regulations of ncRNA regulators might subsequently mediate pathway dysregulation and lead to the development and progression of different types of breast cancer.
In this study, we integrated multiple omic data sets to identify the epigenetically dysregulated ncRNAs. There is much scope to improve our understanding of the ncRNA regulation in human complex diseases. The TSSs of miRNAs or lncRNAs used in our analyses were obtained independently from those of mRNAs, which were also supported by the enrichment of H3K4me3 signals (Supplementary Figure S2). In the present analyses, the aberrantly epigenetically dysregulated miRNAs, lncRNAs and protein coding genes were identified separately by overlapping the DHMRs or DMCs to their promoter regions. We computed the proportion of epigenetically dysregulated sites that were overlapped with both miRNAs (or lncRNAs) and mRNAs, and found on average 7.30% and 7.04% of epigenetic altered sites in luminal and basal subtypes, respectively. This observation suggests that most of the miRNAs might be regulated by independent epigenetic sites. Of these overlapping sites, only 22.0% and 15.6% sites were associated with the changes of transcriptional levels for both mRNAs and miRNAs in luminal and basal subtypes. On the other hand, the proportions for lncRNAs in these two subtypes were similar to those for miRNAs. Thus, integrating the transcriptome data sets to further identify the epigenetically and transcriptionally dysregulated ncRNAs may support the regulation of these epigenetic sites to ncRNAs. We believed that identifying the more sophisticated TSSs of ncRNAs in the future will enhance our understanding of ncRNA regulations.
In summary, our data demonstrate the suitability of integrating multiple omics data sets to identify novel epigenetically dysregulated ncRNAs (lncRNAs and miRNAs) in different breast cancer subtypes. This study presents the aberrant epigenetic patterns of ncRNAs, thus providing a highly valuable resource for further investigation of the epigenetic regulation of breast cancer subtypes.
Systematic comparative analyses of epigenetic regulation in two breast cancer subtypes revealed a large number of epigenetically dysregulated ncRNAs.
Various types of epi-modifications might alter synergistically or antagonistically to modulate the transcription of miRNAs and lncRNAs in both breast cancer subtypes.
We highlighted the complementary dysregulation of epi-modifications of members within the same miRNA family, which were converged to the same directed alterations as a result of diverse epi-modifications.
Analysis of epigenetic modification-mediated miRNA regulatory networks revealed that cancer progression is associated with specific miRNA-gene modules in both cancer subtypes.
Funding
The National High Technology Research and Development Program of China (863 Program, Grant No. 2014AA021102), the National Program on Key Basic Research Project (973 Program, Grant No. 2014CB910504), the funds for Creative Research Groups of the National Natural Science Foundation of China (Grant No. 81421063), the National Natural Science Foundation of China (Grant Nos. 91439117, 61473106, 31571331 and 61502126), the China Postdoctoral Science Foundation (Grant Nos. 2016T90309, 2014T70364, 2015M571436 and LBH-Z14134), Natural Science Foundation of Heilongjiang Province (Grant No. QC2015020), Weihan Yu Youth Science Fund Project of Harbin Medical University, Harbin Special Funds of Innovative Talents on Science and Technology Research Project (Grant No. RC2015QN003080).
Juan Xu is an associate professor in the College of Bioinformatics Science and Technology at Harbin Medical University, China. Her research activity is focused on ncRNA regulation and epigenetic in complex diseases.
Zishan Wang is a MS student in the College of Bioinformatics Science and Technology at Harbin Medical University, China.
Shengli Li is a MS student in the College of Bioinformatics Science and Technology at Harbin Medical University, China.
Juan Chen is a MS student in the College of Bioinformatics Science and Technology at Harbin Medical University, China.
Jinwen Zhang is a MS student in the College of Bioinformatics Science and Technology at Harbin Medical University, China.
Chunjie Jiang is a MS student in the College of Bioinformatics Science and Technology at Harbin Medical University, China.
Zheng Zhao is a MS student in the College of Bioinformatics Science and Technology at Harbin Medical University, China.
Jing Li is a professor in The 1st Affiliated Hospital of Heilongjiang University of Chinese Medicine, China.
Yongsheng Li is an associate professor in the College of Bioinformatics Science and Technology at Harbin Medical University, China. His research interests focus on epigenetic regulation and bioinformatic methods development.
Xia Li is a professor in the College of Bioinformatics Science and Technology at Harbin Medical University, China. Her research interests focus on computational system biology and ncRNA regulation in human diseases.
References
Author notes
Juan Xu, ZiShan Wang and Shengli Li authors contributed equally to this work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}