





Abstract
Chromosome instability, a hallmark of lung cancer, is a driving mechanism for hexavalent chromium [Cr(VI)] carcinogenesis in humans. Cr(VI) induces structural and numerical chromosome instability in human lung cells by inducing DNA double-strand breaks and inhibiting homologous recombination repair and causing spindle assembly checkpoint (SAC) bypass and centrosome amplification. Great whales are long-lived species with long-term exposures to Cr(VI) and accumulate Cr in their tissue, but exhibit a low incidence of cancer. Data show Cr(VI) induces fewer chromosome aberrations in whale cells after acute Cr(VI) exposure suggesting whale cells can evade Cr(VI)-induced chromosome instability. However, it is unknown if whales can evade Cr(VI)-induced chromosome instability. Thus, we tested the hypothesis that whale cells resist Cr(VI)-induced loss of homologous recombination repair activity and increased SAC bypass and centrosome amplification. We found Cr(VI) induces similar amounts of DNA double-strand breaks after acute (24 h) and prolonged (120 h) exposures in whale lung cells, but does not inhibit homologous recombination repair, SAC bypass, or centrosome amplification, and does not induce chromosome instability. These data indicate whale lung cells resist Cr(VI)-induced chromosome instability, the major driver for Cr(VI) carcinogenesis at a cellular level, consistent with observations that whales are resistant to cancer.
Hexavalent chromium [Cr(VI)] is a global environmental contaminant and a known human lung carcinogen (Straif et al., 2009). To understand how Cr(VI) affects health, it is important to consider multiple perspectives by using a One Environmental Health approach (Pérez and Pierce Wise, 2018). Whales are of particular interest because they are the closest marine relative to humans, have long lifespans, breathe air, and accumulate high levels of Cr (Wise et al., 2009, 2015, 2019). Having extended lifespans and 1000-fold the number of cells humans have, whales would be expected to have higher rates of cancer than humans. Yet, species such as bowhead whales have been estimated to live over 200 years and reports of cetacean cancers are rare (Keane et al., 2015; Newman and Smith, 2006). This incongruity is known as “Peto’s Paradox” (Caulin and Maley, 2011; Nunney, 1999; Peto et al., 1975) and makes whales good candidates for comparative analyses to determine how 2 species with unique environmental adaptations respond differently to toxicants. The longevity of whales while living in an extreme environment strongly suggests whales have effective mechanisms conferring resistance to cancer caused by environmental carcinogens (de Magalhães, 2015; Keane et al., 2015; Ma and Gladyshev, 2017; Seim et al., 2014).
A key driving mechanism in Cr(VI) carcinogenesis is chromosome instability (Hirose et al., 2002; Holmes et al., 2008; Kondo et al., 1997; Proctor et al., 2014; Salnikow and Zhitkovich, 2008; Wise and Wise, 2010). Chromosome instability has 2 main categories: structural chromosome instability, featuring chromosome breaks, gaps, and translocations, and numerical chromosome instability characterized by the loss or gain of entire chromosomes (Funk et al., 2016; Negrini et al., 2010; Palumbo and Russo, 2016). Numerous studies show Cr(VI) induces structural chromosome instability (Wise et al., 2002, 2003, 2006a; Xie et al., 2005, 2009) and its underlying lesion DNA double-strand breaks (Ha et al., 2003, 2004; Wakeman et al., 2004; Xie et al., 2005, 2008). Prolonged particulate hexavalent chromium exposure suppresses the RAD51 response in human lung cells (Bryant et al., 2006; Gastaldo et al., 2007). Studies in human lung cells show acute (24 h) Cr(VI) exposure induces double-strand breaks and increases homologous recombination repair in a canonical manner (Browning et al., 2016; Qin et al., 2014; Xie et al., 2009). In contrast, prolonged (120 h) Cr(VI) exposure increases DNA double-strand breaks, but actually inhibits homologous recombination repair by targeting RAD51, a key recombinase in homologous recombination repair (Browning et al., 2016; Qin et al., 2014).
Numerical chromosome instability is also a key effect of Cr(VI) and the most common form of chromosome instability in cancer (Brinkley, 2001; Holmes et al., 2006; Vasudevan et al., 2021; Wise et al., 2018; Xie et al., 2009). Gain or loss of chromosomes causes gene imbalances that perturb pathways critical to genomic stability, such as DNA repair, cell cycle regulation, and DNA segregation (Duijf and Benezra, 2013; Pavelka et al., 2010; Stingele et al., 2012; Vasudevan et al., 2021). How numerical instability arises is not fully understood but it is known that mitotic disruption can cause asymmetrical segregation of chromosomes, resulting in aneuploid daughter cells. Cr(VI) causes centrosome amplification and premature centromere division, which are both drivers of mitotic disruption leading to numerical chromosome instability (Holmes and Wise, 2010; Holmes et al., 2006, 2010; Martino et al., 2015).
How whales evade carcinogenesis is unknown. Studies show whale cells are more resistant to Cr(VI)-induced chromosome aberrations than human cells (Li Chen et al., 2009, 2012; Wise et al., 2015), suggesting whales may have cellular mechanisms to evade Cr(VI)-induced chromosome instability. This study focused on the mechanisms underlying Cr(VI)-induced chromosome instability to determine which aspects whale cells can protect against.
