




Abstract
The ability to sequence the DNA of an organism has become one of the most important tools in modern biological research. Until recently, the sequencing of even small model genomes required substantial funds and international collaboration. The development of ‘second-generation’ sequencing technology has increased the throughput and reduced the cost of sequence generation by several orders of magnitude. These new methods produce vast numbers of relatively short reads, usually at the expense of read accuracy. Since the first commercial second-generation sequencing system was produced by 454 Technologies and commercialised by Roche, several other companies including Illumina, Applied Biosystems, Helicos Biosciences and Pacific Biosciences have joined the competition. Because of the relatively high error rate and lack of assembly tools, short-read sequence technology has mainly been applied to the re-sequencing of genomes. However, some recent applications have focused on the de novo assembly of these data. De novo assembly remains the greatest challenge for DNA sequencing and there are specific problems for second generation sequencing which produces short reads with a high error rate. However, a number of different approaches for short-read assembly have been proposed and some have been implemented in working software. In this review, we compare the current approaches for second-generation genome sequencing, explore the future direction of this technology and the implications for plant genome research.
INTRODUCTION TO CROP GENOMES
Many crop genomes are large, complex and often polyploid, making genome sequencing a major challenge. With the decreasing cost of second-generation DNA sequencing technologies, genome size in itself would not prevent the application of whole genome sequencing approaches. However, the large size of many crop genomes is predominantly due to the amplification of repetitive elements as well as whole or partial genome duplication. Estimates of the dispersed repetitive fraction of the human genome, range from 35% [1] to 45% [2]. One study on the repetitive elements of the rice genus found the percentage of repetitive elements in the samples to range from 25% of reads in Oryza coarctata (HHKK) to 66% of reads in Oryza officinalis (CC) [3]. A similar study for maize estimated that the repetitive content ranges from 64 to 73% [4]. It is the resolution of repetitive sequences and the discrimination of duplicated regions of genomes that provides the greatest challenge for crop genome sequencing and assembly.
OVERVIEW OF SEQUENCING TECHNOLOGIES
Second-generation sequencing
The ability to sequence genomes is being advanced by increasingly high throughput technology. This is driven by what has been termed next or second-generation sequencing. Second-generation sequencing describes platforms that produce large amounts (typically millions) of short DNA sequence reads of length typically between 25 and 400 bp. While these reads are shorter than the traditional Sanger sequence reads, all current sequence reads are dwarfed by the size of many crop genomes which frequently consist of several thousand million base pairs of DNA.
The first approach to second-generation sequencing was pyrosequencing, and the first successful pyrosequencing system was developed by 454 Life Sciences and commercialised by Roche as the GS20. This was capable of sequencing over 20 million base pairs in just over 4 h [5]. The GS20 was replaced during 2007 by the GS FLX model, capable of producing over 100 million base pairs of sequence in a similar amount of time. More recently, this technology has advanced to produce more than 400 Mbp with the introduction of the Titanium chemistry. Two alternative ultra high throughput sequencing systems now compete with the Roche GS FLX; the SOLiD system from Applied Biosystems (AB); and Solexa Genome Analyser technology, which is now commercialised by Illumina. For a summary of the current (June 2009) data produced by each of these technologies, see Table 1.
Data produced by current DNA sequencing technologies.
| Sanger | GSFLX | AB SOLiD | Illumina GAII | |
|---|---|---|---|---|
| Nucleotides per run | 70 kbp | 400 Mbp | 20 Gbp | 50 Gbp |
| Read length | 750 bp | 400 bp | 50 bp | 75 bp |
| Sanger | GSFLX | AB SOLiD | Illumina GAII | |
|---|---|---|---|---|
| Nucleotides per run | 70 kbp | 400 Mbp | 20 Gbp | 50 Gbp |
| Read length | 750 bp | 400 bp | 50 bp | 75 bp |
Data produced by current DNA sequencing technologies.
