




Abstract
Repurposing and repositioning drugs—discovering new uses for existing and experimental medicines—is an attractive strategy for rescuing stalled pharmaceutical projects, finding treatments for neglected diseases, and reducing the time, cost and risk of drug development. As this strategy emerged, academic researchers began performing high-throughput screens (HTS) of small molecules—the type of experiments once exclusively conducted in industry—and making the data from these screens available to all. Several methods can mine this data to inform repurposing and repositioning efforts. Despite these methods' limitations, it is hopeful that they will accelerate the discovery of new uses for known drugs, but this hope has not yet been realized.
INTRODUCTION
Finding new uses for approved medicines and experimental medicines that fail approval in their initial indication—drug ‘repurposing’ and ‘repositioning’, respectively—has emerged as an attractive strategy for rescuing stalled pharmaceutical projects, finding therapies for neglected diseases and reducing the time, cost and risk of drug development [1–6]. Encouraging success stories from both pharmaceutical companies and academia include sildenafil for the treatment of impotence [1], thalidomide for the treatment of multiple myeloma and a painful complication of leprosy [1, 4], and aminobisphosphonates for the treatment of progeria [7]. These stories highlight the immense financial payoff and humanitarian impact of successful repurposing; sildenafil grosses several billion dollars each year, and thalidomide and aminobisphosphonates ameliorate rare or neglected diseases that were previously untreatable.
At the same time that repurposing and repositioning emerged as drug-development strategies, academic researchers began performing high-throughput screens (HTS) of small molecules—the type of experiments once exclusively conducted in industry—and making the data from these screens available to all [8]. The National Institute of Health (NIH) administers the largest repository of screening data: PubChem. This repository holds the data from several hundred biochemical and phenotypic screens with several more deposited each month [9, 10]. Several additional screens are available through repositories like ChemBank [11] and Collaborative Drug Discovery's database [12], but most NIH funded work is deposited in PubChem after a brief embargo period.
Mining these public screens may prove an effective strategy for repositioning or repurposing drugs. The most promising strategy is to mine phenotypic screens—those that measure a disease-relevant response in a whole-cell or organism system—because they can identify molecules that work on any target involved in the disease process modeled by the assay. The screens' design does not fix ahead of time the targets that active molecules must act upon, so phenotypic screens can uncover unexpected relationships between drugs, targets and diseases. It is also possible, but less likely, that biochemical screens—which focus on a single protein target—could inform repurposing efforts in a similar way by uncovering unexpected interactions between drugs and important targets.
Mining HTS data is cheaper and quicker than directly testing molecules in assays [8]. Experimentally testing all known drugs in medically-relevant assays requires a large investment in both HTS infrastructure and expertise in the many assays of interest. Existing investments in HTS infrastructure at most screening centers can easily handle the relatively small number of drug molecules (Figure 1). The key limiting factor in this strategy, however, is the expertise, time, and resources required to develop each assay, a process that has not been multiplexed in any way. In contrast with the direct experimental approach, once the data from a screen is available, computational mining of this data can proceed without requiring investment in assay-specific expertise, often for a fraction of the cost of the original screen. Therefore, it is conceivable that a small team of computational scientists could mine thousands of screens in a small amount of time and for little more cost than their salaries. In order to verify predictions, some molecules must eventually be experimentally tested, but the real value of mining is that it would direct resources toward those assays most likely to yield positive results, dramatically reducing the resources required for experiments.

HTS infrastructure. This robot shuffles hundreds of thousands of molecules in 384-well plates to execute some of the small-molecule screens at the Broad Institute of Havard/MIT. HTS infrastructure like this has only recently become widely available to academics.
At a high-level, each mining strategy works in a similar way; they use data from a phenotypic HTS as input to predict which drugs might be treat diseases relevant to the phenotype measured by the screen's assay. Before explaining these HTS mining techniques, we first explain the details of how HTS projects are structured. These details are important because they elucidate why different strategies might be used in different cases. After explaining these details, we move to discussing several different strategies for mining HTS, their limitations, and in what cases they might be most successfully applied.