Materials and methods
Chemicals and reagents
Zinc chromate (CAS no. 13530-65-9; 99.7% purity) was purchased from Pfaltz and Bauer (Waterbury, Connecticut). DMEM/F12 (1×), phosphate-buffered solution (PBS) 1× with calcium and magnesium, PBS 1× without calcium or magnesium, glutagro supplement (200 mM), penicillin/streptomycin solution, tissue culture dishes, flasks, and plasticware were purchased from Corning, Inc. (Corning, New York). Sodium pyruvate (100 mM) was purchased from Lonza (Allendale, New Jersy). Trypsin-EDTA (0.25%), cosmic calf serum was purchased from Hyclone (Logan, Utah). Prolong Diamond Antifade Mountant with DAPI were purchased from Life Technologies (Grand Island, New York). FNC Coating Mix was purchased from AthenaES (Baltimore, Maryland). Demecolcine, potassium chloride (KCl), triton X-100, 5-bromo-2′-deoxyuridine (BrdU), ethylene glycol tetraacetic acid (EGTA), fish skin gelatin, and glycerol were purchased from Sigma-Aldrich (St Louis, Missouri). 4% paraformaldehyde in PBS was purchased from Alfa Aesar (Ward Hill, Massachusetts). Tween-20, sodium dodecyl sulfate, nitric acid, and micro cover glass were purchased from VWR International (Radnor, Pennsylvania). Piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES) was purchased from Alfa Aesar (Ward Hill, Massachusetts). Sodium azide was purchased from Amresco, Inc. (Solon, Ohio). Magnesium sulfate (MgSO4) was purchased from J.T. Baker (Phillipsburg, New Jersy). Giemsa stain was purchased from Biomedical Specialties Inc. (Santa Monica, California). Acetic acid, methanol, tris-base, EDTA, glycine, and sodium acetate were purchased from J.T. Baker (Phillipsburg, New Jersy). HALT protease and phosphatase inhibitor cocktail, RIPA buffer, Nunc Lab Tek II glass chamber slides, Scientific Super Up-Rite microscope slides, cytoseal 60, and Gurr’s buffer tablets were purchased from Thermo Fisher Scientific Inc. (Waltham, Massachusetts). Mini-Protean TGX gels, 4× protein sample loading buffer, and Odyssey blocking buffer (TBS) were purchased from LI-COR (Lincoln, Nebraska). Gentamicin was purchased from IBI Scientific (Dubuque, Iowa). RNase A was purchased from Fisher BioReagents (Hampton, New Hampshire). Rat serum was purchased from StemCell Technologies (Cambridge, Massachusetts). Fetal bovine serum was purchased from Atlanta Biologicals (Flowery Branch, Georgia).
Cell line development and cell culture
Primary bowhead whale lung fibroblast cells were derived from a male whale obtained during subsistence hunts in Barrow, Alaska. Whale fibroblasts were isolated from tissue and maintained according to our published methods (Wise et al., 2011). Briefly, tissue was immersed for at least 30 min in tissue buffer containing 2% penicillin/streptomycin and 0.4% gentamicin. Tissue was sliced into small pieces using a sterile scalpel and placed in tissue culture flasks with 500 µl of DMEM/F12 media containing 15% cosmic calf serum, 1% Glutagro, 1% penicillin/streptomycin, and 0.1% sodium pyruvate. Flasks were inverted and incubated at 33°C. After 48 h, flasks were turned upright and flooded with media then monitored for fibroblast migration from explants. After colonization of the tissue culture flask, tissue was removed and cells were released with 0.25% trypsin-EDTA. Whale lung cells were incubated in 5% CO2 at 33°C, medium was replaced every 2 days and cells were subcultured every 6 days.
WTHBF-6 is an immortalized human lung cell line derived from normal human bronchial fibroblasts from a 67-year-old Caucasian male’s normal lung (Wise et al., 2004). All cells were maintained as subconfluent monolayers in DMEM/F12 media supplemented with 15% cosmic calf serum, 1% penicillin/streptomycin, 1% Glutagro, and 0.1% sodium pyruvate. Human lung cells were incubated in 5% CO2 at 37°C, medium was replaced every 2 days and cells were subcultured every 4 days using 0.25% trypsin-EDTA.
Treatment with particulate Cr(VI) compound
All experiments were performed on logarithmically growing cells. The whale lung fibroblasts have an approximate doubling time of 36 h (Wise et al., 2011) and were allowed 72 h to enter logarithmic growth phase before treatment. According to published methods (Xie et al., 2009), zinc chromate was prepared by washing twice with deionized H2O to remove water soluble contaminants, rinsed twice with acetone to remove organic contaminants, and thoroughly dried. Washed zinc chromate was suspended in sterile deionized water and stirred overnight at 4°C. Before treatment, fresh media was added to cell culture dishes and a suspension of zinc chromate particles was applied at environmentally relevant concentrations (as explained in Toyoda et al., 2023) of 0, 0.1, 0.2, 0.3, or 0.4 µg/cm2. Treatment durations were 24 or 120 h to represent acute or prolonged Cr(VI) exposure, respectively, because RAD51 and homologous recombination repair activity differ in human lung cells after 24 and 120 h exposures to 0.1–0.3 µg/cm2 zinc chromate (Browning et al., 2016; Qin et al., 2014).
Clonogenic survival assay
Cytotoxicity was determined by a clonogenic survival assay following previously published methods (Wise et al., 2002). Cells were seeded on 6-well plates and then treated for 24 or 120 h with zinc chromate. At the end of the treatment period, media were removed and cells were rinsed with 1× PBS and released from the plate with 0.25% trypsin-EDTA. From each treatment condition, 1000 cells were seeded onto each of four 100 mm tissue culture dishes. Cells were maintained in culture, without any further treatment, and media were changed every 5 days until colonies formed. Colonies were stained with crystal violet. Colonies were counted in each dish and averaged across all dishes for each treatment. Average colony growth per treatment group was reported relative to the control group. Three independent experiments were performed.
Flow cytometry for cell cycle arrest and γ-H2AX staining
In order to determine cell cycle stage and quantify DNA double-strand breaks during distinct cell cycle phases, flow cytometry for cell cycle analysis and γ-H2AX staining was used according to published methods (Huang and Darzynkiewicz, 2006). Briefly, cells were harvested after zinc chromate treatment and fixed with 4% paraformaldehyde for 15 min on ice. Cells were washed with 1× PBS, centrifuged and transferred to a new tube containing cold 70% ethanol. Samples were stored at −20°C for at least a week before analysis. After fixation and permeabilization, cells were washed with FACS Buffer (1× PBS, 10 mM sodium azide, 5% FBS). Cells were then blocked with 2% rat serum for 15 min followed by incubation with primary anti-γ-H2AX-AF647 (pS140) antibody (BD Biosciences, 560447) at the manufacturer’s recommended amount of 0.5 µg per sample for 1 h at room temperature. An isotype control sample was incubated with Mouse IgG1 kappa-AF647 Isotype antibody (BD Biosciences, 557714) at the manufacturer’s recommended concentration of 0.5 µg per sample for 1 h at room temperature. Samples were washed with FACS buffer before incubation for 30 min in the dark with 1 ml propidium iodide (PI) staining solution. The PI staining solution was as follows per sample: 1 ml PBS, 1 mg RNase A, and 5 µl PI (1 mg/ml in water) (Invitrogen, P3566). Samples were analyzed by flow cytometry (Attune NxT V6, Thermo Fisher Scientific). Data were stratified by phase of the cell cycle based on PI fluorescence into G1, S, and G2/M phases. Within each phase of the cell cycle, DNA damage was quantified by calculating the percentage of cells staining positive for γ-H2AX normalized to control fluorescence. Data were analyzed using FlowJo Software (version 10.8) and expressed as the percentage of cells in each phase of the cell cycle and the percent γ-H2AX positive cells in each phase of the cell cycle normalized to control for each phase.