| Sanger | GSFLX | AB SOLiD | Illumina GAII | |
|---|---|---|---|---|
| Nucleotides per run | 70 kbp | 400 Mbp | 20 Gbp | 50 Gbp |
| Read length | 750 bp | 400 bp | 50 bp | 75 bp |
| Sanger | GSFLX | AB SOLiD | Illumina GAII | |
|---|---|---|---|---|
| Nucleotides per run | 70 kbp | 400 Mbp | 20 Gbp | 50 Gbp |
| Read length | 750 bp | 400 bp | 50 bp | 75 bp |
The Solexa Genome Analyzer (currently the GAIIx) system uses reversible terminator chemistry to generate up to 50 000 million bases of usable data per run. Sequencing templates are immobilised on a flow cell surface, and solid phase amplification creates clusters of identical copies of each DNA molecule. Sequencing then uses four proprietary fluorescently labelled nucleotides to sequence the millions of clusters on the flow cell surface. These nucleotides possess a reversible termination property, allowing each cycle of the sequencing reaction to occur simultaneously in the presence of the four nucleotides. Illumina sequencing has been developed predominantly for re-sequencing, with more than 10-fold coverage usually ensuring high confidence in the determination of genetic differences. In addition, each raw read base has an assigned quality score, assisting assembly and sequence comparisons. The GAII is used for transcriptome profiling (RNA Seq) and Chromatin ImmunoPrecipitation sequencing (ChIP Seq), and the relatively low error rate of this system supports de novo sequencing applications. This technology has now been applied for the sequencing of several crop species including cucumber [6] and Brassica rapa.
The AB SOLiD System (currently version 3) enables parallel sequencing of clonally amplified DNA fragments linked to beads. The method is based on sequential ligation with dye labelled oligonucleotides and can generate more than 20 gigabases of mappable data per run. The system can be used for tag-based applications such as gene expression and Chromatin ImmunoPrecipitation sequencing, where a large number of reads are required and where the high throughput provides greater sensitivity for the detection of lowly expressed genes. The AB SOLiD system is predominantly used for re-sequencing where comparison to a reference and high sequence redundancy enables the identification and removal of erroneous sequence reads.
In contrast to the short-read sequencers, the Roche 454 FLX system produces read lengths on average 300–500 bp and is capable of producing over 400 Mbp of sequence with a single-read accuracy of >99.5%. Amplification and sequencing is performed in a highly parallelised picoliter format. Emulsion PCR enables the amplification of a DNA fragment immobilised on a bead from a single fragment to 10 million identical copies, generating sufficient DNA for the subsequent sequencing reaction. Sequencing involves the sequential flow of both nucleotides and enzymes over the beads, which converts chemicals generated during nucleotide incorporation into a chemiluminescent signal that can be detected by a CCD camera. The light signal is quantified to determine the number of nucleotides incorporated during the extension of the DNA sequence. Newbler software has been developed by 454 life sciences specifically to assemble this type of data and it has been successfully applied for the assembly of a bacterial genome [7].
Third-generation sequencing
There has been a dramatic increase in second-generation sequencing capability over the last 2–3 years and it is clear that we are still in the early stages of what can be achieved. It is expected that the Illumina and AB systems will be producing up to 100 Gbp of data per run by the end of 2009 due to an increase in read length and number of reads. In addition, several companies plan to bring to market what is termed third-generation sequencing, taking DNA sequence production to a further level of scale and dramatically reducing costs. The first of these technologies to come to market uses single molecule sequencing, commercialised by Helicos Biosciences (Cambridge, MA, USA). Termed ‘True Single-Molecule Sequencing’, this method has been used to sequence the genome of the virus M13 [8]. Pacific biosciences have developed a single molecule real time sequencing system that promises to produce several Gbp of relatively long reads (>1 kbp) [9]. Other companies such as VisiGen Biotechnologies (Houston, TX, USA, http://www.visigenbio.com); Complete Genomics (Mountain View, CA, USA, www.completegenomics.com/); NABsys (Providence, Rhode Island, USA, www.nabsys.com); ZS Genetics (North Reading, MA, USA, www.zsgenetics.com) and Oxford Nanopore Technologies (Oxford, UK, www.nanoporetech.com) are also working on third-generation sequencing systems and it is likely that more advanced sequencing will be available in the relatively near future. These changes will revolutionise genome sequencing and as data production becomes facile, the challenge increasingly becomes the ability to analyse this data and in particular, the ability to associate variation in sequences with heritable traits.
