




Abstract
RNA structure plays a crucial role in gene maturation, regulation and function. Determining the form and frequency of RNA folds is essential for a better understanding of how RNA exerts its functions. Low-throughput studies have focused on RNA primary sequences and expression levels, but with an emphasis on relatively small numbers of transcripts. However, with the recent advent of high-throughput technologies, it is realistic to begin analyzing RNA secondary structures on a genome-wide scale. Here, we review genome-wide RNA secondary structure profiles as well as advances in computational structure predictions. We further discuss the novel characteristics of RNA secondary structure across messenger RNAs. Probing RNA secondary structure by high-throughput sequencing will enable us to build atlases of RNA secondary structures, an important step in helping us to understand the versatility of RNA functions in diverse cellular processes.
Introduction
Insights into the RNA structure–function relationship have caused a major shift in our understanding of the RNA molecule and its biological role. The sequence of RNA is much more than the mediator of genetic code between DNA and protein in the central dogma. Rather, through modulating folding, it has profound consequence for a transcript’s function [ 1 ]. RNA structures influence almost every step of a transcript’s life including transcription, splicing, cellular localization, translation and turnover of the RNA [ 2–7 ]. RNA structures can provide crucial insights into their functions in multiple cellular processes. RNA structure is divided into three levels, i.e. primary, secondary and tertiary. The primary structure of an RNA molecule is the sequence of nucleotides, over the four letters of the alphabet (A, U, C, G), along the polynucleotide chain. By convention the sequence is given from 5′ to 3′. The secondary structure of an RNA molecule is the pattern of hydrogen bonding (base pairing) between bases along the polynucleotide chain. The tertiary structure of an RNA molecule is the final 3D shape into which it folds, defined by both the secondary structure hydrogen bonding and other interactions between nucleotides. As a consequence, secondary structure prediction is a much more tractable problem for computational biologists than tertiary structure prediction. The vast majority of algorithms designed to tackle RNA structure prediction focus solely on secondary structure.
The secondary structure of several classes of RNAs, such as ribosomal RNAs, transfer RNAs and riboswitches, have been studied in detail [ 2 ]. Classical approaches, generally, use small molecules, ions or strand-specific enzymes to cleave or modify the RNA and then detect the cleavages or modifications via labeled primer extension, reverse transcription (RT) and polyacrylamide gel electrophoresis analysis [ 2 ]. However, the structural information for most RNAs has been missing owing to the difficulty in probing long RNAs and the low-throughput characteristic of traditional methods.
High-throughput sequencing is being applied in many fields, such as DNA–protein interactions (ChIP-seq), mapping RNA binding sites on a genome-wide scale (CLIP-seq), transcriptome annotation (RNA-seq), gene discovery (DNA-seq) and DNA-methylation detection (MeDIP-seq). Recently, several approaches have combined RNA structural methods with high-throughput sequencing to obtain genome-wide RNA structural information at single nucleotide resolution. These include Parallel Analysis of RNA Structure (PARS) [ 8 ], Frag-seq [ 9 ], selective 2′-hydroxyl acylation analyzed by primer extension sequencing (SHAPE-seq) [ 10 ], Structure-seq [ 11 ], dimethyl sulphate-seq (DMS-seq) [ 12 ] and Mod-seq [ 13 ]. These methods can provide information about secondary structures of thousands of transcripts in the cell at a time.
The idea of computationally predicting RNA secondary structure is not new, though it has become more feasible on a genome-wide scale as processing power has increased and our understanding of the folding process has developed. Many algorithms have been developed for predicting RNA secondary structure, and their prediction accuracies are steadily improving. The Vienna RNA package [ 14 ] is a comprehensive collection of tools that provides many algorithms for computation, comparison and analysis of RNA secondary structures and properties, which are based on dynamic programming algorithms. The core of the package aims at predicting RNA secondary structure with minimum free energies as well as at computations of the equilibrium partition functions and base-pairing probabilities. The package includes a series of tools, such as RNAfold (structure prediction of single sequences), RNAalifold (consensus structure prediction on a set of aligned sequences), RNAinverse (sequence design), RNAcofold (computing the hybridization energy and base-pairing pattern of two RNA sequences), RNAup (predicting RNA–RNA interactions) and the barriers program (considering kinetics of RNA folding).
As some long RNAs may have multiple structures, it is interesting to represent the ensembles of probable structures. Sfold [ 15 ], a statistical sampling algorithm, has been developed for taking a representative sample of structures from the Boltzmann ensemble of RNA secondary structures. The resulting structures are clustered into sets of similar structures and they identify the cluster centroids as representative of the most likely structure of the RNA, rather than the minimize the free energy (MFE) structure. The probability of any structural motif can be estimated by this algorithm. The sampling strategy has a wide application, such as prediction of messenger RNA (mRNA) secondary structure and target accessibility. RNAshapes [ 16 , 17 ] is a software package that based on the abstract shapes approach, which is a viable alternative to Sfold. RNA shape abstraction maps structures to a tree-like domain of shapes, which retains adjacency and nesting of structure features, but disregards helix lengths. Shape abstraction can be applied during structure prediction rather than afterward as it integrates well with dynamic programming algorithms.
Databases providing information about the structures of nucleic acids and proteins are important resources in bioinformatics and molecular biology. This importance is reflected by the increasing number of databases being developed and made publicly available such as Rfam [ 18 ]. It is the major resource today to model structural RNAs. It is represented by multiple sequence alignments and covariance models (CMs). This database integrates structural RNA alignments into a common structure-annotated format, uses CM software and provides a system for automatically analyzing and annotating sequences. In Rfam, a seed alignment contains known representative members of family and is hand-curated and annotated with structural information. It is also used to build a CM which is then used to search a nucleotide sequence database using the CMSEARCH program. The program reports scores for matches to the model, and family-specific threshold is chosen and then the matches are aligned to models using the program CMALIGN.
With the advent of technologies such as high-throughput sequencing, genome-wide RNA structural measurements can be integrated into computational algorithms to obtain more accurate structural features. Recently, an integrative approach, Seqfold [ 19 ], for tackling this issue has been developed, which can combines RNA secondary structure profiling data, obtained by PARS, FragSeq, SHAPE-seq and so on, with computational prediction algorithms.
The advent of these integrative approaches is providing insights into the genome-wide relationships between RNA structures and functions. Here, we review advances in genome-scale RNA secondary structure profiles and then focus on computational structure predictions. We also discuss novel characteristics of RNA secondary structure across mRNAs.
Genome-wide and high-throughput methods for RNA secondary structure detection
Classical low-throughput approaches
Traditionally, the chemicals and enzymes that either cleave or modify single- or double-stranded regions can be used to probe the secondary structure of an RNA molecule of interest in solution, one molecule at a time [ 20–22 ]. RNA secondary structure was commonly measured using the nucleases cleavage method. Generally, single-stranded regions of RNA have high probabilities to be the specific cleavage sites of nucleases, such as nuclease S1, nuclease P1, RNase T1, RNase U2 and RNase A. RNase V1 prefers to specifically cleave double-stranded regions of RNA [ 23 ], and so is a popular probing method to interrogate helical regions directly. Additionally, RNase V1 can also cleave stacked but single-stranded nucleotides.