Neutral comet assay
The comet assay (ie, single-cell gel electrophoresis) is a method for measuring DNA strand breaks in eukaryotic cells. Neutral comet assay was used to measure DNA double-strand breaks as previously described with slight adjustments (Xie et al., 2005). Logarithmically growing cells were seeded and treated with zinc chromate. At the end of the treatment, the cells were washed, collected, and resuspended in PBS. Then the cell suspension was mixed with agarose at a ratio of 1:10 and spread evenly on Trevigen Comet slides. After complete solidification at 4°C, slides were immersed in a freshly prepared lysis solution (2.5 M NaCl, 100 mM EDTA, 10 mM Tris, pH 10) containing an additional 1% Triton X-100 at 4°C for 30 min, followed by 1 mg/ml proteinase-K incubation for 2 h at 37°C. Electrophoresis was carried out at 21 V (1 V/cm) for 20 min in a freshly prepared electrophoresis buffer (300 mM sodium acetate, 100 mM Tris, pH 9.0). Finally, slides were immersed in DNA precipitation buffer (0.15 M NH4Ac, ethanol) for 20 min followed by fixation in 70% ethanol and stained with SYBR green. All the steps described above were conducted under reduced light to prevent spurious DNA damage. Comet images were captured using an Olympus fluorescence microscope equipped with a SensiCam. Image analysis was carried out by Comet Assay IV software (Perceptive Inc., UK). The tail intensity was adopted as a measurement to quantify DNA damage. One hundred randomly selected cells were analyzed for each treatment concentration. Three independent experiments were conducted.
Immunofluorescence staining
DNA damage response proteins, such as γ-H2AX and RAD51, accumulate and/or modify near chromosomal DNA double-strand breaks to form nuclear foci visible under a fluorescence microscope, which is helpful for the study of DNA damage and repair. Immunofluorescence staining was conducted as previously described with minor alterations (Xie et al., 2005). Briefly, cells were seeded on glass chamber slides coated with FNC, allowed to re-enter logarithmic growth, and treated with zinc chromate for either 24 or 120 h. At harvest, cells were fixed with 4% paraformaldehyde for 10 min, permeabilized with 0.2% Triton X-100 for 10 min, and blocked with 10% goat serum and 5% BSA in PBS for 1 h. For RAD51 foci, cells were fixed with 4% paraformaldehyde for 10 min, permeabilized with 0.5% Triton X-100 for 30 min. Cells were then incubated with anti-γ-H2AX (Cell Signaling, 2577L; 1:100) or anti-RAD51 (Santa Cruz, sc-8349; 1:200) antibodies at 4°C overnight, washed with PBS, and incubated with Alexa Fluor 488 (Life Technologies, A11034; 1:2000) for 1 h. Cells were washed with PBS and coverslips were mounted with DAPI. Slides were analyzed by fluorescent microscopy on Olympus BX51 and representative images were obtained by Applied Spectral Imaging GenASIS software version 7.1.0.1277. Nuclear foci were scored in 100 cells per concentration/timepoint using fluorescence microscopy. Results were expressed as the percentage of cells with >28 or >5 foci based on background levels such that negative controls had 5% or less of cells with this level. Images of 100 cells per concentration/timepoint were obtained with an Olympus microscope. Three independent experiments were conducted.
Centrosomes were analyzed by immunofluorescence as published previously (Holmes et al., 2006). Bowhead whale cells were seeded on glass, FNC-coated chamber slides. Cells were allowed to re-enter logarithmic growth, and treated with zinc chromate at final concentration of 0 or 0.2 µg/cm2 zinc chromate. After 24 or 120 h exposure, media were aspirated and cells were washed twice with microtubule stabilizing buffer (3 mM EGTA, 50 mM PIPES, 1 mM MgSO4, and 25 mM KCl), fixed with −20°C methanol for 10 min and allowed to air dry completely. Cells were rehydrated for 3 min in 0.05% Triton X-100, followed by 30 min of blocking with centrosome buffer (5% normal goat serum, 1% glycerol, 0.1% bovine serum albumin, 0.1% fish skin gelatin, 0.04% sodium azide in PBS). Cells were incubated with anti-γ-tubulin antibody (Sigma, T6557; 1:1000) and anti-α-tubulin-FITC antibody (Sigma, F2168; 1:250) for 1 h each, washing in PBS 3 times between each incubation. Cells were incubated with isotype-specific Alexa Fluor 555 (Life Technologies, A21422; 1:800) for γ-tubulin visualization. Cells were washed and aged overnight before mounting coverslips with Prolong Diamond Antifade Mountant with DAPI. Slides were analyzed by fluorescent microscopy on an Olympus BX51 microscope and representative images were obtained by Applied Spectral Imaging GenASIS software version 7.1.0.1277. Centrosome number was counted in 1000 interphase and 100 mitotic cells per treatment. Two independent experiments were analyzed.
Sister chromatid exchange assay
Sister chromatid exchanges (SCEs) were quantified as an assessment of homologous recombination repair activity according to our published methods (Browning et al., 2017). Cells were seeded into 100 mm dishes, allowed to re-enter logarithmic growth, and treated with zinc chromate for 24 or 120 h. Then, cells were incubated with 0.6 µg/ml BrdU for 72 h prior to harvesting. Five hours prior to the end of treatment, 0.1 µg/ml demecolcine was added to arrest cells in metaphase. Cells were then harvested, resuspended in 0.075 M KCl for 17 min and fixed with 3:1 methanol: acetic acid. Then cells were dropped on clean, wet slides. Slides were aged overnight, immersed in PBS for 5 min and then stained with 0.5 µg/ml Hoechst 33258 pentahydrate solution for 10 min at room temperature. Slides were incubated in 25 µg/ml Hoechst 33258 pentahydrate solution under 27 W fluorescent lights for 11 h in a humidity chamber. After incubation, slides were washed with distilled water and soaked in 2× sodium chloride/sodium citrate solution for 15 min at 60°C. Finally, slides were washed with distilled water, stained with 4% Giemsa stain in Gurr’s buffer and coverslipped. The total number of SCEs was counted in 50 metaphases per concentration/timepoint. Three independent experiments were conducted.