DE NOVO GENOME ASSEMBLY APPROACHES
Assembling the data
One challenge to sequencing crop genomes is the vast difference in scale between the size of the genomes and the lengths of the reads produced by the different sequencing methods. While there may be a 10–500× difference in scale between the short reads produced by second-generation sequencing and modern Sanger sequencing, this is still dwarfed by the difference between Sanger read length and the lengths of complete chromosomes. Human chromosomes vary between 47 and 245 million nucleotides in length, around 50 000–250 000 larger than average Sanger reads, and it was not considered feasible to sequence and de novo assemble the human genome using Sanger alone due to the complexity of assembly. It is therefore counter intuitive to consider sequencing more complex genomes using shorter reads. However, the ability to produce vast quantities of paired read sequence data gives significant power over traditional Sanger data, enabling the adoption of novel strategies to sequence complex plant genomes.
The first sequence fragment assembly algorithms were developed beginning in the late 1970s [10, 11]. Early sequencing efforts focussed on creating multiple alignments of the reads to produce a layout assembly of the data. Finally, a consensus sequence could be read from the alignment and the DNA sequence was inferred from this consensus. This approach is referred to as the overlap-layout-consensus approach and culminated in a variety of software applications such as CAP3 [12] and Phrap (Phil Green, Phrap Documentation, www.phrap.org). From the early to mid 1990's, research began to focus on formalising, benchmarking and classifying fragment assembly algorithm approaches. Two papers [13, 14] formalised the approach of placing sequence reads or fragments of reads in a directed graph. In this graph, each node represents a read or fragment and any two nodes are joined by an edge if there is an overlap on the left-hand side of one read with the right-hand-side of another. The resulting DNA sequence is inferred by walking along the edges of the graph. These two papers have formed the foundation for modern second-generation sequence assembly approaches. Idury and Waterman introduced the concept of Eulerian tours of a graph of equal length sequence fragments, while Myers formalised the concept of a fragment assembly string graph, where the read fragments need not be of the same length. These ideas were implemented in software such as Euler [15], and all modern sequence fragment assemblers are extensions of these concepts and in many cases they use some combination of the two.
Problems of assembling complex genomes
One challenge of genome sequencing lies in the fact that only a small portion of the genome encodes genes, and that these genes are surrounded by repetitive DNA that are often difficult to characterise. Repeats can cause ambiguity with fragment assembly and thus pose the greatest challenge when assembling genomic data.
The most robust method to overcome the problems of repeats when assembling shotgun reads is to increase the read length to a point that every repeat is spanned by at least one read. However, increasing sequence read length has proven technically problematic and most second-generation sequencing technologies compromise read length to deliver an increased number of reads.
Modifications of second-generation sequencing methods attempt to overcome the problem of short reads by producing read pairs, where two reads are produced with a known orientation and approximate distance between them, increasing the ‘effective’ read length. If the distance between the paired ends is large enough, repeats will be spanned by pairs of reads removing ambiguity from the assembly (Figure 1). It is expected that the ability to produce increased quantities of paired read sequence data, combined with custom bioinformatics tools, will provide the foundation to sequence large and complex plant genomes.
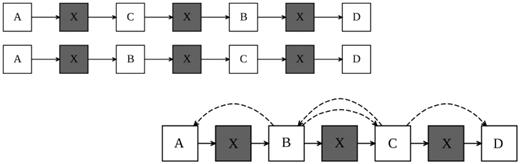
Repetitive regions (X) in genomes confound sequence assembly using the overlap consensus approach where the reads do not span the repeats. The use of paired reads that span the repetitive regions provide a reference for the spatial relationship between contigs and permit the ordering and orientation of unique sequences.
Current second-generation de novo sequence assembly tools
There have been a number of sequence assembly tools published in recent years. Successive tools have increasingly improved in terms of both the quality of the output assemblies as well as robustness in the assembly of more complex genomes [16]. There has been a general trend of improving elements of the algorithm underlying the Eulerian approach.
SSAKE
SSAKE was one of the first short-read assemblers released [17]. When SSAKE is run, all reads that can be joined using the method are returned as contigs and all reads not incorporated are returned in a separate file. The user has the option to set the size of the minimum overlap allowable between reads, and SSAKE can be set to stop whenever there are two or more candidates for extension (strict mode). When SSAKE is not run in strict mode, extension can still proceed in the face of multiple candidates. SSAKE does not incorporate data from paired ends and thus has no way to identify and differentiate between repetitive regions. Indeed in non-strict mode, SSAKE will effectively collapse multiple repetitive regions into one contig, and in strict mode any reads at the boundary of a repetitive region will break the contig and errors in the dataset will also break contigs.