THE ANATOMY OF A SCREEN
The structure of screening experiments exposes both the technical limitations of HTS as well as reasons to hope that mining HTS data can unearth valuable information. At a high level, screening workflows are often conceptualized as a multi-stage funnel, where, at each stage, an experiment filters out uninteresting molecules and forwards the rest on to the next stage (Figure 2). Molecules are filtered by predefined thresholds—based on potency, selectivity, structure, or ADME properties—in order to satisfy project-specific goals.
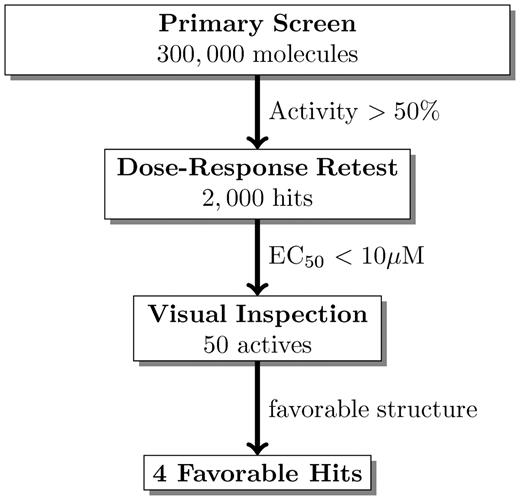
A screening funnel. In this hypothetical example, a primary screen is followed by a dose–response confirmatory test, and finally the confirmed actives are visually inspected to identify active molecules with chemically favorable structures. The numbers in this example are similar to those observed in typical NIH funded screens deposited in PubChem. More complex workflows include additional assays which might ensure molecules are selective or work by a particular mechanism.
Screens are designed to find molecules that satisfy narrowly defined and project-specific goals. Consequently, all funnels will, by design, exclude molecules useful for other purposes. For example, one screener might look for potent and selective activators of a protein, while another might look for allosteric inhibitors with sufficiently different structures than those of known drugs. In particular, screeners commonly aim to find molecules with novel structures [13]. Therefore, they sometimes filter out known drugs—some of which may be apparently active in the initial screen—without sending them for confirmatory testing. This example highlights the key issue: the most interesting molecules for repurposing efforts may be ignored by the original screener, and key experiments useful for repurposing efforts may not be performed.
Furthermore, each stage depends on predefined cutoffs, because important properties—such as solubility, potency, selectivity, drug-likeness—are conceptually clear but imprecisely delineated. For example, a noisy initial screen might identify a list of ‘hits’ with inhibitory activity greater than 50% at a single dose. These hits are commonly sent for confirmatory testing at several doses to ensure they yield a dose–response curve consistent with true actives. Confirming hits by measuring their potency in a dose–response experiment is important because the potency of a hit is not well correlated with activity in the initial screen measured at a single dose [14] (Figure 3). The successfully confirmed hits with potencies of at least 10 μM might be further filtered by additional experiments designed to ensure they are sufficiently specific to the targeted phenotype. In this example, the 50% activity and 10 μM potency thresholds are somewhat arbitrary—another screener might have chosen 25% activity and 1μM—and additional actives can be discovered by adjusting these thresholds to advance more molecules through the workflow [15].
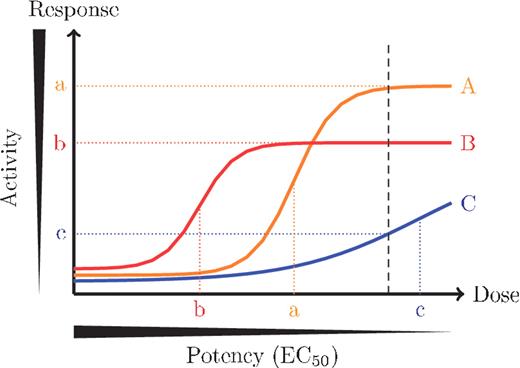
Potency does not always correlate with single-dose activity. Each line corresponds with the behavior of a different molecule in the assay at different doses. HTS screens are usually performed at a single dose, shown as the black, dashed line, and the response at this dose is used to rank molecules in the primary screen for follow up (y-axis). High potency molecules reach 50% maximal activity (EC50) at lower doses (x-axis), but potency can only be measured by testing the molecule at several doses. As in this example, activity at a single dose does not always correlate with potency, and activity in the primary screen does not always correlate with the potency measured in dose–response follow-up experiments.