Chemical reagents are much smaller in size than nucleases and so suffer less steric hindrance and can probe more nucleotides in a folded RNA [ 9 ]. Chemical probing methods have been widely applied to measure RNA secondary structures [ 2 , 24 ]. Generally, chemical probes are preferred for single-stranded regions of RNAs. There are many chemicals used for probing RNA secondary structures. Metal ions are the smallest and simplest reagents, and are particularly useful for unconstrained nucleotides. For instance, Mg 2+ -assisted in-line probing was used to study riboswitches [ 25 ]. There are two major categories of chemicals widely used today. One type is base-specific, including DMS, which reacts with unconstrained adenine and cytosine, kethoxal, which primarily targets guanosine, and CMCT, which mainly modifies uracil. Members of the other type react with the RNA backbone and so allow all four nucleotides to be detected at the same time in a single experiment [ 20 , 26 , 27 ]. These include hydroxyl radicals and SHAPE-reagents, containing N -methylisatoic anhydride, 1-methyl-7nitroisatoic anhydride (1M7) and benzoyl cyanide. Hence, combining different experiments with multiple chemical probes can provide complementary information on RNA secondary structures.
Traditionally experimental probing of RNA molecules involved chemically modifying RNA by the reagents and quantifying the fragments resulting from either RNA cleavage or primer extension in gel electrophoresis or capillary electrophoresis. The location of cleaved or modified nucleotides typically indicates the pattern of base-pairing or RNA secondary structure. The classical techniques have yielded many important insights to our understanding of RNA structure–function relationships. However, the steps required to read out the information are time-consuming and labor-intensive. Furthermore, de novo probing of RNAs of unknown sequence is impossible with the classical approach because the identity and sequence content of the RNA needs to be known to design primers for RT [ 28 ].
Given the limitation of traditional technologies to measure RNA secondary structure by nucleases cleavage or chemical reagents, there is a need for novel methods to enable genome-wide information to be obtained. The arrival of next-generation sequencing technologies, which can produce a huge amount of data cheaply and quickly, makes it easy to obtain genome-scale information [ 29 ]. We are now seeing experimental protocols merging chemicals experiments with high-throughput sequencing technology to enable genome-scale measurement of RNA secondary structures ( Table 1 ). This is leading to a better understanding of the global features of RNA secondary structures.
The summary of high-throughput sequencing of RNA secondary structure
| Methods | Types | Reagents | Conditions | Species | Year | Reference |
|---|---|---|---|---|---|---|
| PARS | Nucleases digestion | Nuclease V1 and S1 | In vitro | yeast | 2010 | [ 8 ] |
| Fragseq | Nucleases digestion | Nuclease P1 | In vitro | mouse | 2010 | [ 9 ] |
| SHAPE-seq | Chemicals modification | 1M7 | In vitro | Bacillus subtilis | 2011 | [ 10 ] |
| Structure-seq | Chemicals modification | DMS | In vivo | Arabidopsis | 2014 | [ 11 ] |
| DMS-seq | Chemicals modification | DMS | In vivo | human | 2014 | [ 12 ] |
| Mod-seq | Chemicals modification | DMS | In vivo / in vitro | yeast | 2014 | [ 13 ] |
| Methods | Types | Reagents | Conditions | Species | Year | Reference |
|---|---|---|---|---|---|---|
| PARS | Nucleases digestion | Nuclease V1 and S1 | In vitro | yeast | 2010 | [ 8 ] |
| Fragseq | Nucleases digestion | Nuclease P1 | In vitro | mouse | 2010 | [ 9 ] |
| SHAPE-seq | Chemicals modification | 1M7 | In vitro | Bacillus subtilis | 2011 | [ 10 ] |
| Structure-seq | Chemicals modification | DMS | In vivo | Arabidopsis | 2014 | [ 11 ] |
| DMS-seq | Chemicals modification | DMS | In vivo | human | 2014 | [ 12 ] |
| Mod-seq | Chemicals modification | DMS | In vivo / in vitro | yeast | 2014 | [ 13 ] |
The summary of high-throughput sequencing of RNA secondary structure
| Methods | Types | Reagents | Conditions | Species | Year | Reference |
|---|---|---|---|---|---|---|
| PARS | Nucleases digestion | Nuclease V1 and S1 | In vitro | yeast | 2010 | [ 8 ] |
| Fragseq | Nucleases digestion | Nuclease P1 | In vitro | mouse | 2010 | [ 9 ] |
| SHAPE-seq | Chemicals modification | 1M7 | In vitro | Bacillus subtilis | 2011 | [ 10 ] |
| Structure-seq | Chemicals modification | DMS | In vivo | Arabidopsis | 2014 | [ 11 ] |
| DMS-seq | Chemicals modification | DMS | In vivo | human | 2014 | [ 12 ] |
| Mod-seq | Chemicals modification | DMS | In vivo / in vitro | yeast | 2014 | [ 13 ] |
| Methods | Types | Reagents | Conditions | Species | Year | Reference |
|---|---|---|---|---|---|---|
| PARS | Nucleases digestion | Nuclease V1 and S1 | In vitro | yeast | 2010 | [ 8 ] |
| Fragseq | Nucleases digestion | Nuclease P1 | In vitro | mouse | 2010 | [ 9 ] |
| SHAPE-seq | Chemicals modification | 1M7 | In vitro | Bacillus subtilis | 2011 | [ 10 ] |
| Structure-seq | Chemicals modification | DMS | In vivo | Arabidopsis | 2014 | [ 11 ] |
| DMS-seq | Chemicals modification | DMS | In vivo | human | 2014 | [ 12 ] |
| Mod-seq | Chemicals modification | DMS | In vivo / in vitro | yeast | 2014 | [ 13 ] |
High-throughput approaches for measuring RNA secondary structures in vitro
An approach, termed PARS, was developed to profile the secondary structure of all mRNA in budding yeast [ 8 ]. The structural information of >3000 distinct transcripts was obtained for a single point of time. The method first extracts polyadenylated transcripts from the log phase and renatures these transcripts in vitro , before using a strand-specific nuclease to treat the resulting library. RNase V1 preferentially cleaves double-stranded RNAs (dsRNAs) while RNase S1 tends to cleave single-stranded RNAs (ssRNAs). Subsequently, V1- and S1-generated fragments are separately captured and undertake deep sequencing. A nucleotide-resolution score is obtained by combining the two complementary experiments ( Figure 1 A). The higher the score, the more possibility to be double stranded [ 8 ].