VCAKE
VCAKE was released in 2007 and is an extension of SSAKE that can handle erroneous reads [18]. VCAKE begins exactly as SSAKE but handles the cases where there are no candidates or multiple candidates for extension differently. In the case where there is no read to extend with, VCAKE will allow a read with at most one mismatch after the first 11 bases to be used in the extension. In the case where there are multiple candidates, VCAKE employs a majority vote method based on read depth at the base in question to decide which read to extend with. In the case that the second most occurring read is present more than a set number of times, extension is terminated, as it is likely that the read is at the border of a repetitive region. In tests between VCAKE and SSAKE, SSAKE and VCAKE perform equally well on simple viral sequences but VCAKE appears to perform much better on more complicated sequences [18]. VCAKE does not make use of paired end data and so also cannot resolve ambiguities caused by repetitive regions.
SHARCGS
SHARGCS builds on the methods used in SSAKE and VCAKE [19]. SHARGCS employs a three-phase algorithm to assemble the data, including filtering, naïve assembly and refinement. Filtering comprises two separate operations. First, any reads not appearing a minimum number of times are removed, as are any reads which do not meet a minimum quality standard. When there are different quality values for a base on reads with the same orientation, the maximum is taken as the quality value for that base, and when the reads have reversed orientations, the sum of the quality values is taken. A second filtering step removes all reads that do not overlap with at least one read on either side. The default behaviour for SHARCGS produces three assemblies with different filtering levels. SHARCGS then attempts to merge contigs from different rounds of assembly to produce a more accurate set of contigs which are returned to the user with quality values. As with both SSAKE and VCAKE, SHARCGS does not include paired end data in the algorithm and as a result has an increased probability of collapsing repetitive regions.
Edena
Edena is the first short-read assembly algorithm to be released that uses the traditional overlap-layout-consensus approach [20]. Edena does not include an error correction phase before assembly, which leads to the formation of an overly complex sequence graph; however, it does include an error correction phase which cleans the graph before assembly begins. The first phase of the algorithm removes duplicate reads, keeping only the original read and the number of times it has been seen. Next, it uses the reads to construct an overlap graph where the reads are represented as nodes. Two nodes are joined by an edge if there is an overlap between them. The resulting graph contains erroneous edges that need to be removed. First Edena removes transitive edges using the method described by Myers [14]. Following this, dead end paths of consecutive nodes shorter than 10 reads, that are attached to the main body of the graph on only one side are removed. Finally Edena removes P-bubbles. P-bubbles occur when two sub-sequences are almost identical. This causes the graph to first fork and then rejoin, giving the appearance of a bubble in the graph. Where this is caused by single nucleotide polymorphisms (SNPs) between repetitive regions, each side of the bubble would have a similar topology and copy number. Where a P-bubble is caused by an error, one side of the bubble would have sparse topology and lower copy number (Figure 2). Edena removes the side of the bubble with the lowest copy number/sparsest topology. Once the graph has been cleaned, an assembly is produced and the resulting contigs returned to the user. Edena does not make use of paired end data.

An example of a P-bubble most likely caused by an error. The reads making up the lower sequence have a low copy number and the overlaps are short. This phenomenon can also be caused by low copy number repeats.
Velvet
Velvet [21] uses an Idury/Waterman/Pevzner model [13, 15] to make an initial graph. Like Edena, Velvet does not include an initial error correction phase but instead uses a series of error correction algorithms analogous to Edena to clean up the graph. Unlike Edena, Velvet uses an Euler-like strategy, which while efficient in terms of memory use, appears to complicate P-bubble removal. Edena removes P-bubbles by investigating topology and read depth and removing erroneous reads in their entirety. In an Euler-like algorithm, kmers can belong to both perfect and imperfect reads and there is a non-trivial amount of housekeeping to be done when removing individual kmers. Velvet includes an algorithm called Tour Bus that traverses the graph looking for P-bubbles, and uses a combination of copy number and topographical information to remove the erroneous edges. Velvet then removes all remaining low copy number edges. Velvet has recently been extended to include the use of paired reads using the Pebble and BreadCrumb algorithms.