Mitotic index analysis
To determine whether cells accumulate during mitosis, the mitotic index was measured as described previously (Xie et al., 2008). Cells were seeded, allowed to re-enter logarithmic growth, and treated with various concentrations of zinc chromate for 24 or 120 h. After treatment, cells were harvested and incubated in 0.075 M KCl hypotonic solution for 10 min. Then cells were fixed, dropped on slides as described in SCE assay method. Slides were uniformly stained using a 5% Giemsa stain in Gurr’s buffer. The mitotic index was determined as the number of mitotic cells per 5000 total relative to control cells using light microscopy. Three independent experiments were conducted.
Chromosome instability assay
Measurement of chromosomal aberrations was used to determine clastogenicity and changes in chromosome number according to our published methods (Wise et al., 2002). Briefly, cells were seeded, allowed to re-enter logarithmic growth, and treated with various concentrations of zinc chromate for 24 or 120 h. Five hours for whale lung cells and 1 h for human lung cells prior to the end of treatment, 0.1 µg/ml demecolcine was added to arrest cells in metaphase. Cells were then harvested, fixed, and dropped on slides as described in SCE assay method. Finally, slides were stained with 5% Giemsa stain and scored for chromosome aberrations using previously defined criteria (Wise et al., 2002). One hundred metaphases were analyzed for each treatment and 3 independent experiments were conducted. Any metaphases with greater or fewer than the normal chromosome complement were scored as aneuploid. Evidence of spindle assembly bypass including centromere spreading, premature centromere division, and premature anaphase was recorded. A minimum of 100 diploid metaphases per concentration were analyzed and abnormal centromere separation events were recorded in all metaphases encountered during analysis.
Cell-equivalent Western blot analysis
Cells were seeded in tissue culture dishes and treated with zinc chromate as described above. Cell-equivalent whole cell protein analysis was performed as published previously (Speer et al., 2021). At the end of Cr(VI) exposure period, cells were released from dishes by incubation in 0.25% trypsin-EDTA. Cells were collected, washed in PBS, and counted with Beckman Coulter Multisizer 4e. Cells were lysed in RIPA buffer supplemented with 100× HALT protease and phosphatase inhibitor cocktail. Cell lysis was centrifuged 10 min at 14 000 rpm and supernatant was collected and prepared with 4× protein sample loading buffer. Protein samples in loading buffer were heated for 10 min at 70°C and stored at −20°C. Prepared sample volumes equivalent to 50 000 lysed cells (antibody-dependent) was loaded into Mini-Protean TGX gels. After electrophoresis, protein was transferred onto 0.45-µm nitrocellulose membrane in ice-cold transfer buffer with 10%–20% methanol. Membrane was probed with anti-securin mouse monoclonal antibody (Abcam, ab3305; 1:500) and anti-α-tubulin rabbit monoclonal antibody (Cell Signaling, 2125; 1:1500) at 4°C overnight and then IRDye 800CW (Li-Cor, 925-32210; 1:15 000) and IRDye 680RD near-infrared fluorescent secondary antibodies (Li-Cor, 925-68071, 1:15 000) at room temperature for 1 h. Membranes were scanned using Odyssey CLx scanner and analyzed with Li-Cor Image Studio software (version 5.2). Three independent experiments were analyzed.
Statistics
Statistical analyses were performed using GraphPad Prism 9.4.0. All data were expressed as the mean ± SEM (standard error of the mean). A 1-way ANOVA with Dunnett’s multiple comparisons test was used when comparing the means of 2 variables at the same timepoint or same species. A 2-way ANOVA with Tukey’s multiple comparisons test was performed when comparing human lung SCE data. A 2-way ANOVA with Dunnett’s multiple comparisons test was used when comparing the means of 2 variables in cell cycle data. A 2-way ANOVA with Bonferroni’s multiple comparisons test was used when comparing the means of 2 variables in different species at the same timepoint. A student t-test was used when comparing the means of 2 variables in centrosome amplification in interphase cells and in mitotic cells in human lung cells. Statistical significance was determined to be a p value less than .05. We also included p values of less than .1 to provide a more complete picture of outcomes that are meaningfully different but that do not yet reach the statistical level of p < .05, but likely would with an increased number of samples as in some setting such outcomes are being described as not meaningfully different from controls, which is an incorrect conclusion. Thus, we provide the reader with this option to consider the meaning of the outcomes.
Results
Cr(VI) induces DNA double-strand breaks and causes cell cycle arrest in whale cells
We measured the ability of Cr(VI) to induce DNA double-strand breaks in bowhead whale lung cells using γ-H2AX foci formation and the neutral comet assay. By both measures, we found particulate Cr(VI) induced a concentration-dependent increase in DNA double-strand breaks (Figure 1). For γ-H2AX foci formation, 0, 0.1, 0.2, 0.3, and 0.4 µg/cm2 zinc chromate induced 5%, 17%, 22%, 33%, and 44% of cells with γ-H2AX foci after acute exposure and 5%, 11%, 20%, 27%, and 23% after prolonged exposure, respectively (Figs. 1A and 1B). A similar pattern was observed for the comet assay. Specifically, 0.1, 0.2, 0.3, and 0.4 µg/cm2 zinc chromate induced 150%, 153%, 174%, and 184% relative tail intensity increases after acute exposure and 145%, 177%, 191%, and 206% after prolonged exposure, respectively (Figure 1C). Using the comet assay, we compared break levels after prolonged exposure in whale and human lung cells and found particulate Cr(VI)-induced essentially equal amounts of DNA double-strand breaks (Figure 1D).