Approaches for measuring RNA structures via nuclease digestion. ( A ) Overview of PARS. In PARS, nuclear RNA is folded in vitro and RNase V1 or S1 is used to digest the double- and single-stranded regions, respectively. After random fragmentation, deep sequencing is performed. A PARS score is calculated at each base, with a positive PARS score indicating that a base is double stranded, and a negative PARS score indicating that a base is single stranded. ( B ) Overview of Fragmentation sequencing (Frag-seq). Nuclear RNA is folded in vitro and treated with P1 endonuclease that cleaves at single-stranded regions, which leaves a 5′P group. This 5′P is captured by adaptor ligation followed by RT and high-throughput sequencing. Controls of endogenous 5′P and 5′OH are also prepared in parallel. A cutting score can be calculated, at each base, that incorporates reads from P1 nuclease and reads from controls. A positive cutting score indicates that the base is single stranded.
FragSeq is another high-throughput approach to probe short noncoding RNA (ncRNA) secondary structures in mouse cell lines in vitro [ 9 ]. It uses nuclease P1 to cleave ssRNAs followed by deep sequencing ( Figure 1 B). The products of endonuclease P1 are 5′ monophosphate and 3′ OH. In this method, three treatments are carried out: one is nuclease P1 treatment, one is control (without nuclease digestion), and the other is polynucleotide kinase (PNK) and ATP treatment. The control treatment was used to exclude the endogenous 5′ phosphate, and the PNK and ATP treatment was used to estimate endogenous breaks without leaving 5′ phosphate and 3′ OH. The cutting scores for each site represent the probability of being single stranded. FragSeq revealed RNA structural characteristics and provided meaningful insights into their relationship between RNA structures and their functions [ 9 ].
PARS and FragSeq both measure RNA secondary structures via nucleases digestion, while SHAPE-seq is a high-throughput method for detecting RNA secondary structures by using chemical modification [ 10 , 30 ]. SHAPE-seq combines SHAPE chemistry with next-generation sequencing of primer extension products to obtain RNA secondary structure information ( Figure 2 A). SHAPE reagents react with the RNA backbone without nucleotide bias, probing the four nucleotides simultaneously [ 10 , 31–33 ]. The reactivities calculated by maximum likelihood estimation at each nucleotide can be scaled and used in RNA prediction programs such as RNAstructure [ 32 ] to infer secondary structure. Sequencing the 3′ RNA bar codes can determine simultaneously structural information about multiple RNAs. Additionally, SHAPE-seq has the ability to detect subtle conformational changes in RNA secondary structures owing to single nucleotide point mutations. The structural changes resulting from mutations can be measured by comparing the reactivities of the mutation sites obtained via SHAPE-seq or capillary electrophoresis (SHAPE-CE) separately. SHAPE-seq was a large breakthrough toward the measurement of RNA secondary and tertiary structures [ 10 ].
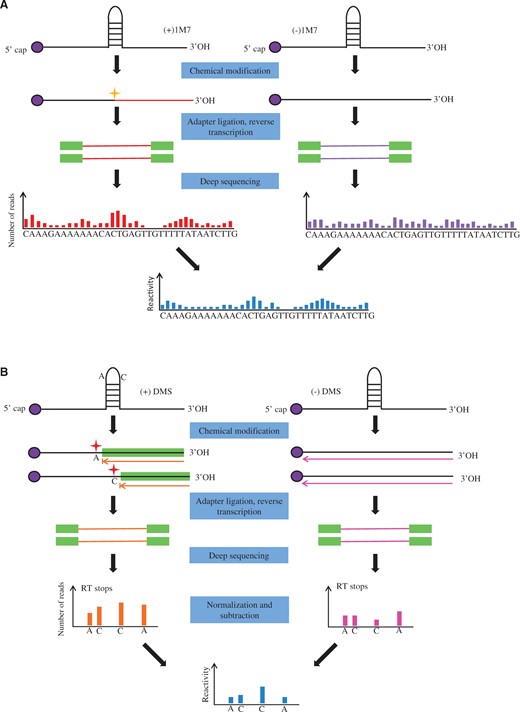
Approaches for measuring RNA structures via chemical probing. ( A ) Overview of SHAPE-Seq. Nuclear RNA is folded in vitro and modified by 1M7. Adapter ligation and RT is followed by SHAPE chemistry, sequencing library generation and next-generation sequencing. Raw read counts at each nucleotide position in the (+) and (−) treatments are calculated to determine the reactivities at each nucleotide. ( B ) Overview of structure-seq. Arabidopsis seedlings are treated with DMS. RT is performed after adaptor ligation. Reverse transcriptase stalls one nucleotide before DMS-modified A s and C s. Double-stranded DNA is generated by PCR. (−)DMS library is prepared in parallel. Deep sequencing is performed with different indices for (+)DMS and (−)DMS libraries. Counts of the reverse transcriptase (RT) stops are normalized and subtracted.
All these three approaches mentioned above have some limitations. It may be challenging to perform PARS on organisms with highly repetitive genomes due to the difficulty of mapping the sequencing reads to the individual transcripts accurately. FragSeq is designed to target short ncRNAs. Hence, the structural information of longer RNAs in the transcriptome cannot be determined. The limitation in SHAPE-seq is that it is impossible to probe novel, unknown RNA structure in the cell because the sequence of the RNA needs to be known to design primers and clone the transcript.
Note that different strategies were applied to deal with the multiple mapped reads in PARS and FragSeqs [ 28 ]. The mapping in PARS should be uniquely mapped, while in FragSeq multiple mapping reads were allowed and counted together with unique mapping reads. SHAPE-seq can be a good candidate for probing the structure of several highly similar RNA sequences owing to the unique barcode on each RNA, enabling the sequencing reads to be accurately assigned to each transcript.
A stochastic model within SHAPE-seq is parameterized by adducts probabilities ( ), the Poisson rate for the number of adducts per molecule ( c ), and the natural polymerase drop-off probabilities in the control experiment ( ) and is used to infer the quantities of interest by maximum likelihood estimation [ 34 ]. The number of modifications per RNA is modeled as a Poisson process of an unknown rate c . The maximum likelihood of observing the entire data from both channels establishes the model parameters that best explain the observed data and thus best reflect underlying structural properties of the RNA molecule. SHAPE reactivities are normalized to a uniform scale. A normalized reactivity of 1.0 is defined as the average intensity of the top 10% reactivities, excluding a few highly reactive nucleotides taken to be outliers. According to different system under study, different methods are selected to remove the outliers. In a simple normalization scheme, the approach of 2–8% was chosen to exclude the top 2% of reactivities and the next 8% are averaged. Finally, all reactivities are normalized via dividing by this average value. This heuristic rule is based on general experience in Kevin Weeks’ laboratory [ 32 ]. Another normalization approach is box-plot normalization scheme, which removes the peaks >1.5 times the interquartile range (numerical distance between the 25th and 75th percentiles) above the 75th percentile. After that, the next 10% of the intensities are averaged and then all reactivities are divided by this average to be normalized. However, only if the sequence is long enough for meaningful statistics to be calculated (generally >300 reactivity measurements), it is wise to use the box-plot method.
High-throughput methods for obtaining RNA in vivo structural information
The cellular environment is complicated and it is possible that RNA structural conformations may be significantly different between in vitro and in vivo systems. Hence, genome-wide in vivo RNA structural profiling data can provide more accurate information for a better understanding of how RNA secondary structures exert function in living cells [ 11 , 12 ]. The advancement of in vivo RNA structural methods is likely to give rise to a research boom and produce an explosion of RNA structural data in the near future.