EulerSR
The latest addition to the EULER family of algorithms [15] is EULER SR [22] which has been optimised to assemble short reads. The original EULER algorithm was designed as an implementation of the Idury and Waterman algorithm but included a novel method for error correction. Short reads are broken down into even shorter k-tuples. Pevzner describes a k-tuple as ‘weak’ if it appears less than a set number of times and ‘solid’ otherwise. For any read which contains weak k-tuples, the algorithm tries to find the minimum number of base changes which will make these k-tuples solid. If that number is less than an amount defined by the user, then the changes are made, otherwise the sequence is discarded. Pevzner shows that this method corrects over 86% of errors for the dataset he analysed [15]. After the reads have been corrected, a de Bruijn graph is constructed and a set of contigs produced and returned to the user. Like Velvet, Euler can make use of paired read data to assist in the assembly.
AllPaths
ALLPATHS [23] begins by correcting errors using an EULER like method, and then makes a large set of assemblies, referred to as unipaths. ALLPATHS then uses paired read information to sort unipaths into localised groups. For each localised group of unipaths, the number of paths is trimmed until, ideally only one path remains. This method reduces the complexity in the overall graph by making local optimisations. The results from the local optimisations are stitched together to produce one long sequence graph. The final phase of the algorithm attempts to fix errors that may have been incorporated in earlier stages. Unlike most other assemblers, AllPaths returns the assembly as a graph, complete with ambiguities which can arise from limitations of the data set and also from polymorphism in diploid genomes. This graph can be used in further downstream analysis.
ABySS
ABySS is the newest next-generation short-read assembler and the first to natively embrace parallel architectures [24]. The algorithm behind ABySS is almost identical to that used in other popular Eulerian assemblers. The distributed elements of ABySS use the Message Passing Interface (MPI) protocol for communication between nodes.
Summary
Currently, the most popular tools for genome assembly using short read data are Velvet, ABySS and EulerSR. These programs offer different advantages; for example Velvet can use a mixture of both short reads and traditional long (Sanger) reads while ABySS has been designed to run on a distributed system. Generally, the older algorithms that do not use paired end data will not be capable of accurately assembling the genomes of organisms more complex than bacteria. There continue to be advances in the current published algorithms, and additional approaches are under development. We expect that this field of research will continue to advance to face the challenge of assembling currently intractable large and complex genomes such as wheat and barley.
Sequencing approaches
Sequencing technology has undergone a major revolution since the first Eukaryotic genomes were sequenced using traditional Sanger methods. The ability to produce gigabases of DNA sequence data has allowed researchers to consider sequencing genomes which were previously considered intractable. The approaches to genome sequencing have also changed, moving from laborious BAC by BAC to whole genome shotgun (WGS) methods. The most appropriate genome sequencing approach depends on the genome size and complexity as well as the technology available at the time.
Genome sequencing using bacterial artificial chromosomes
The most robust method for genome sequencing is known as BAC by BAC sequencing. The idea behind this method is to cut a large genome into smaller overlapping sections (usually ∼100–150 kbp in length). The DNA fragments are maintained as bacterial artificial chromosomes (BACs) enabling the production of sufficient quantities of DNA for sequencing. The next step is usually to order these fragments using physical and genetic mapping, and then sequence a minimal tiling path of these fragments that represents the complete genome. BAC sequencing can use traditional Sanger, 454 pyrosequencing or Illumina GAII short read sequencing, or potentially combinations of these methods. Assembly of the sequence reads to produce the representation of the BAC fragment could use a range of assemblers specific to the data type. Examples include Phrap, CAP3 [12], Newbler, or Velvet [21]. Several additional assemblers are available or under development. The benefit of BAC by BAC genome sequencing is the reduction of complexity, as only relatively short regions are assembled at one time, and this approach is still favoured for genomes which contain a large number of repetitive elements. However, the cost of producing and mapping the BAC library as well as the significant cost associated with the sequencing of large numbers of BACs makes BAC by BAC sequencing approaches increasingly unfavourable, and several genome sequencing projects which started with the BAC by BAC approach have now moved to WGS methods.
WGS
An alternative approach to BAC by BAC sequencing is the WGS method. This approach avoids the costly process of generating a BAC library and minimum tiling path by randomly cutting the entire genome into many smaller reads which are then individually sequenced. Computational algorithms order and assemble the vast number of reads to resolve the complete genome sequence. There are often time and cost savings of using WGS over a BAC by BAC approach; however these benefits are offset by the difficulty in assembling the reads. With large and complex cereal genomes, WGS remains unfeasible, as the majority of the reads will represent repetitive elements in the genome. However, the ability to produce increasing volumes of data along with the development of advanced algorithms for the assembly of this data is making WGS increasingly popular.
Alternative approaches
While the BAC by BAC approach remains expensive and WGS is difficult for complex genomes, alternative methods have been developed to reduce the genome complexity for sequencing. Some methods attempt to sequence a portion of the genome. These include restriction analysis, where genomic DNA is digested with restriction endonucleases and fractionated to remove repeat abundant fractions [25]. An alternative method for reduced complexity sequencing involves the isolation of specific chromosomes or portions of chromosomes by chromosome sorting [26–28]. Other methods enrich for gene rich regions of the genome. These include methyl filtration, where genomic sequences are enriched for hypomethylated low copy regions [29, 30], and CoT based cloning [31–33] which uses DNA renaturation kinetics to enrich for low abundance sequences. These methods all reduce the complexity of the sample being sequenced and provide an alternative to BAC by BAC and WGS approaches.
Re-sequencing
The process of whole genome re-sequencing involves aligning a set of reads to a reference genome that is similar to the subject being sequenced. Re-sequencing has proved to be a valuable tool for studying genetic variation and with the re-sequencing of the genomes of James Watson and Craig Venter [34, 35], the task of re-sequencing has largely been conquered and large scale re-sequencing of human populations is now underway [36]. Although re-sequencing can be very useful, the quality of the re-sequenced genome is highly dependant on the quality and relatedness of the reference sequence.
EXAMPLES OF PLANT GENOME SEQUENCING PROJECTS
Arabidopsis [37] and rice [38, 39] were the first plant genomes to be sequenced, applying standard Sanger sequencing of tiled genomic fragments maintained in BAC vectors. Plant genome sequencing projects are rapidly changing pace with the new technology and researchers are quickly adopting second-generation sequencing to gain insight into their favourite genome. Roche 454 technology is being used to sequence the 430 Mbp genome of Theobroma cacao [40], while a combination of Sanger and Roche 454 Sanger sequencing and 454 (4× versus 12× coverage, respectively) is being used to interrogate the apple genome [41, 42]. A similar approach is being applied to develop a draft consensus sequence for the 504 Mbp grape genome [43] where a combination of 6.5× Sanger paired read sequences and 4.2× unpaired Roche 454 reads were assembled into 2093 metacontigs (ordered and aligned scaffolds of contiguous sequence with some gaps) representing an estimated 94.6% of the genome. This study also identified 1.7 million SNPs which were mapped to chromosomes. Illumina Solexa and Roche 454 sequencing has also been used to characterise the genomes of cotton [44]. Roche 454 sequencing has been used to survey the genome of Miscanthus [45], while Sanger, Illumina Solexa and Roche 454 sequencing is being used to interrogate the banana genome [46].
Ossowski et al. [47] reported the re-sequencing of reference Arabidopsis accession Col-0 and two divergent strains, Bur-0 and Tsu-1 using Illumina Solexa technology to produce between 15× and 25× coverage. As well as finding over 2000 potential errors in the reference genome sequence, they identified more than 800 000 unique SNPs and almost 80 000 1–3 bp indels. Longer indels are more difficult to identify from short read data without the use of paired reads, however, they did identify more than 3.4 Mbp of the divergent accessions that is dissimilar to the Col-0 reference. The re-sequencing of rice and Medicago truncatula has also been undertaken using the Illumina Solexa for SNP discovery [48, 49]. As increasing numbers of reference genomes become available, it is expected that whole genome re-sequencing of crop genomes will become common, providing insights into crop genome structure and diversity. While Brassica shares extensive synteny with Arabidopsis thaliana, there is not enough similarity at the microsynteny level to approach Brassica genome sequencing as a re-sequencing of Arabidopsis. The Multinational Brassica Genome Project (MBGP) steering committee selected B. rapa as the first Brassica species to be fully sequenced, and established a BAC by BAC approach, though recently, llumina GAII sequencing has been applied to generate 70× coverage of this genome in a collaboration with the Beijing Genome Institute in Shenzhen (BGI) and the Institute of Vegetables and Flowers (IVF) in Beijing (X Wang, pers. comm.). It is expected that the combination of the BAC sequence data and the assembled contigs from the Illumina GAII data will lead to the release of a high quality genome sequence during 2009. The sequencing of Brassica rapa follows from the success of sequencing the cucumber genome, where a hybrid strategy consisting of 4× Sanger, 68× Illumina Solexa, as well as 20× and 10× coverage from end sequenced fosmid and BAC libraries were used to assemble more than 96% of the cucumber genome [6].