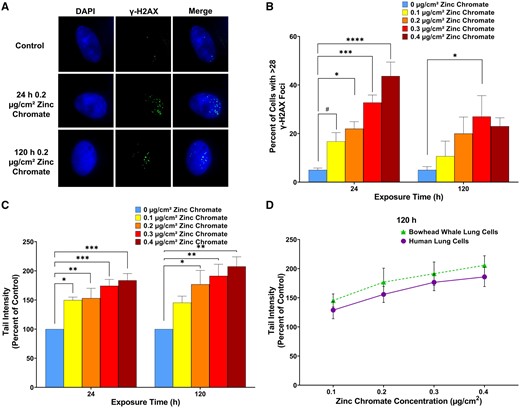
Particulate Cr(VI) induces DNA double-strand breaks in bowhead whale lung cells. Data reflect the mean of at least 3 independent experiments. Error bars = Standard error of the mean. *Significantly different from untreated cells at the same timepoint (#p < .1, *p < .05, **p < .01, ***p < .001, ****p < .0001). A, Representative images of γ-H2AX foci in bowhead whale lung cells. B, Breaks measured as γ-H2AX foci. C, Breaks measured with the neutral comet assay. D, Breaks measured in bowhead whale compared with human lung cells measured with the neutral comet assay after 120 h particulate Cr(VI) exposure. Human data originally published as tail moment in Qin et al. (2014), reanalyzed here as tail intensity.
We also assessed cell cycle arrest and where these breaks occurred in the cell cycle. We did not find a clear increase in the number of cells in any particular cell cycle phase by flow cytometry in bowhead whale lung cells after zinc chromate exposure (Figs. 2A–D). However, standard flow cytometry does not distinguish between cells in G2 and M, as their DNA amounts are equivalent, and thus both appear together as the G2/M peak. Therefore, we measured the mitotic index to determine if there was a difference in M phase. We found particulate Cr(VI) reduced the number of cells in M phase in a concentration-dependent manner after 24 or 120 h exposure (Figure 2E). Specifically, the mitotic index after 0, 0.1, 0.2, 0.3, and 0.4 µg/cm2 zinc chromate treatment was 0.0166, 0.0129, 0.0100, 0.0085, and 0.0076 after acute exposure and 0.0101, 0.0090, 0.0065, 0.0055, and 0.0041 after prolonged exposure, respectively (Figure 2E). Altogether, the data indicate the G2/M levels are predominately driven by cells in G2 indicating Cr(VI) also induced G2 phase arrest in whale cells.
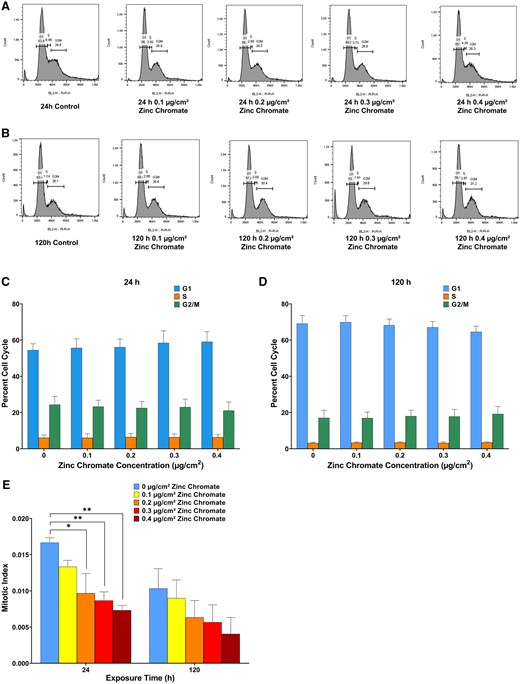
Particulate Cr(VI) induces cell cycle arrest in bowhead whale lung cells. Data reflect the mean of at least 3 independent experiments. Error bars = Standard error of the mean. *Significantly different from untreated cells at the same timepoint (*p < .05, **p < .01). (A) and (B) are the flow cytometry histograms of cell cycle profiles. A, Cell cycle profiles after 24 h zinc chromate. B, Cell cycle profiles after 120 h zinc chromate exposure. C, Flow cytometry gating analysis after 24 h zinc chromate exposure. D, Flow cytometry gating analysis after 120 h exposure. E, Mitotic index after 24 or 120 h zinc chromate exposure in bowhead whale lung cells.
In order to understand in which cell cycle phase Cr(VI)-induced DNA double-strand breaks occur, we measured the percent γ-H2AX-positive cells in each cell cycle phase using flow cytometry. In bowhead whale lung cells, Cr(VI)-induced DNA double-strand breaks were found in the S phase and G2/M phase. Only the highest concentration was statistically significant for 120 h but there is a dose-dependent trend (especially after prolonged exposure) (Figs. 3A and 3B). This outcome was more pronounced after 120 h exposure. For human lung cells, we found with increasing zinc chromate concentrations, more DNA double-strand breaks occurred in S and G2/M phases (Figs. 3A and 3B).

Particulate Cr(VI) induced-DNA double-strand breaks occur in the S and G2/M phases in bowhead whale lung cells and human lung cells. Analysis of γ-H2AX in cell cycle phase by flow cytometry. Data reflect the mean of at least 3 independent experiments. Error bars = standard error of the mean. *Significantly different from untreated cells at the same timepoint (*p < .05, **p < .01). A, 24 h exposure in bowhead lung cells. B, 120 h exposure in bowhead lung cells. C, 24 h exposure in human lung cells. D, 120 h exposure in human lung cells.
Homologous recombination repair is active in whale lung cells after acute and prolonged Cr(VI) exposure
Cr(VI)-induced DNA double-strand breaks are principally repaired by homologous recombination repair. We measured homologous recombination repair in bowhead whale lung cells with RAD51 foci formation and SCE formation (Figure 4). We found Cr(VI) induced a significant increase in RAD51 nuclear foci formation (Figs. 4A and 4B). Specifically, 0, 0.1, 0.2, 0.3, and 0.4 µg/cm2 zinc chromate induced >5 RAD51 nuclear foci in 8%, 14%, 20%, 25%, and 30% of cells after 24 h exposure, and 3%–9%, 16%, 21%, and 22% of cells after 120 h exposure, respectively (Figure 4B).