Structure-seq uses DMS to methylate unpaired adenine and cytosine residues of RNA in loops, bulges, mismatches and joining regions [ 11 ]. The modified sites are then identified by deep sequencing. This method was first applied to Arabidopsis seedlings, and obtained their genome-wide RNA structural information at nucleotide resolution from >10,000 transcripts. Before deep sequencing is performed, DMS treatment and control libraries are prepared separately. Arabidopsis seedlings are treated with DMS, and then RT using random hexamer (N 6 ) with adaptors is performed. This is followed by ssDNA ligation, polymerase chain reaction (PCR) and finally next-generation sequencing with different indices for treatment and control libraries ( Figure 2 B). As RT stalls one nucleotide before DMS-modifications, structural information is inferred through the base 1 nt upstream of the mapped base. Counts of the reverse transcriptase stops at each position are normalized and subtracted, which provides a measure of the DMS reactivity of that site.
Another approach, termed DMS-seq [ 12 ], for globally monitoring RNA secondary structure in vivo with single-nucleotide precision has also been developed. It uses DMS to modify the unpaired adenines and cytosines to obtain the structure of rRNA and mRNA in yeast under different conditions— in vivo and in vitro at five different temperatures (30, 45, 60, 75 and 95 degrees centigrade). DMS-seq also adapts to probe the RNA secondary structure in mammalian cells. Comparison between RNA in vivo and in vitro structural information obtained by DMS-seq reveals that, in rapidly dividing cells, mRNA in vivo are far less structured than in vitro . The result also shows that many structures of mRNA regions apparently in vitro do not exist in vivo . Additionally, the result of probing the RNA secondary structure in yeast under ATP-depleted conditions shows that energy-dependent processes are strongly modulating RNA secondary structure in vivo , contributing to the predominantly unfolded state of mRNAs in native cells. It is also shown that the thermodynamic stability plays an important, but only partial, role in determining RNA secondary structure in vivo .
Mod-seq is also a method for investigating RNA secondary structure [ 13 ], which is applicable for both in vivo and in vitro and can be used with any small molecule that reacts with RNA and blocks RT. This approach also uses DMS to probe the secondary structure of yeast total RNA in vivo , and analyses all four ribosomal RNAs and 32 additional ncRNAs. RNAs extracted from DMS-treated and untreated yeast cells are randomly fragmented and specific 5′ and 3′ adapter oligonucleotides are ligated to RNA fragments. This is followed by RT, PCR and finally next-generation sequencing. A data analysis pipeline, ‘Mod-seeker’, was also provided to enable detection of modification sites at each nucleotide from treated and untreated cells [ 13 ]. A comparison of these two samples enables an estimate of the fraction of RT stops at each site. Mod-seq can also be applied to identify the binding site of an RNA-binding protein in vivo . There is no RNA length limitation in this method and so it is ideal for studying long RNAs and complex RNA mixtures.
A comparison of the DMS-seq, Structure-seq and Mod-seq protocols reveals several common steps: (1) All three approaches use DMS to treat the RNAs followed by next-generation sequencing to probe the RNA secondary structure in vivo . (2) The sequence reads are mapped to the genome, transcriptome or RefSeq, and then calculate the DMS reactivity in each transcript by identifying the modification sites (RT stops) in the treatment and control separately. (3) The normalized DMS reactivities are determined by comparing the treatment and control, which can be used to analyze multiple genome-wide RNA secondary structure features.
There are also differences in DMS-seq, Structure-seq and Mod-seq protocols. (1) The process of library preparation is different among three methods. The size-selected cDNA are circularized to PCR amplification and sequencing in DMS-seq and Mod-seq. However, it is not the case in Structure-seq. (2) DMS-seq is not single-kinetic reaction that will cause multiple modification hits on the RNAs. Thus, DMS-seq performed the RNA fragmentation. In comparison, Structure-seq is single-kinetic reaction which will cause at most one modification hit on the RNAs. Thus, Structure-seq does not require the RNA random fragmentation. Also the structure-seq used the random primer for the RT, which will not introduce the bias of mapping regions. (3) After mapping the short reads onto the genome, Mod-seq counts the number of modification stop reads at each position in each gene in replicates of both treatment and control samples. And then, significant sites of modification in treatment sample were identified by using Cochran-Mantel-Haenszel tests, which requires the number of samples should be multiple of four. This approach cancelled out the experimental noise by using data from replicate experiments. To serve the same purpose, Structure-seq calculated the DMS reactivity by subtracting the normalized number of RT stops for the nucleotide in each transcript between DMS treatment and control libraries. However, the significance of modification sites was not examined as the method used in Mod-seq.
Although the high-throughput approaches have made many successful achievements, they also have some limitations. First, the cost and efforts are still expensive to achieve. Additionally, these methods are unable to calculate the metastable states of the RNA molecules. Hence, there is still a long way to go to comprehensively explore RNA secondary structure.
Computational and experimental information incorporated approaches for predicting RNA secondary structure
Researchers have turned to computational approaches to circumvent the difficulties and low-throughput of experimental methods for determining RNA secondary structures. The development of bioinformatics analysis is an important issue because the data itself is difficult to interpret without the help of mathematical methods. In particular, computational approaches are required to search and analyze the rapidly increasing volumes of structural data. Multiple algorithms have been developed for predicting RNA secondary structure from primary sequence [ 35–39 ]. These methods can be used to predict the structure of any interesting RNA, including a designed mutant RNA. Many biological features and patterns have been discovered via computational predictions.
There would be little point in attempting to computationally predict the secondary structure of an RNA in isolation, if it were actually dependent on the tertiary interactions in vivo . Fortunately there is evidence that structure formation is hierarchical and secondary structure can safely be determined in ignorance of the tertiary structure [ 40 ]. As RNA conformation is hierarchical, the local interactions occur first which then provide a scaffold to the native tertiary structure [ 41–43 ]. Predicting the secondary structure of RNA can provide very useful insights into its tertiary structure and the relationship between structure and function [ 41 , 44 ].
Structure prediction from base-pairing rules
RNA secondary structure is defined by the base-pairing within that sequence. The premise of many RNA secondary structure prediction algorithms is that an RNA molecule will fold into the secondary structure, which minimizes its free energy. Algorithms that attempt to predict the structure of a single RNA sequence apply thermodynamic principles to determine the most likely set of base-pairings for the sequence. Energy must be put into the system to break chemical bonds and is released when they are formed, so a simple approach to predicting the lowest free energy secondary structure for an RNA sequence is to maximize the number of paired bases, according to known base-pairing rules. Early attempts at determining secondary structure involved manually trying to identify the largest possible set of compatible base-pairing [ 45 ]. Dot-plots, which were at the time called ‘Tinoco plots’ and reviewed in [ 46 ], are a helpful aid for this task. However, this approach is impractical for longer sequences or situations involving many sequences, in which case a computational approach is required.
Various attempts were made to automate the process of maximizing the number of base-pairings for a sequence; however, the dynamic programming approach proposed by Nussinov et al . [ 47 ] provided the first efficient computational method ( Table 2 ). Dynamic programming is simply a particular way of solving optimization problems and is widely used even in the most recent structure prediction algorithms. The Nussinov algorithm to maximize base-pairing has been superseded by more complex free-energy minimization algorithms, but it serves as a simple illustration of the application of dynamic programming and many of the modern algorithms are inspired by this approach.
The summary of computational methods for predicting RNA secondary structure
| Methods | Examples | Principles |
|---|---|---|
| Dynamic programming approach | Nussinov algorithm etc. | To maximize the number of paired bases, according to known base-pairing rules |
| Minimizing free energy algorithm | mFold, RNAfold, RNAstructure etc. | RNA secondary structure with the lowest Gibbs free energy of folding will be the most prevalent |
| Machine learning approach | CONTRAfold, CentroidFold etc. | To optimize model parameters by using statistical learning algorithm |
| Comparative sequence analysis | RNAalifold, ComRNA etc. | RNA structure is more conserved than sequence |
| Integrative approaches | RNAstructure, SeqFold, etc. | To incorporate experimental data into computational algorithms |
| Methods | Examples | Principles |
|---|---|---|
| Dynamic programming approach | Nussinov algorithm etc. | To maximize the number of paired bases, according to known base-pairing rules |
| Minimizing free energy algorithm | mFold, RNAfold, RNAstructure etc. | RNA secondary structure with the lowest Gibbs free energy of folding will be the most prevalent |
| Machine learning approach | CONTRAfold, CentroidFold etc. | To optimize model parameters by using statistical learning algorithm |
| Comparative sequence analysis | RNAalifold, ComRNA etc. | RNA structure is more conserved than sequence |
| Integrative approaches | RNAstructure, SeqFold, etc. | To incorporate experimental data into computational algorithms |
The summary of computational methods for predicting RNA secondary structure
| Methods | Examples | Principles |
|---|---|---|
| Dynamic programming approach | Nussinov algorithm etc. | To maximize the number of paired bases, according to known base-pairing rules |
| Minimizing free energy algorithm | mFold, RNAfold, RNAstructure etc. | RNA secondary structure with the lowest Gibbs free energy of folding will be the most prevalent |
| Machine learning approach | CONTRAfold, CentroidFold etc. | To optimize model parameters by using statistical learning algorithm |
| Comparative sequence analysis | RNAalifold, ComRNA etc. | RNA structure is more conserved than sequence |
| Integrative approaches | RNAstructure, SeqFold, etc. | To incorporate experimental data into computational algorithms |
| Methods | Examples | Principles |
|---|---|---|
| Dynamic programming approach | Nussinov algorithm etc. | To maximize the number of paired bases, according to known base-pairing rules |
| Minimizing free energy algorithm | mFold, RNAfold, RNAstructure etc. | RNA secondary structure with the lowest Gibbs free energy of folding will be the most prevalent |
| Machine learning approach | CONTRAfold, CentroidFold etc. | To optimize model parameters by using statistical learning algorithm |
| Comparative sequence analysis | RNAalifold, ComRNA etc. | RNA structure is more conserved than sequence |
| Integrative approaches | RNAstructure, SeqFold, etc. | To incorporate experimental data into computational algorithms |
Structure prediction based on experimentally derived energies of RNA folding
When maximizing the number of base pairs, it is likely that multiple conformations will have the same, optimal number of pairings. It is biologically meaningless to arbitrarily select one of them. One option would be to weight the different pairings according to the strength of the bonds, perhaps GC = 3, AU = 2, GU = 1, but even this is likely to get many equally optimal pairings on a sequence of any significant length. Another problem with the Nussinov algorithm is that base-pairing is not the only contributor to the free energy of an RNA secondary structure: there are other parameters to be considered, such as stabilizing interactions between neighboring base pairs (stacking energies) and the destabilizing effects of loops. More attempts to predict structure use experimentally derived energies for RNA secondary structures. As the Nussinov algorithm stands, there is no obvious way to include information about these energy parameters. Zuker and Sankoff [ 48 ] proposed a modification to the algorithm to MFE of the structure, rather than maximizing the number of base pairs. To use a dynamic programming approach to MFE of the structure, Zuker & Sankoff make the assumption that the free energy of a structure is the sum of the free energies of its substructures.
The MFE algorithm has been implemented by Mfold [ 35 ], RNAstructure [ 49 ], RNAfold [ 50 ] and others ( Table 2 ). These methods use efficient dynamic programming algorithms combined with experimentally derived energy parameters to determine the global landscape of RNA secondary structure conformation and to establish the most thermodynamically stable structure. The prediction accuracy is high for short RNA sequence (<100 nt), and ∼70% of the bases can be correctly predicted. However, the accuracy drops to ∼20–60%, with increasing RNA lengths, when compared with either high-resolution crystal structures or structures obtained by comparative analysis [ 2 , 51–53 ]. Hence, a great challenge is to predict accurately the folds of long sequences, such as long noncoding RNAs (lncRNAs).
One of the major problems with identifying the minimum free energy structure is that even for relatively short sequences there are often many structures with free energies close to the minimum. Choosing the lowest may amount to an almost arbitrary choice given the small distinctions between them. A possible solution is to add constraints to the folding process—enzyme cleavage, cross-linking or chemical modification data. Additionally, there are also other fundamental limitations of secondary structure prediction algorithms. For example, they assume entropy is additive. Also, they neither deal well with non-canonical base pairs nor handle posttranscriptional modifications.
The Zuker and Stiegler [ 54 ] algorithm is an optimal computer method for folding large RNA molecules using thermodynamics and other auxiliary information. It is in essence a fusion of the two approaches: Nussinov algorithm and Waterman & Smith algorithm [ 55 ]. The latter takes into account stacking and destabilizing energies. The Zuker and Stiegler algorithm allows folding of sequences up to 600 nucleotides with a significantly reduced computation time. Additionally, the chemical modification, enzyme cleavage or even phylogenetic data on secondary structure can be used in this algorithm to better predict the RNA secondary structure. After which, the prediction of suboptimal solutions [ 56 , 57 ] gave rise to the ability to not only predict the state of the RNA molecule from the energetics point of view, but also to perform time-dependent folding predictions. Some RNA molecules form long-lived metastable structures rather than directly forming the thermodynamic ground state. Thus, the equilibrium view is not always sufficient, and one has to resort to methods that consider kinetics of RNA folding [ 58 , 59 ], such as Kinfold [ 60 ], Kinefold [ 61 ], Barriers [ 58 ].
Rather than considering only the minimum free energy structure, it is possible to consider the distribution of molecules of an RNA across all its possible structures. In principle, it was possible to calculate the partition function as soon as it was possible to enumerate all possible base-pairings (and thus all possible structures) and their energy contributions. However, it was not until McCaskill [ 62 ] (1990) developed a dynamic programming approach that calculating the partition function became computationally feasible. McCaskill (1990) takes a similar approach as that taken in the Zuker algorithm. The free energy of a structure is deemed to be the sum of the free energies of the substructures and the structure is considered to comprise a series of nested loops, enclosed by base-pairings.
Structure prediction based on machine learning approaches
Except for the approaches based on experimentally measured energy parameters mentioned above, alternative methods are based on probabilistic frameworks ( Table 2 ), for example stochastic context-free grammars (SCFGs), which use fully automated machine learning algorithms to derive model parameters [ 51 ]. The CONTRAfold [ 37 ], another RNA secondary structure prediction method, is based on conditional log-linear models, which is a flexible class of probabilistic models generalized on SCFGs via using a discriminative training set and flexible feature representations. It relies on statistical learning techniques to optimize model parameters. This approach bridges the gap between probabilistic and thermodynamic models by incorporating most of the features observed in typical thermodynamic models. Hence, the statistical learning algorithm can serve as an important alternative to determination of the thermodynamic parameters for RNA secondary structure prediction. CentroidFold web server is an implementation of the γ-centroid estimator [ 63 ], a kind of posterior decoding method based on statistical decision theory, for predicting RNA secondary structure [ 64 ]. It can accept either a single RNA sequence or a multiple alignment of RNA sequences. The server predicts (common) secondary structure with the most accurate prediction engine and returns a base-pair notation and a graph representation.
Structure prediction from sequence and phylogeny
It is reasonable to assume that a functional RNA secondary structure will be shared in related sequences and so using phylogenetic information to inform the structure prediction process can be helpful. Unfortunately, identifying conserved regions of structure relies on a good alignment of the related sequences. Even fairly closely related RNAs can be quite divergent in sequence, so traditional alignment methods are unreliable. An understanding of the folding of related sequences can inform the alignment, but as folding without any phylogeny information is unreliable we have a circular problem.
A comparative sequence analysis method, initially demonstrated on the 5S ribosomal RNA, is the most powerful and reliable method for dissecting the helical composition of RNAs [ 65 , 66 ]. This approach relies on the fact that many of the known functional RNA secondary structures are conserved in evolution. It determines RNA secondary structures by aligning the conservation patterns of base pairs among orthologous or paralogous genes. When enough homologous sequences are available, RNA secondary structure can be determined by comparing the pattern of conserved pairings ( Table 2 ). It is powerful in predicting the presence of helices of ribosomal and riboswitch RNAs with high confidence, as several thousand sequences are available and covariance analysis can be performed [ 2 , 67 ].
A dynamic programming algorithm was proposed by Sankoff [ 68 ], which solves both the structure and alignment problems simultaneously, but it is computationally expensive and impractical for more than a couple of sequences. The original Sankoff algorithm imposes a number of biologically motivated constraints on the search and a number of variations have been proposed, which use further constraints to limit the search space such that the algorithm runs in a practical time. Other methods ignore energy parameters entirely and base their consensus structures solely on some measure of co-variation of base pairs in an alignment, generally through use of iterative improvements to the alignment and predicted consensus structure. The covariance-based methods can work well, but they require a reasonably large set of sequences, covering enough evolutionary distance to provide enough co-variations. For small sets of closely related sequences, it may be necessary to combine thermodynamic parameters with the evolutionary information to find a structure. Several approaches have been developed to predict RNA secondary structure from sequence and phylogeny, such as Sankoff DP algorithm [ 68 ], CARNAC [ 69 ] and ComRNA [ 70 ].
High-throughput structural information constrains the prediction of RNA secondary structure by integrative approaches
The computational and experimental approaches both have some limitations in characterizing RNA secondary structure. However, the limitations in many cases can be overcome by combining these two complementary methods together. In recent years, many integrative approaches have been developed to predict RNA secondary structure by incorporating the experimental probing data ( Table 2 and Figure 3 ).
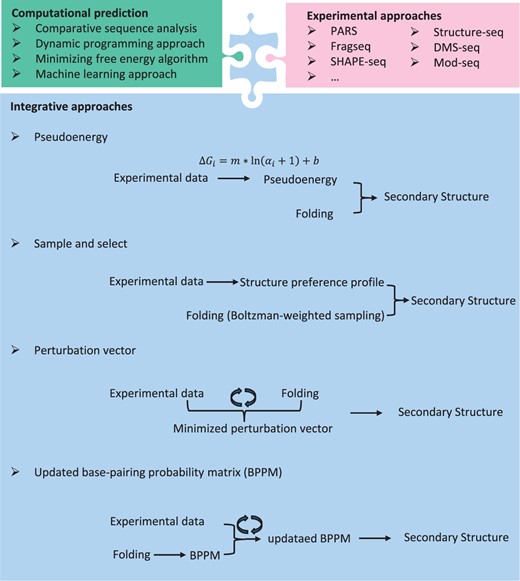
Overview of the RNA secondary structure prediction methods. Integrative approaches are computational and experimental information incorporated approaches for predicting RNA secondary structure. There exists several algorithms, including psuduoenergy-based algorithm, sample and select approach, perturbation vector minimization and updated BPPM.
Typically, one commonly used algorithm was based on an empirical energy model [ 71 ]. Many programs were developed to predict RNA secondary structure via minimizing free energy, such as mfold [ 72 ], RNAstructure [ 73 ] and RNAfold [ 74 ]. Free energy minimization plays an important role in determining RNA secondary structure, which can not only help to identify important regions during comparative analysis but also can be used when only a single sequence is available. Another method is to add a ‘pseudo-energies’ term in the energy model ( Figure 3 ), which is implemented in the program RNAstructure.
where is the SHAPE value for base i ; i = 1 . . . n, where n is the sequence length; Using the secondary structural information obtained by comparative sequence analysis, the two free parameters m and b were parameterized against 23S rRNA (defaults set to m = 2.6 and b = −0.8 kcal mol −l ). This pseudoenergy approach was reported to yield secondary structure prediction accuracies of >96% for Escherichia coli rRNA. Zarringhalam et al . proposed a new approach to minimize a Manhattan distance between the modified pseudoenergy and the predicted structure [ 75 ].
where R is the gas constant (0.001986 kcal mol −1 K −1 ) and T is the absolute temperature in Kelvin.
Washietl et al . [ 77 ] proposed a formal method, expanding the idea of incorporating experimental data as pseudo-energies into energy-based folding algorithms, to well combine the experimental data with the computational prediction in the partition function. This algorithm is different from the former method mentioned above. They use a perturbation vector ( Figure 3 ) to minimizes a weighted sum of perturbation energies and discrepancies between experimentally determined and computationally predicted base-pairing probabilities, which is on the assumption that both experimental probing data and the thermodynamic energy parameters are noisy approximation to the physical ground truth. In addition, the perturbation vector modifies the energy model only when necessary. For example, no pseudo-energies are need when experiment and thermodynamic model are in perfect agreement.
The base-pairing probability matrix (BPPM) of an RNA sequence stores all the probabilities for possible base pairs in an RNA sequence, which plays an important role in multiple algorithms in RNA informatics, such as RNA secondary structure prediction, multiple alignment of RNA sequences, RNA–RNA interaction predictions, RNA motif search and microRNA (miRNA) gene identifying. A novel algorithm was proposed to perform iterative updates of the BPPM ( Figure 3 ) according to marginal probability constrains after formulating the problem mathematically [ 78 ]. The algorithms were based on the RAS algorithms, which were used in macroeconomics. In particular, this algorithm is robust to missing data and noise and can be applied in any condition as long as the BPPM of the RNA sequence can be found.
Recently, SeqFold [ 79 ] has been developed to reconstruct RNA secondary structures on a genome scale, by using sample and select approach ( Figure 3 ). It can incorporate current high-throughput RNA secondary structure profiling data, including PARS, FragSeq, SHAPE-seq, Structure-seq and DMS-seq. On the one hand, SeqFold transforms experimental signals from RNA secondary structure profiling data into a structural preference profile (SPP) for all the bases of a given RNA. On the other hand, each transcript is modeled as 1000 structures by Sfold using a Boltzmann-weighted sampling procedure and these are grouped into distinct clusters for each RNA. Given an SPP, the centroid of the selected cluster is taken as the predicted RNA secondary structure. Then the accessibility of each base can be estimated by the cluster average. The result indicates that higher (or lower) average accessibilities of the bases tend to be single stranded (or double stranded) [ 79 ]. The sample and select approach requires that the correct structure be sampled from the thermodynamic ensemble.
We expect that with an increasing amount of experimental RNA secondary structure information in vivo , there will be a trend to develop new algorithms or tools to reveal dynamic conformations of RNA secondary structure by integrating in vivo profiling data.
Novel characteristics of RNA secondary structure across mRNAs
Improvements in measuring RNA secondary structures will enable exploration of the relationships between RNA secondary structure and function. The functions of almost all classes of RNAs, such as mRNAs and ncRNAs (riboswitches, ribozymes, lncRNAs and miRNAs), can be influenced by their secondary structures. Recently RNA secondary structure of high-throughput sequencing revealed that they play important roles in many biological processes, including transcription, splicing, RNA localization, translation and RNA decay.
Global structural features across coding and noncoding regions
The analysis of overall structure features in the coding and noncoding regions of mRNAs show that there is a significant variation in the structural content of 5′ UTRs and 3′ UTRs compared with coding regions. In addition, the structure patterns of coding and noncoding regions also differ among different species ( Figure 4 A). In yeast and Arabidopsis, coding regions tend to be more structured than untranslated regions. However, in contrast, one or both UTRs were much more structured than the coding region within HIV-1, fruit fly, nematode and human mRNAs [ 8 , 80–83 ]. These conflicting results may be affected by the diversity of RNA-binding proteins and miRNAs that evolved within each species, or they could reflect functional differences in these regions.
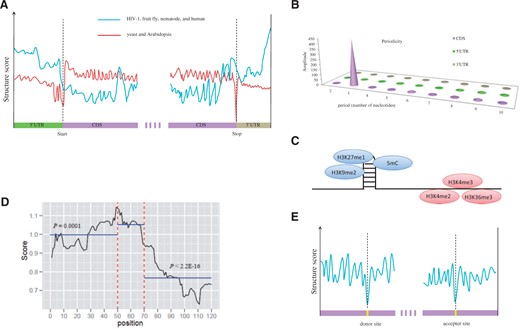
Schematic diagrams show novel structural features revealed by the RNA secondary structure profiles. ( A ) In HIV-1, fruit fly, nematode and human, the UTRs tend to be more structured than CDS regions. Whereas, there is a reverse pattern in yeast and Arabidopsis where UTRs are poorly structured compared with CDS regions. ( B ) A 3 nt periodicity is found only in CDS regions in yeast and Arabidopsis. ( C ) In Arabidopsis, repressive chromatin modifications (H3K9me2 and H3K27me1), as well as DNA cytosine methylation (5mC), were enriched at the highly structured regions, while the activating (H3K4me2, H3K4me3 and H3K36me3) histone modifications were enriched on less structured regions. ( D ) The miRNA binding regions (shown as position 50–70) are more structured than their flanking regions (50 nt) in Arabidopsis. ( E ) Compared with the flanking sequences, a dramatic decrease was observed in secondary structure upstream of the splice donor and acceptor sites in Arabidopsis.
The overall architectures of the UTRs are not conserved. However, the regions near the start and stop codons of the coding sequence have a high probability of being single stranded, indicating the increased accessibility of the regions when protein translation begins and ends for five eukaryotic species (yeast, Arabidopsis, fruit fly, nematode and human) [ 8 , 80–82 ]. These findings are consistent with previous computational predictions for both mouse and human genes [ 84 ]. Additionally, unstructured regions around the start codons are enriched in high translation efficiency mRNAs, but are absent in low translation efficiency mRNAs. The less structured regions near start codons may possibly facilitate ribosome binding and increase translational efficiency [ 8 , 11 , 85 ]. Hence, the structure of start codons may be a major factor for translation initiation.
Analysis of the coding regions reveals that a 3 nt periodicity of secondary structure exists in yeast and Arabidopsis. That is, the first nucleotide of each codon is least structured and the second nucleotide is most structured ( Figure 4 B). However, this periodicity was absent in UTR regions. This RNA structural pattern significantly influences in vivo polyribosome association and may directly or indirectly be attributed to translation [ 8 , 11 , 86 , 87 ].
The Structure-seq results reveal that the region −15 to −2 nt upstream and −1 to 5 nt downstream of known [ 88 ] alternative polyadenylation cleavage sites have significantly lower and higher DMS reactivity [ 11 ]. This structured–unstructured pattern near the cleavage site may facilitate the regulation of alternative polyadenylation.
RNA structural hot and cold spots and epigenetic modifications
The results from dsRNA sequencing and ssRNA sequencing data reveal several key structural features and functions of RNA folding on a genome-wide scale for Arabidopsis, fruit fly and nematode. The genomic regions that were significantly enriched in either paired RNAs or unpaired RNAs are identified individually as dsRNA hot spots or ssRNA hot spot, individually. Most of the dsRNA hot spots mapped to known highly structured RNAs, such as rRNAs, snRNAs, snoRNAs and smRNA-producing regions in the three species. A large proportion of dsRNA reads also correspond to transposable elements (TEs) in fruit fly and Arabidopsis. This is consistent with the knowledge that highly structured regions always have low transcription activation. However, this is not the case in nematode, in which only a small fraction of dsRNA reads corresponded to TEs. In contrast, ssRNA-seq reads mostly correspond to protein coding mRNAs and pseudogenes in three species [ 80 , 81 ].
The dsRNA and ssRNA hot spots are closely related to distinct epigenetic modifications [ 80 , 89–91 ]. The dsRNA hot spots are enriched in repressive chromatin modifications. For example, H3K9me2 and H3K27me1 modifications, as well as DNA cytosine methylation (5mC), were enriched in Arabidopsis ( Figure 4 C). H3K9me3 was enriched in fruit fly. However, the activating and euchromatic H3K4me2, H3K4me3 and H3K36me3 histone modifications were enriched in ssRNA hot spots in Arabidopsis. H3K4me3 and H3K9 acetylation were enriched in fruit fly [ 81 ]. But a similar analysis in nematode showed that activating euchromatic modifications H3K4me2, H3K4me3 and H3K79me1 were enriched in both paired and unpaired RNAs, which may be the consequence of most worm hot spots being encompassed by protein-coding mRNAs [ 80 , 81 ]. These findings indicated that protein-coding transcripts prefer to be single stranded and possibly take part in epigenetic regulatory pathways.
RNA localization and stability
Recent evidence indicates that most mRNAs are localized in specific cell types, which plays a crucial role in regulating cell migration and differentiation [ 92 , 93 ]. The PARS study in yeast revealed biological coordination of increases in RNA secondary structure, especially in coding regions. These RNAs encode proteins localized to distinct cellular domains, suggesting that structural localization elements are present in different mRNAs [ 8 ]. The transcripts that encode secretory proteins contain a signal sequence in the 5′ end of the coding region, which helps to promote RNA nuclear export [ 8 , 85 , 94 ]. Also, the PARS data revealed that 5′ UTRs and the first 30 coding nucleotides of signal peptide mRNAs were less structured than other transcripts, suggesting that the specific RNA secondary structure in these regions may guide the cytotopic localization of these mRNAs and their resulting proteins [ 8 , 85 ].
Previous studies showed that RNA secondary structure and folding energies play an important role both in translation and the stability of an mRNA under various cellular stress conditions. Early research on how bacteria are affected by temperature changes indicated that 5′ UTRs containing thermodynamically unstable structures can initiate translation [ 95 ]. Subsequent studies found that RNA thermometers may also exist in eukaryotes [ 96–101 ].
If only the 3′ end of the RNA was structured or contained a short 3′ overhang, the transcript was protected from degrading [ 102 ]. However, there is a significant anti-correlation between RNA secondary structure and mRNA abundance in Arabidopsis. Highly structured mRNAs tend to be less abundant in the Arabidopsis transcriptome than less structured transcripts because highly structured RNAs have more possibility to be degraded or processed into smRNAs [ 81 ].
The structural feature on miRNA binding sites
Analysis of the structure patterns of miRNA target sites shows that the target sites have higher average DMS score compared with the flanking regions (50 nt), indicating that the miRNA target sites in transcripts are less structured than their flanking regions in Arabidopsis thaliana ( Figure 4 D). These results are consistent with the previously predicted results [ 103 ]. Additionally, there are similar findings in Caenorhabditis elegans [ 104 ]. This may contribute to miRNAs to interact with their target mRNAs [ 39 , 103 , 105–107 ].
RNA splicing
RNA secondary structure has long been proposed to regulate alternative splicing events [ 108 ]. 5′ splice site with significant lower DMS reactivity are strongly structured and this correlates with unspliced events. However, this structural pattern was absent in 3′ splice site. The structural features in splice sites may have a regulatory effect for alternative splicing [ 11 ].
Recently, the PIP-seq of nuclear RNA transcriptome [ 109 ] allows the detection of the RNA secondary structure pattern at the splice donor and acceptor sites of unmature transcripts. A dramatic decrease in secondary structure upstream of the splice donor and acceptor sites ( Figure 4 E), revealing that this region is more accessible to intermolecular RNA pairing than flanking sequences, perhaps facilitating binding of splicing factors. Further analysis revealed the distinct structural profiles for different subtypes of alternative splicing (cassette exons, intron retention, and constitutive introns) indicates that RNA secondary structures are required for their proper regulation.
Comparison of RNA secondary structures among in vitro, in vivo and in silico prediction
Genome-wide in vivo RNA structural analysis is now possible. The genome-wide in vivo structure probing on the Arabidopsis, yeast and human transcriptome was successfully carried out via DMS, which made it possible to explore the native RNA secondary structures under complicated cellular environments. A good review also talked about the relationship among RNA sequence, structure and function [ 110 ]. Although the in silico structure prediction or in vitro probing have made improvements in our understanding of RNA, these structures may not represent the true structure in the native cells. In vitro structures are more similar to the structures predicted by in silico than that in vivo structures. Another interesting result is that the degree of DMS modification has a greater excess than expected, indicating that these structures are more dynamic or tend to be less structured than those observed in vitro .
The mRNAs in Arabidopsis encoding proteins related to stress and stimulus responses tend to be more single-stranded in vivo [ 81 ]. There is a significant difference in their in vivo probing and in silico -predicted RNA secondary structures of these mRNAs. The plastic and dynamic structure features of these stress-response mRNAs may be responding to changes in cellular conditions. By contrast, little difference exists in the mRNAs involved in basic biological functions, such as gene expression, protein maturation and processing and peptide metabolic processes, suggesting that these mRNAs minimize large structural changes to maintain homeostasis [ 11 , 85 ].
Perspectives
The study of RNA secondary structure has progressed from low-throughput experimental methods that explore RNA secondary structures one transcript per experiment, to computational approaches for RNA secondary structures prediction on a larger scale, to high-throughput and genome-wide methods to measure RNA secondary structures, such as Structure-seq, DMS-seq. The change from in vitro to in vivo enables the determination of native RNA secondary structures, providing new perspectives when probing real RNA secondary structures on a genome-wide scale. Integrative methods such as SeqFold have been developed to combine the high-throughput profiling data into computational predictions, greatly improving the accuracy of RNA secondary structure prediction.
An explosion of RNA structural profiling data will be produced in the next few years with the advancement of in vivo RNA secondary structure analysis methods. Although many discoveries and progresses have been obtained, much remains to be explored. Firstly, it may be possible to accurately predict the pseudoknots or long-range and tertiary-structure interactions with the advancement of computational strategies. Secondly, if the in vivo and dynamic RNA structural maps can be constructed, they will play a crucial role in understanding how RNA secondary structures change in different cellular environments. This will be achieved by carrying out deep-sequencing and in vivo RNA structurome analysis under normal conditions, as well as under different stresses, such as changes in temperature, ion concentration and others. Another direction will be to compare the RNA structurome across species. These observations will result in increasingly meaningful regulatory networks and some biological pathways, which will feed on to clinical research. The analysis of structural features of longer transcripts such as lncRNAs will reveal the common motifs and regulatory patterns conserved at the structure level that have been overlooked before. In summary, there are many directions to explore, and this is an exciting time to be studying RNA secondary structure.
Combining nucleases or chemical probes with next-generation sequencing provides a new perspective for genome-wide measurement of RNA secondary structure.
The accuracy of computational prediction of RNA secondary structure may be greatly improved through incorporating high-throughput structural information.
Structural patterns in mRNA relate to many processes including epigenetic modifications, RNA localization and stability, RNA splicing and structural feature on miRNA binding sites. Their study is further complicated by the differences between in vivo and in vitro structures.
Acknowledgments
We are grateful to Sharon Aviran in University of California at Davis for helpful comments and discussion.
Funding
The work was supported by the National Natural Sciences Foundation of China (31371328, 30971743); National Science and Technology Project of China (2008AA10Z125); and the Henry Lester Trust, UK.
References
Author notes
*These authors contributed equally to this work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}