Moreover, screens are noisy. In well-designed screens, the assay readout has high fidelity, but several technical details can cause false positives and false negatives. Some molecules degrade because they are chemically unstable under the conditions of the HTS experiment or library storage [16, 17]. Likewise, some molecules directly interfere with the readout or are thought to promiscuously bind to several proteins [18]. Consequently, several studies have demonstrated many molecules that appear inactive in the initial screen actually are active when retested [19–21, 15].
Consequently, as noisy artifacts of complex workflows with project-specific goals, HTS data does not comprehensively survey the chemical libraries on which they are run. Rather than accurately annotating the activity of all tested molecules, HTS instead aims to identify and characterize only a small number of molecules with narrowly and subjectively delineated properties. Interesting molecules are often missed [15, 22]. Although they are not widely used, several strategies—based on systematic error correction [23, 24], structural similarity [19, 20], machine learning [21] and even economics [15]—can recover some of these missed actives.
The complexity, subjectivity and noise in HTS projects creates both a challenge and an opportunity. On one hand, mining a HTS screen can be difficult, requiring the application of sophisticated algorithms capable of integrating noisy and irregularly sampled data. On the other hand, there is hope that careful mining can unearth diamonds: molecules that do not satisfy the initial project's thresholds or goals but can still inform repositioning efforts. In the easiest cases, mining these molecules can be straightforward; however, the hardest cases require complex algorithms that integrate and analyze data from several sources.
MAKING CONNECTIONS
In the simplest case, an approved drug is confirmed active in a medically-relevant screening project. HTS projects often aim to find entirely new chemical structures, so these high value actives are often ignored because they are not chemically novel. In this case, we have only to make the connection, which can be directly confirmed in a simple experiment and then moved immediately to animal trials when sensible.
Public databases store most of the key information required to make these connections. For example, DrugBank—the most comprehensive publicly available database of known drugs [25–27]—catalogues approved, experimental, withdrawn and illicit small-molecule drugs, annotating them by indication, intended targets and cross references to PubChem and PubMed. As useful as DrugBank and related resources are, they are far from complete. Furthermore, only about 460 of DrugBank's drugs are regularly screened by the NIH. In the near future, the release of CandiStore—a much larger and comprehensive database of drugs—may improve the situation [28]. In the meantime, proprietary databases like Pharmaprojects may prove useful to some.
Several tools use DrugBank and other public resources to identify known drugs that are confirmed active in public screening data, making connections that can inform repurposing efforts [29, 30]. For example, Chem2Bio2RDF links several screening databases with several drug databases using a standards-driven interface, enabling complex queries to be succinctly described and submitted to their server. While its interface is not easy for the typical biologist to use, the developers provide many clear example queries, which can be easily edited so as to yield useful results. The real utility of integrative methods may be best seen in more user-friendly tools like BioEclipse [31] and the Web Engine for Non-obvious Drug Interactions (WENDI) [30]. The next generation of tools—like WENDI—may prove most useful when integrating data from several sources to easily answer high level questions like ‘what drugs have been confirmed active in a recent phenotypic screen?’
As useful as these tools may become, their approach—integrating databases to identify known drugs among confirmed actives—is limited. First, only a fraction of known drugs are directly tested on a regular basis (Table 1), and this approach cannot identify untested drugs.