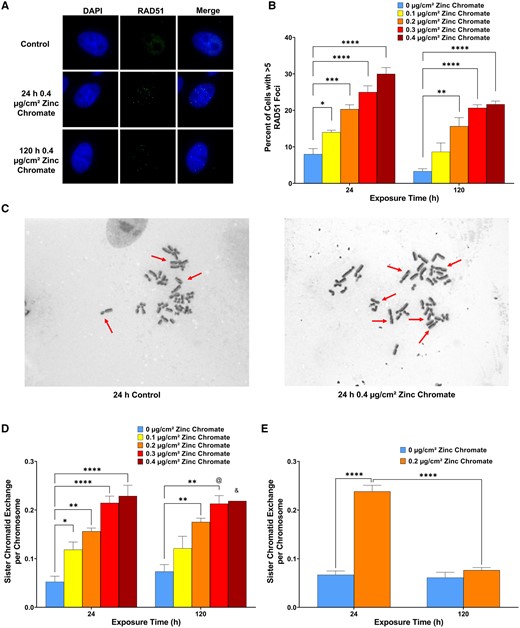
Particulate Cr(VI) induces homologous recombination repair activity in bowhead whale lung cells. Data represent an average of 3 experiments. Error bars = standard error of the mean. *Significantly different from untreated cells at the same timepoint (*p < .05, **p < .01, ***p < .001, ****p < .0001). & data from 1 experiment, 2 experiments produced no metaphases. @ data from 3 experiments but 1 experiment only had 28 metaphases. A, Representative images of RAD51 foci bowhead lung cells. B, RAD51 foci formation. C, Representative images of SCEs, indicated by red arrows, in bowhead lung cells. D, SCE formation in bowhead lung cells. E, SCE formation in human lung cells.
We also found particulate Cr(VI) induced a significant concentration-dependent increase in SCE formation after both 24 and 120 h exposures in bowhead whale lung cells (Figs. 4C and 4D). Specifically, 24 h exposure to 0, 0.1, 0.2, 0.3, and 0.4 µg/cm2 zinc chromate induced an average of 0.05, 0.12, 0.16, 0.21, and 0.23 SCEs per chromosome, whereas 120 h exposure induced an average of 0.07, 0.12, 0.18, 0.21, and 0.22 SCEs per chromosome, respectively. Altogether these data indicate whale lung cells retain functional homologous recombination that is responding to the DNA double-strand breaks after acute and prolonged Cr(VI) exposure.
Because SCEs have not been measured after prolonged particulate Cr(VI) exposure in human lung cells, we measured SCEs after zinc chromate treatment (Figure 4E). We found particulate Cr(VI) induced a significant concentration-dependent increase in SCE formation after acute exposure but inhibited SCE formation after prolonged exposure in human lung cells (Figure 4E). Specifically, 24 h exposure to 0 and 0.2 µg/cm2 zinc chromate induced an average of 0.07 and 0.24 SCEs per chromosome, whereas 120 h exposure induced an average of 0.07 and 0.08 SCEs per chromosome, respectively. Thus, prolonged Cr(VI) exposure inhibited homologous recombination repair measured as SCE formation, consistent with reports showing prolonged particulate Cr(VI) exposure inhibits homologous recombination repair using other measures (Browning et al., 2016; Qin et al., 2014).
Whale cells maintain securin levels and centrosome fidelity after acute and prolonged Cr(VI) exposure
We examined 2 major driving forces of numerical chromosome instability, spindle assembly checkpoint (SAC) bypass and centrosome amplification in whale cells. Prolonged Cr(VI) induces both outcomes in human lung cells (Holmes and Wise, 2010; Holmes et al., 2006, 2010; Martino et al., 2015; Wise et al., 2006b). We found Cr(VI) did not induce SAC bypass (Figs. 5A and 5B) or centrosome amplification in bowhead whale lung cells (Figs. 5C–F). Securin is a central regulating protein for both centrosome amplification and SAC bypass (Chao et al., 2006; Jallepalli et al., 2001; Zou et al., 1999). Consistent with the absence of SAC bypass and centrosome amplification, prolonged Cr(VI) exposure did not significantly affect securin levels in whale lung cells (Figs. 5G and 5H).
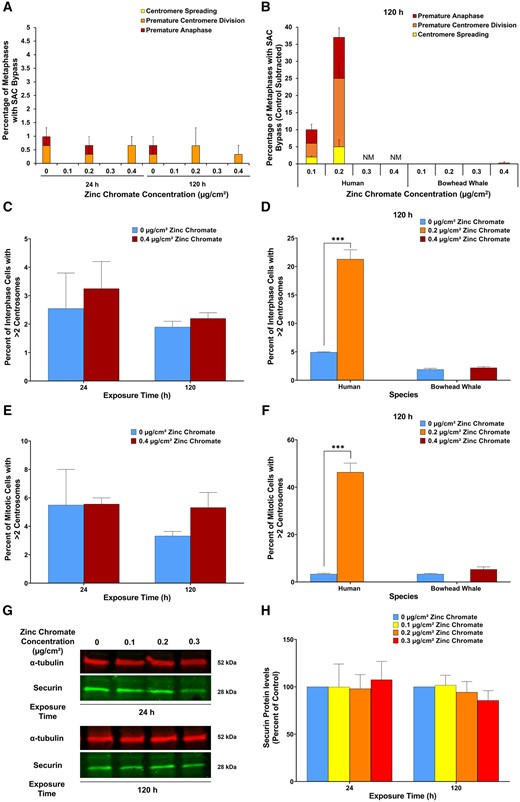
Cr(VI) does not alter securin levels and does not induce spindle assembly checkpoint (SAC) bypass or centrosome amplification in whale cells. Human data are added here for ease of comparison and were originally reported in Holmes et al. (2010). A, SAC bypass shown as the sum of stacked columns of centromere spreading, premature centromere division, and premature anaphase. Data reflect the mean of 3 independent experiments +/− the standard error of the mean. B, SAC bypass shows as the sum of stacked columns of centromere spreading, premature centromere division, and premature anaphase in bowhead whale lung cells compared with human lung cells. Data for bowhead whale lung is the same as panel A, graphed here on a scale that fits the human data. NM = not enough metaphases to analyze treatment. Data reflect the mean of 3 independent experiments +/− the standard error of the mean. C, Centrosome amplification in interphase cells. Data reflect the mean of 2 independent experiments. D, Centrosome amplification in interphase cells in bowhead whale lung cells compared with human lung cells. Data for bowhead whale lung is the same as panel C, graphed here on a scale that fits the human data. Human data reflect the mean of 3 independent experiments +/− the standard error of the mean. E, Centrosome amplification in mitotic cells. Data reflect the mean of 2 independent experiments. F, Centrosome amplification in mitotic cells in bowhead whale lung cells compared with human lung cells. Data for bowhead whale lung is the same as panel E, graphed here on a scale that fits the human data. Human data reflect the mean of 3 independent experiments +/− the standard error of the mean. G, Representative Western blot for securin. α-tubulin was used as a loading control. H, Securin whole cell protein levels. Data are expressed as percent of untreated control cells and reflect the mean of 3 independent experiments +/− the standard error of the mean.