The large cereal genomes remain elusive, but the advances in technology are starting to make de novo sequencing of these genomes feasible. Roche 454 has been applied for the sequencing of complex BACs from barley [50, 51] and this has been complemented by repeat characterisation using WGS Illumina Solexa data [52]. However, the size of the wheat genome is much larger (17 000 Mbp) than related cereal genomes such as barley (Hordeum vulgare, 5000 Mbp), rye (Secale cereale, 9100 Mbp) and oat (Avena sativa, 11 000 Mbp). The size and hexaploid nature of the wheat genome creates significant problems in elucidating its genome sequence and it may take several years before sequencing technology is fast and cheap enough to readily determine the whole genome sequence. A pilot project led by the French National Institute for Agricultural Research (INRA) was initiated in 2004. A total of 68 000 BAC clones of a 3B chromosome specific BAC library [53] have been fingerprinted and there are plans to sequence a minimal tiling path of BACs for this chromosome using Roche 454 technology. Expectations are that by the end of 2010, the majority of the gene-rich regions of hexaploid wheat will have been sequenced. However, these expectations are likely to be modified by the rapid changes in sequencing technology and the ever-reducing cost of producing sequence data.
CONCLUSIONS, FUTURE DIRECTIONS AND CHALLENGES
The advances in genome sequencing technology have provided a means to sequence increasingly large and complex genomes. Data generation is now no longer limiting complex genome sequencing, and the sequencing and re-sequencing of previously intractable plant genomes is now becoming feasible. The ability to analyse and assemble this data is currently limited by a lack of dedicated bioinformatics tools that are designed to cope with the repetitive nature of many crop genomes. We have also seen that with the release of each new assembly tool we see an improvement, even for static datasets, in assembly statistics. These include amongst others: an increase in contig sizes, a decrease in the total number of contigs and a decrease in the number of assembly errors. While it has not been formally proven that complex repeats can always be resolved from the data currently produced simply using novel algorithms, newer algorithms have continually demonstrated improvements in the overall quality of assemblies and this is largely due to the ever increasing sophistication in their methods of handling repeats. However sophisticated the current methods for handling repeats are now, they still represent a nascent stage in the evolution of these algorithms. There are many directions this improvement can take and we are confident that the greatest increases in the ability to accurately resolve repeat structures in crop genome assemblies will stem from improvements made to the assembly algorithms. Improvements in data quality and quantity will of course also assist in producing better assemblies. As improved algorithms are developed, it may be expected that the sequencing of large and complex genomes will become commonplace, and following the research path of mammalian genomics, focus will move to the detection of sequence variation and the association of this variation with important agronomic traits, with downstream applications for crop improvement.
FUNDING
Grains Research and Development Corporation (Project DAN00117); Australian Research Council (Projects LP0882095, LP0883462, LP0989200); Australian Genome Research Facility (AGRF); the Queensland Cyber Infrastructure Foundation (QCIF); the Australian Partnership for Advanced Computing (APAC); and Queensland Facility for Advanced Bioinformatics (QFAB).
The ability to produce genome sequence data continues to increase at an unprecedented rate.
The size of many plant genomes combined with the large number of repetitive elements and polyploidy creates a challenge for traditional genome sequencing methods.
Current sequencing and assembly approaches cannot produce a finished genome for large and complex cereal genomes.
Novel approaches will be required to enable the sequencing of complex plant genomes using second or third-generation sequencing data.

{kind=link}
{kind=link}