Known drugs in the NIH screening collection
| Class | Number | Included | Neighbors |
|---|---|---|---|
| Small molecule | 4646 | 465 | 2111 |
| Approved | 1493 | 354 | 996 |
| Investigational | 365 | 82 | 202 |
| Experimental | 3243 | 101 | 1103 |
| Illicit | 188 | 6 | 114 |
| Nutraceutical | 71 | 8 | 43 |
| Withdrawn | 65 | 18 | 40 |
| Class | Number | Included | Neighbors |
|---|---|---|---|
| Small molecule | 4646 | 465 | 2111 |
| Approved | 1493 | 354 | 996 |
| Investigational | 365 | 82 | 202 |
| Experimental | 3243 | 101 | 1103 |
| Illicit | 188 | 6 | 114 |
| Nutraceutical | 71 | 8 | 43 |
| Withdrawn | 65 | 18 | 40 |
On a category-by-category basis, the ‘Number’ column records the number of drugs found in DrugBank, the ‘Included’ column records the number of molecules of each category which are included the NIH's screening collection and regularly screened by academic labs, and the ‘Neighbors’ column records the number of drugs with at least one structural neighbor in the NIH's collection. In this table, structural neighbors are defined as molecules with at least 0.8 similarity as computed using the default settings of OpenBabel [34]. Most of the drugs listed in DrugBank are not regularly screened, but many have neighbors which are screened.
Known drugs in the NIH screening collection
| Class | Number | Included | Neighbors |
|---|---|---|---|
| Small molecule | 4646 | 465 | 2111 |
| Approved | 1493 | 354 | 996 |
| Investigational | 365 | 82 | 202 |
| Experimental | 3243 | 101 | 1103 |
| Illicit | 188 | 6 | 114 |
| Nutraceutical | 71 | 8 | 43 |
| Withdrawn | 65 | 18 | 40 |
| Class | Number | Included | Neighbors |
|---|---|---|---|
| Small molecule | 4646 | 465 | 2111 |
| Approved | 1493 | 354 | 996 |
| Investigational | 365 | 82 | 202 |
| Experimental | 3243 | 101 | 1103 |
| Illicit | 188 | 6 | 114 |
| Nutraceutical | 71 | 8 | 43 |
| Withdrawn | 65 | 18 | 40 |
On a category-by-category basis, the ‘Number’ column records the number of drugs found in DrugBank, the ‘Included’ column records the number of molecules of each category which are included the NIH's screening collection and regularly screened by academic labs, and the ‘Neighbors’ column records the number of drugs with at least one structural neighbor in the NIH's collection. In this table, structural neighbors are defined as molecules with at least 0.8 similarity as computed using the default settings of OpenBabel [34]. Most of the drugs listed in DrugBank are not regularly screened, but many have neighbors which are screened.
Second, there are often many false negatives in screening projects—truly active molecules that were not sent for confirmatory testing and incorrectly assumed to be inactive [15]. Consequently, negative results should be treated with caution; a drug may not be confirmed active in a phenotypic screen, and nonetheless be capable of treating the disease for which the screen is designed. This is not to say that all negative results are incorrect, rather more sophisticated methods—discussed in following sections—that take the screen's uncertainty into account may yield more accurate predictions.
Third, even when they are tested, some drugs degrade because they precipitate or are chemically unstable under the conditions of library storage and handling [16, 17]. This type of instability can make testing the drug technically challenging, causing both false positives and false negatives in HTS experiments. Depending on several factors, this instability can happen frequently in some libraries. For example, about 12% of each molecule is precipitated and lost after 10 freeze-thaw cycles [16] and—depending on the exact storage conditions—more than one third of molecules can degrade to <80% purity over a year's time in storage [17]. In practice, many screening centers reduce library degradation by limiting plate-life, capping the number of freeze-thaw cycles, and reducing temperature and humidity in storage. The quality control procedures in screening centers are rarely accessible so it is difficult to know how much these factors impact the data from any specific HTS project. Recent work may be helpful in computationally identifying molecules likely to be problematic [18], but usually this source of error is very difficult to account for in any general methodology.
Finally, about 5–7% of drugs are not active in the form they are administered to patients; they are ‘prodrugs’, which are transformed by the patient's body into their active form [32, 33]. DrugBank and other repositories store the inactive form of prodrugs: the form that is not expected to be active in the screen but may still be useful in the disease. Therefore, while the data-integration approach may uncover unexpected connections between known drug molecules and screens, determining the relevance of these connections still requires careful consideration by human experts.
THE SIMILARITY PRINCIPLE
In a more difficult case, the close structural neighbors of a drug are directly tested in the HTS, but the drug itself is not. Structurally similar molecules often have similar biological properties, so the activities of these structural neighbors predict the activity of the drug to which they are similar. In the simplest strategy, a drug would be predicted active if a structurally similar molecule is experimentally demonstrated active in the screen. More sophisticated strategies use the data from the screen to train predictors—based on, for example, machine learning techniques like support vector machines and neural networks—that then annotate untested drugs with the likelihood they would be active if tested [35–37, 43].
Most of these methods rely on quantitatively computing similarity between molecular structures. A dizzying variety of similarity metrics can do this [35, 38, 39, 40–42]. The most commonly used methods are based on Tanimoto similarity, defined as the proportion of substructures in common between two molecules (Figure 4). There are several ways of enumerating substructures from molecules, and each way yields a different Tanimoto variant [39]. Tanimoto similarity—when computed on linear or circular fragments of the 2D structure of molecules up to a fixed size—correlates surprisingly well with biological activity [35, 43]. In most cases, the path-based molecule similarities computed by open-source tools are just as good as a myriad of proprietary tools [34, 44–46]. Many of the open-source tools are unified by a simple interface: Cinfony [44], which non-experts can easily use.
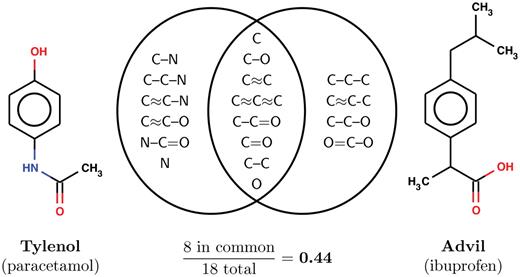
Quantitative structural similarity. The most commonly used structural similarity metric is Tanimoto similarity: the proportion of substructures in common between two molecules. An example using Advil and Tylenol, two over-the-counter pain medications, is shown above. In this example, all linear fragments up to 3 atoms long are extracted from each molecule (with ≈ used to denote aromatic bonds). Dividing the 8 substructures seen in both molecules by the 18 seen in either yields the Tanimoto similarity 0.44. Longer fragments are used in practice, but the formula remains the same.
Several issues arise when using molecular similarity based on 2D descriptors to predict activity. First, although the Tanimoto similarity always ranges from zero to one, its exact value depends on the way substructures are extracted from molecules. Therefore, choosing a fixed cutoff like 0.9 to annotate a molecule's neighbors may not work for all similarity metrics. To make matters worse, because structural similarity is highly dependent on a molecule's size and complexity, it is difficult to choose an appropriate fixed cutoff at which to determine molecules' neighbors [47–49].
Second, while structural similarity is predictive of activity, it is not always accurate. Recent research has focused on ‘activity cliffs’, pairs of molecules that are structurally very similar but have very different activity [50–52]. Some activity cliffs can be traced to deficiencies in 2D-based similarities, which are usually insensitive to stereochemistry and do not work well on large repetitive molecules [47, 53]. However, cliffs are usually difficult to predict. Therefore, structural similarity alone cannot definitively establish the activity of the molecule, and is most useful when used in conjunction with experimental confirmation [43].
LEVERAGING THE TARGET
In the most difficult case, neither the known drug nor its analogs are directly tested in the screen, rendering both direct data-integration and similarity-based methods unusable. Nonetheless, if the active molecules in the screen include molecules known to hit the same target as a known drug, it is possible to infer that this drug would be active too. In this case, confirming the known drug in the screen not only validates the prediction, but also provides experimental evidence that its target—and not some unknown off-target—is the mechanism by which the phenotype is modulated.
This strategy requires knowledge of which molecules interact with known drugs' targets. If several molecules with the same target are confirmed active, it is likely that a drug that hits this target will also be active. Right now, the most comprehensive public database of molecular targets is ChEMBL, which contains the results of over 400 000 experiments gleaned from the literature [28]. This data is lower confidence than the information about known drugs, and much more difficult to sort through. Nonetheless, some report success annotating large databases of molecules with public data [54]. More to the point, one study correctly identified known disease targets from phenotypic screens with 88% accuracy using a similar strategy [55].
Unexpected relationships between targets and diseases can motivate repurposing efforts. For example, genomic studies identified Lamin A (LMNA) as the gene responsible for the premature-aging disease, progeria. Biologists discovered that the diseased form of the protein was post-translationally modified and that inhibiting this modification with aminobisphosphonates—a class of FDA approved cancer drugs—ameliorated progeria in a mouse model [56]. A Phase II trial was initiated in May 2007, the first of its kind for progeria. Further research revealed an additional post-translational modification, modulated by statins, which also affects progression of progeria in mice [7]; this data motivated an additional Phase II trial which will soon begin. In this case study, targets were identified by genomic studies and molecular biology, not computational predictions. Nonetheless, this example illustrates how identifying targets and characterizing the pathways related to a disease can directly and quickly translate into new treatments for disease.
Still, this approach is limited by our incomplete understanding of small molecules' targets. While most bioactive molecules modulate one or a couple of known protein targets, there is no way of knowing whether they modulate additional clinically relevant proteins [57]. This is particularly a problem with certain classes of drugs, like the notoriously promiscuous kinase inhibitors [58], and no strategy handles this problem cleanly. The problem is worse for non-drug molecules because they are less studied. Nonetheless, in most cases the known target is the most likely mechanism of action of a drug for a new purpose, and successful examples of target identification using this approach are encouraging [55].
THE HOPE
The hope is that computational methods will discover unexpected connections between a known drug and a disease, and that this connection will guide focused experimental follow up, leading to successful repurposing. For example, a computational method would predict a drug would be active in a screen had it been tested. This prediction would be confirmed by testing the drug in the assay used in the screen. Next the drug would be tested in an animal model of the disease known to be relevant to the screen, where efficacy in modulating the disease would be demonstrated.
From this point, the next steps forward are complicated, but if the right details line up, the drug can rapidly move to human trials. The marketability, known side-effects, dose, intellectual property concerns and other factors directly affect whether efficacy could be demonstrated and inform whether human trials should be initiated. These issues are extremely important but beyond the scope of this discussion. The real value of HTS mining, however, is in proposing unexpected possibilities, rather than managing a repurposing project to completion.
Unfortunately, no published work clearly demonstrates a case where mining a small-molecule screen directly leads to a successful repurposing effort, so this approach remains an unrealized hope. This is not surprising; academics only recently gained access to HTS infrastructure and data, and the exact pathway used to repurpose a drug in pharmaceutical companies is rarely publicized and the cases that are known can be idiosyncratic [1, 59]. We will probably only know how successful HTS mining can be after at least a decade of academic effort that is just now beginning.
The results of hundreds of medically-relevant screens are publicly available, and the phenotypic screens are most likely to inspire new repurposing hypotheses.
Several web-based tools can identify drugs which are obviously active in medically-relevant screen, but less obvious connections can be made using similarity-based techniques in collaboration with experts.
Data mining efforts will be most effective when coupled with focused experimental follow up to verify predictions in the initial assay and to move confirmed predictions to animal and human studies.
It is hopeful that mining small-molecule screens will speed the discovery of new uses for medications, but this hope has not yet been realized.
Acknowledgements
SJS wrote the manuscript and created the figures. The photograph in Figure 1 was taken by SJS and used with his permission. Bradley T. Calhoun compiled results for the table. BTC and Michael R. Browning provided helpful edits. Pankaj Agarwal provided substantive edits and feedback on the manuscript. The author thanks the Pathology and Immunology Department of Washington University in St Louis for supporting SJS, BTC, and MRB. Marvin was used to generate the chemical structures in Figure 4; Marvin 5.3.5, 2010, ChemAxon (http://www.chemaxon.com). The author declares he has no competing financial interests.

{kind=link}
{kind=link}
{kind=link}
{kind=link}