Whale cells are resistant to Cr(VI)-induced chromosome instability and Cr(VI)-caused toxicity
Cr(VI) damaged chromosomes in a concentration-dependent manner in bowhead whale lung cells (Figs. 6A and 6B), consistent with the induction of DNA-double strand breaks. However, Cr(VI) did not increase the number of cells with multiple aberrations and chromosome instability in whale lung cells. Notably, despite inducing similar levels of DNA double-strand breaks, Cr(VI) induced less chromosome damage in whale lung cells than in human lung cells (Figs. 6C and 6D), consistent with the lack of homologous recombination repair inhibition in whale cells. Similarly, and consistent with the lack of SAC bypass and centrosome amplification, prolonged exposure to Cr(VI) did not induce numerical chromosome instability in bowhead whale lung cells (Figure 6F). Because Cr(VI) did not induce chromosome instability in whale lung cells, we hypothesized the whale lung cells would incur reduced Cr(VI)-induced cytotoxicity than human lung cells. Indeed, we found survival was significantly greater in bowhead whale lung cells than human lung cells at the same zinc chromate concentrations and exposure times (Figure 6G).
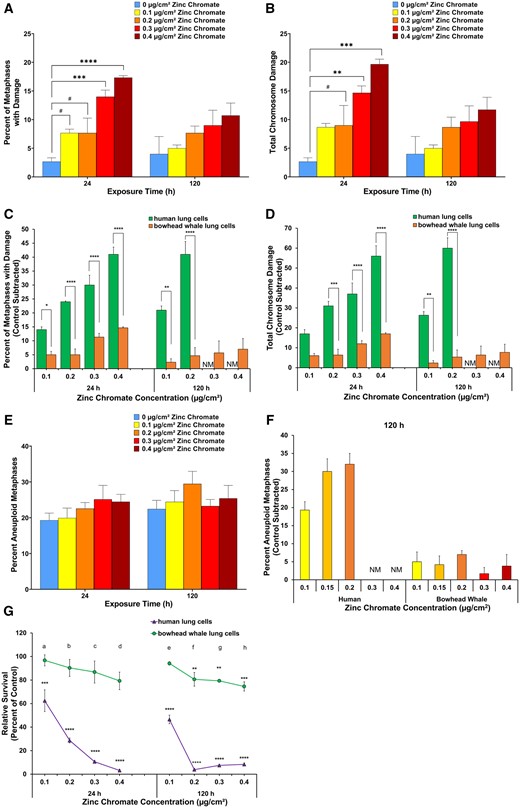
Cr(VI) does not induce structural or numerical chromosome instability and is less genotoxic and cytotoxic in whale lung cells. Data represent an average of at least 3 experiments. Error bars = standard error of the mean. The human chromosome instability data in this figure (panels C–F) are added here for ease of comparison and were originally reported in Holmes et al. (2010). *Significantly different from untreated cells at the same timepoint (#p < .1, **p < .01, ***p < .001, ****p < .0001). NM = not enough metaphases to analyze treatment. A, Structural chromosome instability measured as percent of metaphases with damage. B, Structural chromosome instability measured as total chromosome aberrations in 100 metaphases. C, Structural chromosome instability measured as percentage of metaphases with damage in bowhead whale lung cells compared with human lung cells. Data for bowhead whale lung is the same as panel A, graphed here on a scale that fits the human data. D, Structural chromosome instability measured as total chromosome aberrations in 100 metaphases in bowhead whale lung cells compared with human lung cells. Data for bowhead whale lung is the same as panel B, graphed here on a scale that fits the human data. E, Numerical chromosome instability measured as percent of metaphases with aneuploidy. F, Numerical chromosome instability measured as percent of metaphases with aneuploidy in bowhead whale lung cells compared with human lung cells. Data for bowhead whale lung is the same as panel E, graphed here on a scale that fits the human data. G, Cytotoxicity in bowhead whale cells compared with human lung cells. The significant differences between bowhead whale lung cells and human lung cells at 0.1, 0.2, 0.3, and 0.4 after 24 h exposure are ap < .01, bp < .0001, cp < .0001, and dp < .0001. The significant differences between bowhead whale lung cells and human lung cells at 0.1, 0.2, 0.3, and 0.4 after 120 h exposure are ep < .0001, fp < .0001, gp < .0001, and hp < .0001.
Discussion
Great whales, the really large whales, have low rates of cancer despite having long lives and accumulating elevated levels of carcinogenic metals such as Cr(VI). In fact, Cr levels in whales occur at a level only seen in the lung tissue of workers occupationally exposed to Cr(VI) for over 20 years (Wise et al., 2008). These data suggest whales have evolved cellular and molecular mechanisms to protect themselves against chemical carcinogens such as Cr(VI). The driving mechanism for Cr(VI)-induced human lung carcinogenesis is chromosome instability (Proctor et al., 2014; Wise et al., 2018). Cr(VI)-induced structural chromosome instability is driven by Cr(VI)-induced DNA double-strand breaks combined with an inhibition of DNA double-strand break repair, particularly homologous recombination repair (Browning et al., 2016; Qin et al., 2014; Xie et al., 2009). Specifically, Cr(VI) targets RAD51, the key effector protein in homologous recombination repair to inhibit homologous recombination (Browning et al., 2016; Qin et al., 2014).
Bowhead whales live 200 years and yet cancers have not been found in this species (Keane et al., 2015; Newman and Smith, 2006). This study is the first to consider Cr(VI) in this important species. We found Cr(VI) causes DNA double-strand breaks in bowhead whale lung cells. Notably, for prolonged (120 h) Cr(VI) exposures, the amount of Cr(VI)-induced DNA double-strand breaks in whale lung cells were similar to human lung cells indicating Cr(VI) was equipotent at producing breaks in whales and in humans. This outcome indicates Cr(VI) has the same ability to cause DNA breaks in bowhead whale and human lung cells and any potential differences in Cr intracellular metabolism, break formation or intercellular Cr levels are not affecting the amount of breaks produced.
We found DNA double-strand breaks were similar in bowhead whale lung cells after 24 and 120 h Cr(VI) exposure. This observation is consistent with Cr(VI) inducing persistent break levels in human lung cells and female fin whale skin cells (Meaza et al., 2020; Qin et al., 2014). Interestingly, prolonged exposure to Cr(VI) induced fewer DNA double-strand breaks in male fin whale skin cells (Meaza et al., 2020), suggesting male fin whales may manage these lesions better, although the explanation is uncertain and could be a difference in species. We used male bowhead whale lung cells, which did not show this pattern. Notably, Cr(VI)-induced break levels were higher in both female and male fin whale skin cells than in these male bowhead whale lung cells, suggesting Cr(VI) may have different potencies in whale lung and skin cells, which is consistent with potency differences observed in human lung and skin cells (Xie et al., 2015).
In human lung cells, acute or prolonged exposure Cr(VI) arrests cells in G2 phase with a reduction of cells entering M phase (Holmes et al., 2008, 2010; Xie et al., 2009). Cr(VI)-induced DNA double-strand breaks after acute exposure occur in S and G2, consistent with hypotheses that the breaks are only indirectly formed by stalled replication forks, unrepaired DNA single-strand breaks, failed excision repair or futile mismatch repair (Holmes et al., 2008). The same outcomes occur after prolonged particulate Cr(VI) exposure (Holmes et al., 2010). This study is the first to consider cell cycle arrest and where in the cycle Cr(VI)-induced breaks form in whale cells. Consistent with human lung cells, we found Cr(VI) arrests cells in S and G2 phase with a reduction of cells in M phase in whale cells. Also consistent with human cells, we found Cr(VI)-induced breaks occurred in S and G2, suggesting the breaks in whale cells form by similar indirect mechanisms.
In response to these particulate Cr(VI)-induced DNA double-strand breaks, human lung cells initiate the canonical homologous recombination repair response to fix the breaks in a high-fidelity manner (Browning et al., 2016; Qin et al., 2014). However, prolonged Cr(VI) exposure initiates homologous recombination repair, but then inhibits it by targeting and functionally inactivating RAD51 in the effector step (Browning et al., 2016; Qin et al., 2014). This outcome forces the cells to switch to low fidelity repair mechanism such as single strand annealing and microhomology mediated end joining, repair pathways known to cause chromosome instability (Blasiak, 2021; Jasin and Rothstein, 2013; Yoshioka et al., 2021). Indeed, numerous reports show prolonged Cr(VI) exposure causes chromosome instability (Wise et al., 2002, 2003, 2006a; Xie et al., 2005, 2009) and a recent report shows Cr(VI)-induces microhomology mediated end joining (Haberland et al., 2023).
In contrast, in whale lung cells, particulate Cr(VI) does not target RAD51 and does not inhibit homologous recombination repair indicating bowhead whale cells are resistant to Cr(VI)-induced RAD51 targeting and homologous recombination inhibition. Consistent with the robust homologous recombination repair response, Cr(VI) did not induce structural chromosome instability and was minimally cytotoxic to bowhead whale lung cells. These outcomes are consistent with data in right whale lung cells showing they also resist Cr(VI)-induced RAD51 targeting and homologous recombination inhibition (Browning et al., 2017). Thus, whale cells resist Cr(VI)-induced structural chromosome instability.
This study is also the first to measure numerical chromosome instability and its key underlying events, SAC bypass, and centrosome amplification in whales. Prolonged particulate Cr(VI) potently induces numerical chromosome instability and its underlying events in human lung cells (Holmes et al., 2006). However, in bowhead whale lung cells prolonged Cr(VI) exposure did not induce numerical chromosome instability, SAC bypass, or centrosome amplification. It also did not alter securin levels, a key regulatory protein for both SAC bypass and centrosome amplification. Thus, whale cells also resist Cr(VI)-induced numerical chromosome instability.
Thus, although particulate Cr(VI) induces the same amount of breaks in bowhead whale and human lung cells, it inhibits high fidelity break repair in human lung cells resulting in chromosome instability and lung cancer and does not do so in whale cells. Because Cr(VI) was equipotent at producing breaks in whales and in humans, the lack of repair inhibition in whales cannot be due to any potential differences in Cr intracellular metabolism, break formation, or intracellular Cr levels as these aspects would also affect the production of the breaks. Thus, whale lung cells are able to resist the chromosome instability and subsequently are protected from carcinogenesis due to factors involving repair.
Overall, our data are consistent with the hypothesis that great whales are resistant to cancer including environmental chemical-induced carcinogenesis. We find Cr(VI) induced DNA breaks in whale cells, but whale cells avoid Cr(VI)-induced homologous recombination repair inhibition, SAC bypass and centrosome amplification and consequently, avoid Cr(VI)-induced chromosome instability, the major driver of Cr(VI) carcinogenesis. Our discovery that Cr(VI) affects whale cells differently than human cells has deep and multilayered implications. On the one hand, it helps reveal the mechanism by which whales can escape Cr(VI) carcinogenesis; on the other hand, this difference in turn supports the key points of Cr(VI) carcinogenic mechanism in human cells. Thus, our future work will be to study how whales evade these known impacts of Cr(VI) in human cells. Future work to discern how various whale species, different individuals, and age within the population are affected by carcinogens would also be of value.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Acknowledgments
This work was conducted under National Marine Fisheries Service permits 16305, 21329, and 26774 (J.Wise, PI). We would like to thank Robert Suydam, Craig George, Charlie Brower, the Alaskan Native Whalers, and the department of Wildlife Management, North Slope Borough for the support in obtaining bowhead whale lung tissue samples. We thank Melissa Slotnick, Julieta Martino, Alex Doering, Megan Stevenson, Allison Sample, Jane Murphy, and Britton Goodale for early work in establishing the bowhead whale cells and in developing these assays and bowhead whale cells. We thank Qin Qin for her work establishing the comet assay data in human lung cells published in reference Qin et al. (2014) and adapted here with a different measure.
Funding
Research reported in this publication was supported by the National Institute of Environmental Health Sciences of the National Institutes of Health (R01ES016893 and R35ES032876 to J.P.W.Sr). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
Author notes
Present address for Jennifer H. Toyoda: Mote Marine Laboratory, Sarasota, FL, USA.
Present address for Tayler J. Croom-Pérez: Burnett School of Biomedical Sciences, University of Central Florida College of Medicine Orlando, FL, USA.
Haiyan Lu and Jennifer H. Toyoda are the co-first authors of this study.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments