





Abstract
Emphysema is one of the most important phenotypes for chronic obstructive pulmonary disease (COPD). Apoptosis in alveolar epithelial cells (AECs) causes the emphysematous alterations in the smokers and patients with COPD. Sirtuin 1 (SIRT1) is able to attenuate mitochondrial dysfunction, oxidative stress, and to modulate apoptosis. It has been shown that sodium tanshinone IIA sulfonate (STS), a water-soluble derivative of tanshinone IIA, protects against cigarette smoke (CS)-induced emphysema/COPD in mice. However, the mechanisms underlying these findings remain unclear. Here, we investigate whether and how STS attenuates AEC apoptosis via a SIRT1-dependent mechanism. We found that STS treatment decreased CS extract (CSE)-induced apoptosis in human alveolar epithelial A549 cells. STS reduced oxidative stress, improved mitochondrial function and mitochondrial membrane potential (ΔΨm), and restored mitochondrial dynamics-related protein expression. Moreover, STS promoted mitophagy, and increased oxidative phosphorylation protein levels (complexes I–IV) in CSE-stimulated A549 cells. The protective effects of STS were associated with SIRT1 upregulation, because SIRT1 inhibition by EX 527 significantly attenuated or abolished the ability of STS to reverse the CSE-induced mitochondrial damage, oxidative stress, and apoptosis in A549 cells. In conclusion, STS ameliorates CSE-induced AEC apoptosis by improving mitochondrial function and reducing oxidative stress via enhancing SIRT1 pathway. These findings provide novel mechanisms underlying the protection of STS against CS-induced COPD.
STS attenuates cigarette smoke extract (CSE)-induced apoptosis in alveolar epithelial cells (AECs).
STS reduces CSE-induced AEC apoptosis by reversing mitochondrial dysfunction, mitophagy, and oxidative stress.
Protective effect of STS in AECs is associated with SIRT1 upregulation.
Chronic obstructive pulmonary disease (COPD) encompasses several clinical phenotypes, most prominently emphysema and chronic bronchitis (Chung and Adcock, 2008). It is considered as the third leading cause of mortality worldwide in 2030 by the World Health Organization (Wang et al., 2019). Emphysema is characterized by airspace enlargement and alveolar wall destruction in the lung (Tang et al., 2017). Cigarette smoking is an independent risk factor for the development of COPD/emphysema (Lin et al., 2017). Alveolar epithelial cells (AECs) are in first contact with inhaled cigarette smoke (CS). The induction of AEC apoptosis can lead to the loss of alveoli, which play important roles in mediating emphysematous alterations in COPD patients (Aoshiba et al., 2003). Hence, protection of the AECs from injury and apoptosis by CS would be beneficial for the management of COPD/emphysema.
Oxidative stress, due to the combined effects of excess reactive oxygen species (ROS) and reduced antioxidant enzyme activity, is a major event in the pathogenesis of COPD (Kang and Shadel, 2016). High levels of ROS in the cells cause oxidative damage to cells by eliciting nucleic acids breakage, protein damage, and lipid peroxidation (Pisoschi and Pop, 2015). ROS also act as an important intrinsic signal that modulates cellular apoptosis (Cui et al., 2019). Mitochondria, a crucial source of ROS, play important roles in various physiological and pathological circumstances (Galluzzi et al., 2012; Murphy, 2009). Mitochondrial dysfunction involves in the pathogenesis of COPD due to abnormal redox state and apoptosis (Wang et al., 2019). In addition, mitochondria themselves are prone to oxidative damage, which causes further disturbance of mitochondrial function (Kang and Shadel, 2016). When mitochondria are damaged by external stimuli such as CS, ROS are generated from mitochondrial respiratory chain. Therefore, mitochondrial dysfunction, with increase in oxidative stress, leads to increased apoptosis of AECs (Chung and Adcock, 2008). Mitophagy, a particular form of autophagy that specifically removes damaged mitochondria, plays a key role for maintaining mitochondrial homeostasis. Accumulation of damaged or dysfunctional mitochondria, due to impaired mitophagy, is associated with COPD/emphysema (Cloonan and Choi, 2016; Ito et al., 2015; Sundar et al., 2019; Tsubouchi et al., 2018). Mitophagy is mainly governed by the PTEN-induced putative protein kinase 1 (Pink1)-parkin RBR E3 ubiquitin protein ligase (PRKN) pathway. There are also conflicting reports for the role of Pink1-mediated mitophagy in COPD progression (Araya et al., 2019; Ito et al., 2015; Mizumura et al., 2014; Sundar et al., 2019).
Tanshinone IIA, isolated from the plant Salvia miltiorrhiza Bunge (Dan shen in Chinese), has been widely used in the treatment of cardiovascular disorders in Asian countries (Guan et al., 2018; Jiang et al., 2016; Li et al., 2018). Sodium tanshinone IIA sulfonate (STS) is a water-soluble derivative of tanshinone IIA. Tanshinone IIA or STS modulates a variety of cellular processes with anti-inflammatory and anti-oxidative properties. Gong et al. reported that tanshinone IIA attenuates oxidative damage after spinal cord injury by upregulating miR-124 (Gong et al., 2019). Tanshinone IIA alleviates glutamate-induced oxidative toxicity by preventing mitochondrial dysfunction in SH-SY5Y human neuroblastoma cells (Li et al., 2017). STS also prevents heat stress-induced endothelial cell apoptosis via the PI3K/AKT/eNOS signaling pathway (Cheng et al., 2017). Our previous studies have demonstrated that STS prevents CS-induced emphysema/COPD in mice (Guan et al., 2018; Li et al., 2018). However, for all we know, it is not clear yet whether or not STS can reduce oxidative stress and mitochondrial dysfunction-induced cellular apoptosis in cigarette smoke extract (CSE)-exposed AECs, or if the protective effect of STS is related to Sirtuin 1 (SIRT1) pathway.
In the present study, we demonstrated that STS significantly attenuates CSE-induced apoptosis by improving mitochondrial function and reducing oxidative stress in alveolar epithelial A549 cells. Furthermore, the protective effects of STS were correlated with the upregulation of SIRT1. These findings provide novel mechanistic evidence of STS in preventing CS-induced AEC apoptosis.
MATERIALS AND METHODS
Chemicals and reagents
STS (purity of >98%) was purchased from Chengdu Herbpurify Co., Ltd. (Chengdu, Sichuan, China) and the cigarettes were purchased from Guangdong Tobacco Industry Co., Ltd. (Guangzhou, Guangdong, China). Anti-Bax, anti-Bcl-2 and, anti-β-actin polyclonal antibodies were purchased from Proteintech (Chicago, Illinois). Anti-cleaved caspase 3 polyclonal antibody was purchased from Cell Signaling Technology (California); Anti-SIRT1, anti-NDUFB8, anti-Drp1, anti-Mfn2, anti-SDHB, anti-UQCRC2, anti-Pink1, and anti-COX II polyclonal antibodies were purchased from Abclonal Technology (Wuhan, China). HRP-labeled Goat Anti-Rabbit/Mouse IgG (H + L) were purchased from Abcam Biotechnology (Cambridge, Massachusetts). SIRT1 inhibitor EX 527 (also called Selisistat) was obtained from Selleck Chemicals (Houston, Texas). Mito TEMPOL and N-acetyl-l-cysteine (NAC) were purchased from Sigma-Aldrich (St Louis, Missouri). The poly-vinylidene fluoride membranes were from Millipore Corporation (Billerica, Massachusetts). ECL-Plus detection kit probed was purchased from Tanon Science & Technology Co., Ltd. (Shanghai, China). Other reagents used in this study were all of analytical grade.
Cell culture
Alveolar epithelial A549 cells were obtained from Cell Bank of the Chinese Academy of Sciences (Shanghai, China), and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, gibco, Thermo Fisher Scientific, Massachusetts) with 10% fetal bovine serum (FBS, gibco, Thermo Fisher Scientific, Massachusetts), 100 kU/l penicillin and 100 mg/l streptomycin (gibco, Thermo Fisher Scientific, Massachusetts) in a humidified incubator at 37°C with 5% CO2 atmosphere.
CSE preparation
CSE was freshly prepared from 2 burning cigarettes (Red Roses Label; Guangdong Tobacco Industry Co., Ltd., Guangzhou, China) into 10 ml DMEM within 30 min prior to treatments according to a previously described protocol (Li et al., 2012). In brief, the smoke generated from 2 burning cigarettes was sucked under a constant flow rate (50 ml/10 s) into a syringe and bubbled into a tube containing 10 ml serum-free DMEM medium. The gained CSE solution was sterilized with a 0.22-μm filter (Millipore, Bedford, Massachusetts) and was then adjusted to pH 7.4. This CSE solution was regarded as 100% CSE.
Cell viability assay
A549 cell viability was measured according to the manufacturer’s instructions of CCK-8 Kit (Dojindo, Kumamoto, Japan). Briefly, A549 cells (1 × 104/well) were seeded in a 96-well plate with DMEM containing 1% FBS. After culture overnight, A549 cells were incubated with and without 3% CSE in the presence or absence of 1, 2.5, and 5 μM STS for 48 h. This concentration of STS in A549 cells was chosen based on our previous report, showing that STS decreased CSE-induced inflammation and oxidative stress in RAW 264.7 macrophages (Guan et al., 2018). The medium was removed and cells were incubated in 100 μl medium with 10 μl CCK-8 solution at 37°C for 2 h. The absorbance at 450 nm was read using a microplate absorbance reader (Termo Scientifc, Waltham, Massachusetts).
Apoptotic cell determination
A549 cells were plated in a 6-well plate (2 × 105 cells/well) and treated as indicated for 48 h. The cellular apoptotic rate was determined using Annexin V, FITC Apoptosis Detection Kit by comparing Annexin V-positive cells (R&D Systems, Minneapolis, Minnesota).
Measurement of intracellular ROS
Intracellular ROS production was determined by staining A549 cells with an oxidation-sensitive fluorescent probe 2,7-dichlorodihydrofluorescein diacetate according to the manufacturer’s protocol. A549 cells were treated as indicated for 48 h. Subsequently, the medium was removed, and the cells were incubated with 1 ml of CM-DCFDA (Beyotime Institute of Biotechnology, Haimen, China) solution for 20 min. The dye solution was then removed, and the cells were trypsinized and centrifuged at 500 × g for 5 min at 4°C. Finally, the cells were washed twice with PBS, and kept in the dark on ice. Then the DCF fluorescence was detected by the flow cytometry with a FACSVerse flow cytometer (Becton Dickinson) and the ratio of DCF-positive cells were determined by FlowJo.
Measurement of mitochondrial ROS
Mitochondrial ROS (mtROS) was detected with a MitoSOX Red (ThermoFisher Scientific, Massachusetts) assay, which is based on a redox-sensitive fluorescent probe that is selectively targeted to the mitochondria. Briefly, A549 cells were seeded in 12-well black plates and treated as indicated. Cells were incubated with 5 μmol/l MitoSOX reagent for 10 min at 37°C. The cells were washed 3 times with warm serum-free DMEM, and images were photographed under a fluorescence microscope (EVOS Auto 2, Invitrogen, Washington). The fluorescence intensity of the mtROS was measured by Image J.
Mitochondrial respiration assessment
A549 cells were plated (about 105 cells/well) in an XF24-well plate (Seahorse Bioscience, Massachusetts) and treated as indicated. Immediately before the respiration assay, the medium was changed into Seahorse base DMEM medium containing glucose (10 mM), pyruvate (1 mM) and GlutaMAX (2 mM; Invitrogen). Oxygen consumption rate (OCR) was measured by an XF24 Extracellular Flux Analyzer (Seahorse Bioscience). Then oligomycin (1 μM) was added to inhibit ATP synthesis and to detect respiration linked to ATP production. The uncoupler carbonyl cyanide 4-trifluoromethoxy phenylhydrazone (FCCP, 2 μM) was applied to measure respiratory reserve capacity and maximal respiration. Finally, combination of rotenone plus antimycin A (0.5 μM) was added to completely block mitochondrial OCR.
JC-1 assay
Mitochondrial membrane potential (Δψm) was measured with a commercial JC-1 kit (Beyotime, Shanghai, China) following the manufacturer’s instructions. Briefly, A549 cells were treated as indicated and stained with 10 μM of JC-1 according to the manufacturer instructions (Beyotime, Shanghai, China). Intensity of red (aggregate JC-1)/green (monomeric JC-1) fluorescence was detected with a fluorescence microscope (EVOS Auto 2, Invitrogen, Washington), and the ratio of the red to green fluorescence was proportional to the Δψm.
Immunofluorescence staining
Immunofluorescence was performed using a method described previously (Guan et al., 2019). Briefly, A549 cells were seeded on glass slides in 12-well plates and incubated as indicated for 48 h. The slides were washed 3 times with PBS, fixed in 4% paraformaldehyde for 10 min and then permeabilized with 0.1% Triton X-100 for 20 min. Subsequently, the cells were blocked with 3% BSA at room temperature for 1 h and then incubated with primary antibody (SIRT1, 1:200) at 4°C overnight. After incubation, samples were washed 3 times with PBS and incubated with goat anti-rabbit Cy3-conjugated second antibody (1:200, Beyotime Institute of Biotechnology, Haimen, China) for 1 h and labeled with DAPI for 5 min. Finally, samples were washed 4 times with PBS and mounted in antifade mounting medium, and fluorescence was detected using a fluorescence microscope (Laica, Germany).
Real-time PCR
Real-time PCR was performed as we described previously (Guan et al., 2018). Amplification was conducted using the following primers: 5′-TAGCCTTGTCAGATAAGGAAGGA-3′ (forward) and 5′-ACAGCTTCACAGTCAACTTTGT-3′ (reverse) for human SIRT1, and 5′-GGCTGTATTCCCCTCCATCG-3′ (forward) and 5′-CCAGTTGGTAACAATGCCATGT-3′ (reverse) for human β-actin.
Western blot
Equal amounts of protein (20∼50 μg) from A549 cells were used for Western blot (Guan et al., 2016). Protein bands were scanned and quantified by using the Tanon-5200 infrared scanning system (anon Science & Technology Co., Ltd., Shanghai, China). The intensity analysis was analyzed by a densitometry system named Image J.
Data analysis
Data analysis was performed using SigmaPlot 12.5 (Systat Software, Inc., Chicago, Illinois), and expressed as means ± SEM. Comparison of means among multiple groups was accomplished by 1-way analysis of variance (ANOVA). A 2-sided p value <.05 (p < .05) were considered to be significantly different.
RESULTS
STS Suppressed CSE-Induced Apoptosis in Alveolar Epithelial A549 Cells
A CCK-8 assay was performed to examine the effects of STS on CSE-induced cellular damage. As shown in Figure 1C, 3% CSE significantly decreased cell viability in A549 cells for 48 h exposure, whereas STS treatment (1–5 μM, 48 h) significantly attenuated CSE-induced reduction of cell viability, suggesting the inhibitory effects of STS on CSE-induced cell injury. Flow cytometry assay demonstrated that treatment with STS also reduced CSE-induced cell apoptosis in A549 cells in a dose-dependent manner (Figs. 1A and 1B). Apoptosis is triggered primarily via caspase-mediated signal transduction. Hence, we investigated whether STS inhibits apoptosis via a caspase pathway. Western blot showed that STS significantly decreased the cleaved caspase-3 level in CSE-stimulated A549 cells (Figs. 1D and 1E). Treatment with STS also increased the expression of anti-apoptotic protein Bcl-2 but decreased the pro-apoptotic protein Bax (Figs. 1D and 1E). These results indicate the inhibitory effects of STS on CS-induced AEC apoptosis.
![STS reduced CSE-induced apoptosis in alveolar epithelial A549 cells. A549 cells were cultured with and without 3% CSE and/or 1, 2.5, or 5 μM STS for 48 h. A, Cell apoptosis of A549 cells was examined via the Annexin V-FITC/PI staining assay by flow cytometry. B, The ratio of Annexin V-positive apoptotic cells was statistically analyzed. C, Cell viability was detected by CCK-8 assay. D, E, The protein levels of Bcl-2, Bax, and cleaved caspase 3 were detected by Western blot. Data are presented as mean ± SEM of at least 3 independent experiments, *p < .05, versus untreated cells (3% CSE [−] and STS [−]); #p < .05, versus cells treated with 3% CSE only.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/toxsci/183/2/10.1093_toxsci_kfab087/1/m_kfab087f1.jpeg?Expires=1750322865&Signature=t8FLHlJweZK4DBewAKRAwWL3RTFeedye7O8EzHpuP0S3GVFy-Xr6xn267G9YU0mavEbb-GJ3w4vE4LKnZPuZdmafA2Bz-jL-nStBeWsPLzgiAiOmuW4ckSaqKMkvlNmLJRp0d8BeBMhf2PPtXWffGFcq1GotOWprH9qib7ISQEd-b4SLMh1Iha7V1LRd7aIRFVWMZ0SjQnZ92JVYmkUBrZ5V9jWOgz-u7RCwx2kYWY0oP8fUsPTWennt~LSznmKZAE6DjP7pDkoAqKMZ2eri6~4X7atYOBbSxqoToLT4Y0n-ci9oYm10qJgCJ0Yto9JioiqCnRzNPktTR4PZMD2liw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
STS reduced CSE-induced apoptosis in alveolar epithelial A549 cells. A549 cells were cultured with and without 3% CSE and/or 1, 2.5, or 5 μM STS for 48 h. A, Cell apoptosis of A549 cells was examined via the Annexin V-FITC/PI staining assay by flow cytometry. B, The ratio of Annexin V-positive apoptotic cells was statistically analyzed. C, Cell viability was detected by CCK-8 assay. D, E, The protein levels of Bcl-2, Bax, and cleaved caspase 3 were detected by Western blot. Data are presented as mean ± SEM of at least 3 independent experiments, *p < .05, versus untreated cells (3% CSE [−] and STS [−]); #p < .05, versus cells treated with 3% CSE only.
STS Attenuated CSE-Induced Oxidative Stress in Alveolar Epithelial A549 Cells
Oxidative stress is closely associated with the process of apoptosis (Guan et al., 2019). In fact, treatment of A549 cells with a ROS scavenger NAC significantly inhibited CSE-induced cell apoptosis, as determined by flow cytometry assay (Figs. 2A and 2B). We therefore investigated whether STS reduces CSE-induced oxidative stress in alveolar epithelial A549 cells. As shown in Figures 2C and 2D, CSE significantly enhanced ROS levels by a 3.7-fold, when compared with control cells. STS treatment (1–5 μM) significantly reduced CSE-induced ROS production in a dose-dependent manner, as illustrated by the percentage reduction of DCF-positive cells (19.0%, 23.9%, and 30.2%, respectively). Additionally, quantitative data showed that the mtROS levels in CSE group were 2.7-fold higher than that in the control group. The CSE-induced increase of mtROS levels were also reduced by STS treatment (Figs. 2E and 2F). These data suggest that STS reduces CSE-induced oxidative stress in A549 cells.
![STS-attenuated CSE-induced oxidative stress in alveolar epithelial A549 cells. A, B, A549 cells were incubated with 3% CSE and the ROS scavenger N-Acetyl-l-cysteine (NAC) for 48 h. Cell apoptosis of A549 cells was examined via the Annexin V-FITC/PI staining assay by flow cytometry. Data are presented as mean ± SEM of at least 3 independent experiments, *p < .05, versus untreated cells (3% CSE [−] and NAC [−]); #p < .05, versus cells treated with 3% CSE only. A549 cells were treated with 3% CSE and/or different concentrations of STS (1, 2.5, or 5 μM) for 48 h. C, D, The intracellular ROS was examined by flow cytometry according to the ROS assay kit. E, F, mtROS levels. Data are presented as mean ± SEM of at least 3 independent experiments, *p < .05, versus untreated cells (3% CSE [−] and STS [−]); #p < .05, versus cells treated with 3% CSE only.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/toxsci/183/2/10.1093_toxsci_kfab087/1/m_kfab087f2.jpeg?Expires=1750322865&Signature=FXPpgTrcdJKu1WT7eqS~mj-ahilpv4vWdrAFQYcqvfFt3lACjRfyU-k8KkEIn3~yKdOrtOKr~F9fsDxYBE6cBqGGZUbWjRDgIo42xTfFiAiAESEwORI8oiAkhJ9z-VTV8jZsJOGPLPeekLPl5J4~FWuL47poseNh5oG0LKcANjrAijbP7tUdBtWCQoyuPXXWTUJK~66F6P5CYt~DL-ZDyV~L66MuS-igeHjJ7FxCPkBk~aXCjhGu9HqaYyLT~ZuaSGR6cwDUrVaUkUmkBFS4FlSzSQLwm9-L61kHbRLP8oSPuzxjjuWJ7P-P7Q2NXnxGglmIW~X96EFITTcJzyT83Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
STS-attenuated CSE-induced oxidative stress in alveolar epithelial A549 cells. A, B, A549 cells were incubated with 3% CSE and the ROS scavenger N-Acetyl-l-cysteine (NAC) for 48 h. Cell apoptosis of A549 cells was examined via the Annexin V-FITC/PI staining assay by flow cytometry. Data are presented as mean ± SEM of at least 3 independent experiments, *p < .05, versus untreated cells (3% CSE [−] and NAC [−]); #p < .05, versus cells treated with 3% CSE only. A549 cells were treated with 3% CSE and/or different concentrations of STS (1, 2.5, or 5 μM) for 48 h. C, D, The intracellular ROS was examined by flow cytometry according to the ROS assay kit. E, F, mtROS levels. Data are presented as mean ± SEM of at least 3 independent experiments, *p < .05, versus untreated cells (3% CSE [−] and STS [−]); #p < .05, versus cells treated with 3% CSE only.
STS Ameliorated CSE-Induced Mitochondrial Dysfunction and Improved Mitochondrial Membrane Potential in Alveolar Epithelial A549 Cells
Mitochondria are an important source of ROS within most mammalian cells, we therefore investigated whether mitochondrial damage are also responsible for CSE-induced apoptosis in AECs. Treatment with a mitochondria-targeted antioxidant Mito TEMPOL significantly inhibited CSE-induced apoptosis in A549 cells, as indicated by decreased Annexin V-positive cells (Figs. 3A and 3B). This suggests that mtROS is implicated in CSE-induced apoptosis in alveolar epithelial A549 cells. We then investigated the effects of STS on CSE-induced mitochondrial dysfunction. First, we measured OCR in A549 cells exposed to CSE. Basal respiration, ATP-linked OCR, and maximal respiration were dramatically decreased in CSE-exposed A549 cells, and these were upregulated by STS treatment (5 μM) (Figs. 3C and 3D). There were no discernible differences of OCR between STS-treated cells and control cells. Δψm was determined by JC-1 staining. As shown in Figures 3E and 3F, the ratio of red to green fluorescence was significantly decreased after CSE treatment. However, STS administration prevented CSE-induced loss of Δψm in A549 cells. Therefore, STS protects against reduced mitochondrial respiration and increased mitochondrial depolarization in CSE-exposed AECs.
![STS ameliorated CSE-induced mitochondrial dysfunction and improved mitochondrial membrane potential in alveolar epithelial A549 cells. A, B, A549 cells were incubated with 3% CSE and mitochondria-targeted antioxidant Mito TEMPOL for 48 h. Cell apoptosis of A549 cells was examined via the Annexin V-FITC/PI staining assay by flow cytometry. Data are presented as mean ± SEM of at least 3 independent experiments, *p < .05, versus untreated cells (3% CSE [−] and Mito TEMPOL [−]); #p < .05, versus cells treated with 3% CSE only. A549 cells were cultured with and without 3% CSE and/or 1, 2.5, or 5 μM STS for 48 h. C, The bioenergetic profiles of A549 cells were measured by a Seahorse Extracellular Flux Analyzer, OCR in cells treated with oligomycin, FCCP, and rotenone and antimycin A. D, Quantitative analysis of basal respiration, ATP production and maximal respiratory is shown. E, F, Δψm was determined by JC-1 staining. Data are presented as mean ± SEM of at least 3 independent experiments, *p < .05, versus untreated cells (3% CSE [−] and STS [−]); #p < .05, versus cells treated with 3% CSE only.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/toxsci/183/2/10.1093_toxsci_kfab087/1/m_kfab087f3.jpeg?Expires=1750322865&Signature=ByhvF51zJkz29JTOeNR38EHcMf2DMqL~BL8puMTp6anthhhGg40RLyeEMoml71IUcyvkmPbG9PYlw3MdOfaJKLooWSfgGkMXGpPjgQ56s76KknmgBbSVYEGqEPvDU1ezGragrUy7EVJ0Jh2t57MBXAdJrE5C6fmGbLlBxAJJbHLDj8OvoLRDQkAwVamlPehSd7Bz8fEvAMpDWbO71jvNe24rYPHHVDeeEWQ6O-xjeOeZyx0wU9OA3uVqVJjIIzheHaWHIjQ5pZTnraB92IdI3Fx7H2Cj8j4X~KyNOcXu2Dx7yRkJZBYtOK4ZCXbeDDN6j7LqyvMkIpn1Qp4M3tnxbg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
STS ameliorated CSE-induced mitochondrial dysfunction and improved mitochondrial membrane potential in alveolar epithelial A549 cells. A, B, A549 cells were incubated with 3% CSE and mitochondria-targeted antioxidant Mito TEMPOL for 48 h. Cell apoptosis of A549 cells was examined via the Annexin V-FITC/PI staining assay by flow cytometry. Data are presented as mean ± SEM of at least 3 independent experiments, *p < .05, versus untreated cells (3% CSE [−] and Mito TEMPOL [−]); #p < .05, versus cells treated with 3% CSE only. A549 cells were cultured with and without 3% CSE and/or 1, 2.5, or 5 μM STS for 48 h. C, The bioenergetic profiles of A549 cells were measured by a Seahorse Extracellular Flux Analyzer, OCR in cells treated with oligomycin, FCCP, and rotenone and antimycin A. D, Quantitative analysis of basal respiration, ATP production and maximal respiratory is shown. E, F, Δψm was determined by JC-1 staining. Data are presented as mean ± SEM of at least 3 independent experiments, *p < .05, versus untreated cells (3% CSE [−] and STS [−]); #p < .05, versus cells treated with 3% CSE only.
STS Increased Mitochondrial Oxidative Phosphorylation Protein Complexes in Alveolar Epithelial A549 Cells
Oxidative phosphorylation (OXPHOS) is a process of a flow of electrons in electron transport chain (ETC). We, therefore, investigated whether STS affects the protein levels of different OXPHOS/ETC complexes, that is, Complex I (NDUFB8), Complex II (SDHB), Complex III (UQCRC2), and Complex IV (COX II) in the CSE-treated A549 cells by Western blot. We found that A549 cells exposed to CSE for 48 h showed a remarkable reduction in the protein levels of NDUFB8, SDHB, UQCRC2, and COX II (Figs. 4A and 4B), indicating the CSE-induced impairment of individual OXPHOS complexes. However, these protein levels were significantly increased by STS treatment. These results demonstrated that STS exerts a protective role against mitochondrial dysfunction induced by CSE.
![STS increased mitochondrial OXPHOS protein complexes in alveolar epithelial A549 cells. A549 cells were cultured with and without 3% CSE and/or 1, 2.5, or 5 μM STS for 48 h. A, Western blot was used to analyze the protein levels of Complex I (NDUFB8), Complex II (SDHB), Complex III (UQCRC2), and Complex IV (COX II) in the CSE-treated A549 cells by Western blot. Results are representative of different experiments. B, Densitometric analysis of Complex I (NDUFB8), Complex II (SDHB), Complex III (UQCRC2), and Complex IV (COX II) in the immunoblots using β-actin as the internal reference. Data are presented as mean ± SEM of at least 3 independent experiments, *p < .05, versus untreated cells (3% CSE [−] and STS [−]); #p < .05, versus cells treated with 3% CSE only.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/toxsci/183/2/10.1093_toxsci_kfab087/1/m_kfab087f4.jpeg?Expires=1750322865&Signature=k~Jjw2uIR3D1k-I-qxZ-D0L~Q3fWlSQwmpM3xJLhX~fFR9KQY1skG8r1~dQxCqH7gx6ds9hSdCSTTIRgfJ9OdqOmoTCiDVPFVb9eXmfCNc0lUpf06H3YAXtTz6~Znl1-CHjuCYZeuFcmRYwS5IgTvIWb7VN5kHlTaDcWjxQ4PRn4xVivnpP1HIBNHqBL1mJpNVsmWzMC-ep3TbP84ft3tqPOFNpwsiQ4tH~mqiGCuMx94XmNL1qQzdXEIhNX2CtD2CNdXdhmovCAsmS04NLehCrDzq9~x-BZzQFNEfVm1iQpDpbBu5q-6sQHILf4YFppcibH0~u01LNo0xEnNczqdw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
STS increased mitochondrial OXPHOS protein complexes in alveolar epithelial A549 cells. A549 cells were cultured with and without 3% CSE and/or 1, 2.5, or 5 μM STS for 48 h. A, Western blot was used to analyze the protein levels of Complex I (NDUFB8), Complex II (SDHB), Complex III (UQCRC2), and Complex IV (COX II) in the CSE-treated A549 cells by Western blot. Results are representative of different experiments. B, Densitometric analysis of Complex I (NDUFB8), Complex II (SDHB), Complex III (UQCRC2), and Complex IV (COX II) in the immunoblots using β-actin as the internal reference. Data are presented as mean ± SEM of at least 3 independent experiments, *p < .05, versus untreated cells (3% CSE [−] and STS [−]); #p < .05, versus cells treated with 3% CSE only.
Effects of STS on Mitochondrial Proteins Involved in Mitochondrial Fission/Fusion Process and Mitophagy in CSE-Treated Alveolar Epithelial A549 Cells
To further investigate the mechanisms by which STS alters mitochondrial function in alveolar epithelial A549 cells, we first examined the protein levels of mitochondrial fission and fusion proteins that modulate mitochondrial dynamics. The results showed that CSE treatment to A549 cells induced a significant increase in a mitochondrial fission Drp1 protein, and induced a decrease in a mitochondrial fusion Mfn2 protein (Figure 5), suggesting that CSE disrupates mitochondrial dynamics. STS treatment attenuated these abnormal changes. CSE stimulation in A549 cells also caused a reduction in protein level of Pink1 that plays an important role in the removal of damaged or dysfunction mitochondrial from the cells by mitophagy (Figure 5), implying that impaired mitophagy may directly contribute to CSE-induced mitochondrial dysfunction. However, the downregulation of Pink1 was also attenuated by STS, suggesting that STS promotes mitophagy in epithelial cells.
![Effects of STS on mitochondrial proteins involved in mitochondrial fission/fusion process and mitophagy in CSE-treated alveolar epithelial A549 cells. A, Western blot was used to analyze the protein levels of Pink1, Mfn2, and Drp1 in the CSE-treated A549 cells by Western blot. Results are representative of different experiments. B, Densitometric analysis of Pink1, Mfn2, and Drp1 in the immunoblots using β-actin as the internal reference. Data are presented as mean ± SEM of at least 3 independent experiments, *p < .05, versus untreated cells (3% CSE [−] and STS [−]); #p < .05, versus cells treated with 3% CSE only.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/toxsci/183/2/10.1093_toxsci_kfab087/1/m_kfab087f5.jpeg?Expires=1750322865&Signature=32A-wLfuPDAyiDKCgZltsbbo2b9tu-7yUgID1PnPvuVMsACcEYC65hGpYXLGWVZqkFkM4dhdgSMMp9gO3wqfSByL8x8uHV799~XgC3S48TV0eUsTQdYXFi3N7-5b1J-ZthZSxhIiOHn4atrDfxZikNNmUBp2mjpvuJiYJ-pgJ7oS7L2SllVddqycclvaa9-58XcwQjaJskDXJvRJpu3lUO34~xN00Uduh9X~~XXR0o3rXsn3Vi-2u0zaCK83waP8BNp1lj-7z8VGfQzRNuWqWxF4JnpyJIDdH~Pv9lf1hRSqLA2jpAx~xG4HKUAvyqOrlS8P1hLGVumWUSc9vCYDBg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Effects of STS on mitochondrial proteins involved in mitochondrial fission/fusion process and mitophagy in CSE-treated alveolar epithelial A549 cells. A, Western blot was used to analyze the protein levels of Pink1, Mfn2, and Drp1 in the CSE-treated A549 cells by Western blot. Results are representative of different experiments. B, Densitometric analysis of Pink1, Mfn2, and Drp1 in the immunoblots using β-actin as the internal reference. Data are presented as mean ± SEM of at least 3 independent experiments, *p < .05, versus untreated cells (3% CSE [−] and STS [−]); #p < .05, versus cells treated with 3% CSE only.
STS Upregulated SIRT1 in CSE-Stimulated Alveolar Epithelial A549 Cells
SIRT1, an NAD+-dependent protein, plays an important role in multiple pathophysiological processes, including cell apoptosis, oxidative stress, and mitochondrial dysfunction (Chung and Adcock, 2008; Rajendrasozhan et al., 2008). SIRT1 is also crucial for the turnover of defective mitochondria by mitophagy (Tang, 2016). It has been reported that SIRT1 levels were reduced in lungs of smokers and patients with COPD (Rajendrasozhan et al., 2008), and activation of SIRT1 by SRT1720 ameliorates CS-induced emphysema/COPD in mice (Yao et al., 2012). We therefore examined SIRT1 gene expression and protein levels in the absence or presence of STS treatment in response to CSE stimulation in A549 cells. As shown in Figures 6A and 6B, the mRNA and protein levels of SIRT1 were significantly decreased in CSE-stimulated A549 cells, which were significantly attenuated by STS treatment, as examined by real-time PCR and Western blot assays. Immunofluorescence staining demonstrated that fluorescence intensity of SIRT1 in A549 cells also increased with STS treatment (Figure 6C). These results suggest that the activation of SIRT1 may mediate STS-induced effects on CS-induced AEC apoptosis and emphysema.
![STS-upregulated SIRT1 in CSE-stimulated alveolar epithelial A549 cells. A549 cells were cultured with and without 3% CSE and/or 1, 2.5, or 5 μM STS for 48 h. A, The mRNA levels of SIRT1 in the A549 cells were analyzed by real-time PCR. B, The protein levels of SIRT1 in the A549 cells were analyzed by Western blot. C, Immunofluorescence staining of SIRT1 was performed. Data are presented as mean ± SEM of at least 3 independent experiments, *p < .05, versus untreated cells (3% CSE [−] and STS [−]); #p < .05, versus cells treated with 3% CSE only.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/toxsci/183/2/10.1093_toxsci_kfab087/1/m_kfab087f6.jpeg?Expires=1750322865&Signature=4by50zgzEZPCj3a6FiXkJuPc1om0Z2M81OsvMZVXwglFStcwOax2XB-QMDW7X6Vc7Ai~~KX5RbArTHLRTSMQ~nUXywMO3hloSMeVX0bJBjtVHQY9y0EZvpaPbdhXNaND6nduMG-Et2SWcfE41q5NyldcuRl12ekMkRrHylNz5fSILL3T-bkUc6tyCsAVvwTAcyQikhl2i~sJUSKSgDB9mpbcbTZFdSgkuxfdGLJmEJ5EqUcNNb7p-~t5kkGNfQsiQVVglN2PTtHrlzERuedwpsvB85-AUXStr3LAohF1sDHCD2RIEiWzvGWMIQ-amyTOF1NCvQ69OmttmPneghrOgA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
STS-upregulated SIRT1 in CSE-stimulated alveolar epithelial A549 cells. A549 cells were cultured with and without 3% CSE and/or 1, 2.5, or 5 μM STS for 48 h. A, The mRNA levels of SIRT1 in the A549 cells were analyzed by real-time PCR. B, The protein levels of SIRT1 in the A549 cells were analyzed by Western blot. C, Immunofluorescence staining of SIRT1 was performed. Data are presented as mean ± SEM of at least 3 independent experiments, *p < .05, versus untreated cells (3% CSE [−] and STS [−]); #p < .05, versus cells treated with 3% CSE only.
STS-Mediated Attenuation in Mitochondrial Dysfunction, Oxidative Stress, and Apoptosis Was Associated With SIRT1-Dependent Pathway
To further investigate whether SIRT1 mediates protective effect of STS in AECs, we used EX 527 to inhibited SIRT1. As show in Figures 7A and 7B, when SIRT1 was inhibited, the protection of STS against CSE-induced apoptosis was significantly attenuated, as determined by flow cytometry assay. The Δψm was no longer able to be affected by STS after EX 527 treatment (Figs. 7F and 7G). EX 527 also abolished the inhibitory effect of STS on the production of cytoplasmic (Figure 7C) and mtROS (Figs. 7D and 7E). These data further imply that STS attenuates CSE-induced mitochondrial dysfunction, oxidative stress, and apoptosis via SIRT1 signaling.
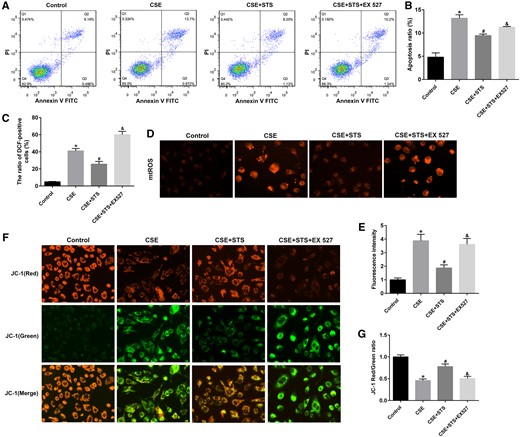
STS had no inhibitory effects on mitochondrial damage, oxidative stress, and apoptosis with SIRT1 inhibition in A549 cells. A549 cells were treated with SIRT1 inhibitor (EX 527, 20 μM) in the absence and presence of 3% CSE and STS (5 μM) for 48 h. A, Cell apoptosis of A549 cells was examined via the Annexin V-FITC/PI staining assay by flow cytometry. B, The ratio of apoptotic cells was statistically analyzed. C, The intracellular ROS was examined by flow cytometry according to the ROS assay kit. D, E, mtROS levels. F, G, The changes in ΔΨm was measured by JC-1 staining. Data are presented as mean ± SEM of at least 3 independent experiments, *p < .05, versus control cells; #p < .05, versus cells treated with 3% CSE only; &p < .05, versus cells treated with CSE and STS.
DISCUSSION
The main and novel findings of the present study are that: (1) STS reduced apoptosis in CSE-stimulated alveolar epithelial A549 cells. (2) STS attenuated CSE-induced mitochondrial dysfunction, mitophagy defects, and oxidative stress in alveolar epithelial A549 cells. (3) Protective effect of STS on mitochondrial dysfunction and oxidative stress-induced cellular apoptosis was likely associated with enhanced SIRT1 pathway.
Apoptosis is involved in the loss of alveolar wall cells in patients with emphysema (Yokohori et al., 2004). Yokohori et al. have observed that in the lungs of patients with emphysema, the level of apoptosis in alveolar wall cells is enhanced (Yokohori et al., 2004). Furthermore, AEC apoptosis in emphysematous lung tissue is associated with an increase in activated subunits of caspase-3 and loss of the anti-apoptotic protein Bcl-2 (Imai et al., 2005; Yokohori et al., 2004). The induction of apoptosis in AECs causes the loss of alveolar wall structures and following emphysematous alterations (Aoshiba et al., 2003). In emphysematous lungs, the apoptosis of AECs may be induced by various stimuli, including inflammation, oxidative stress, and CS (Hoshino et al., 2001; Nakajima et al., 1999). This is corroborated by our findings. The mechanisms underling CS-indcued apoptosis are associated with caspase-3 activation. Treatment with STS significantly increased cell viability and decreased cell apoptosis in CSE-exposed A549 cells. This is consistent with previous observations that STS or tanshinone IIA reduces apoptosis in endothelial cells, umbilical vein endothelial cells, cardiomyocytes, and lens epithelial cells (Cheng et al., 2017; Qi et al., 2019; Zhang et al., 2017; Zhu et al., 2020). Conversely, STS or tanshinone IIA induces apoptosis of human nasopharyngeal carcinoma cells, AtT-20 cells, hepatocellular carcinoma cells, and some other type cells (Li et al., 2017; Liu et al., 2019; Ren et al., 2017). These contradictory results demonstrate that STS exhibits both pro- and anti-apoptotic effects in cancer and noncancer or normal cells, which needs further investigation.
Oxidative stress is a key factor for CSE-induced cellular injury and apoptosis (Luo et al., 2017). Suppression of oxidative stress by SOD mimetic significantly inhibited AEC apoptosis and emphysema in COPD model (Tuder et al., 2003). Numerous studies have demonstrated that STS or tanshinone IIA exerts pulmonary protective effects through its antioxidant properties. For instance, tanshinone IIA attenuates the formation of MDA, inducible nitric oxide synthase, and nitric oxide in bleomycin-induced pulmonary fibrosis in rats (He et al., 2015). STS decreases CSE-induced intracellular ROS production in RAW 264.7 cells (Guan et al., 2018). Consistent with these findings, our results showed that STS protected against CSE-induced oxidative injury in AECs, which was mediated via the reduction in intracellular ROS and mtROS generation. Given the role of oxidative stress in the pathology of cell apoptosis, STS protects against CSE-induced AEC apoptosis, at least in part, by inhibiting oxidative stress.
Mitochondria play important roles not only in cellular respiration but also in other fundamental cellular functions including cellular metabolism and apoptosis/death (Kang and Shadel, 2016). ROS overproduction can damage mitochondrial DNA and membrane proteins, resulting in mitochondrial dysfunction (Valko et al., 2007). Mitochondrial dysfunction, in turn, further causes the disruption of intracellular redox homeostasis, resulting in ROS overproduction and the initiation of apoptosis (Circu and Aw, 2010). Thus, mitochondrial dysfunction, with increase in oxidative stress, results in increased apoptosis of cells (Chung and Adcock, 2008). Increasing evidence suggests that CS exposure can cause impaired mitochondrial function in epithelium, leading to COPD (Hoffmann et al., 2013; Wang et al., 2019). In accordance with this, we found that mitochondrial function was impaired in CSE-induced AEC apoptosis, characterized by a decline in OCR. Although STS treatment significantly improved mitochondrial function in CSE-treated A549 cells. STS also increased ΔΨm and abundance of respiratory chain complexes I-IV in CSE-exposed A549 cells, further suggesting the attenuation of mitochondrial dysfunction in AECs by STS. This is consistent with a previous finding that tanshinone IIA attenuates oxaliplatin-induced neurotoxicity via mitochondrial protection (Cheng et al., 2019), and indicates that the attenuation of emphysema by STS is mediated, at least in part, through the inhibition of CSE-induced mitochondrial dysfunction in AECs.
Fusion/fission protein dysregulation disrupts mitochondrial dynamics and causes per-meabilization of the outer mitochondrial membrane, the release of apoptotic proteins, and subsequently facilitates cell apoptosis/death (Guan et al., 2019). Mfn2 is a major protein required for mitochondria fusion by interacting with the mitochondrial outer and inner membranes, and its reduction can cause mitochondrial fragmentation. Drp1 regulates mitochondrial fission. Mitochondria elongation is induced by either Mfn2 overexpression and/or Drp1 inhibition (Sundar et al., 2019). Here, we found that CSE causes mitochondrial fragmentation as indicated by decreased Mfn2 and increased Drp1 in alveolar epithelial A549 cells. Although STS reduced CSE-induced alterations in the expression of mitochondrial fusion/fission proteins toward mitochondrial fusion. This allows the spread of metabolites, enzymes, and mitochondrial genes throughout the mitochondrial compartments connteracting mitochondrial injury and gene mutation. Therefore, STS may improve mitochondrial function through the orchestration of mitochondrial dynamics.
Mitophagy, a particular form of autophagy, is the quality control mechanism in mitochondria associated with cellular senescence and apoptosis in a stress- and cell-type-dependent manner (Sundar et al., 2019). Mitophagy is activated following oxidative stress to protect the cells from apoptosis, whereas mitophagy defect causes accumulation of oxidative stress damage and cell apoptosis/death (Ornatowski et al., 2020). Pink1 is a key protein that plays an important role in the removal of damaged or dysfunction mitochondrial from the cells by mitophagy. However, there are conflicting reports for the role of Pink1-mediated mitophagy in COPD progression. For instance, increased Pink1-mediated mitophagy promoted program cell death in COPD (Mizumura et al., 2014). In contrast, both Pink1 and Parkin knockdown caused accumulated damaged mitochondria and enhanced ROS production in response to CSE exposure (Ito et al., 2015), and insufficient Pink1-mediated mitophagy aggravated emphysema (Sundar et al., 2019). In this study, we demonstrated that CSE stimulation in A549 cells for 48 h caused a reduction in Pink1 with increased mitochondrial fragmentation. This implies that impaired mitophagy may directly contribute to CSE-induced mitochondrial dysfunction. This is consistent with the findings by Sundar et al. who demonstrated that CSE treatment can cause an insufficient Pink1-mediated mitophagy in primary human lung epithelial cells (Sundar et al., 2019). STS promotes mitophagy in epithelial cells.
The mechanism by which STS inhibits CSE-induced AEC apoptosis remains obscure. SIRT1, a NAD+-dependent histone deacetylase, is a latent gene for modulating apoptosis. For instance, SIRT1 reduced apoptosis after spinal cord injury via microRNA-494 (Yu et al., 2019). SIRT1 also activated its downstream target Foxo1 to exhibit its anti-apoptotic effects (Meng et al., 2017). Recently, reduced level and activity of SIRT1 have been observed in human and experimental COPD (Guan et al., 2019; Rajendrasozhan et al., 2008). The mechanisms underlying SIRT1 reduction may be due to its posttranslational modifications, including phosphorylation and carbonylation (Gao et al., 2011; Zhao et al., 2019). SIRT1 activator SRT1720 protects against emphysema via enhancing oxidative stress and reducing apoptosis of AECs (Gu et al., 2015; Yao et al., 2012). Here, we found that treatment with STS significantly attenuated CSE-induced decrease of SIRT1, and a SIRT1 inhibitor EX 527 blunted the protection of STS against CSE-induced cell apoptosis. It is possible that STS treatment activates SIRT1, leading to increased antioxidant response genes including FOXO3, therefore reducing apoptosis (Yao et al., 2014). We noticed that the role of SIRT1 in cellular apoptosis seems contradictory; it has diverse effects on different types of cells, with both pro- and anti-apoptotic effects. For example, Zhao et al. reported SIRT1 is highly expressed in human melanoma cells, and activation of SIRT1 by resveratrol inhibits p53-dependent apoptosis pathways in these cells (Zhao et al., 2017). Although SIRT1 is downregulated in other tumors, such as breast cancer and hepatocellular carcinoma (Wang et al., 2008). SIRT1 is also downregulated in patients with pulmonary fibrosis, whose expression is negatively related to apoptosis in the lung (Shetty et al., 2017). We suspect that SIRT1 plays different roles depending on its subcellular localization (in both nucleus and cytoplasm), and disease or tissue specificity.
We further investigated whether STS protects against CSE-induced mitochondrial dysfunction and oxidative stress via upregulation of SIRT1. It has been reported that activation of SIRT1 is beneficial for regulation of oxidative stress, apoptosis, and mitochondria biogenesis (Chung et al., 2010). Deficiency of SIRT1 can weaken mitochondrial function and accelerate oxidative stress (Yao et al., 2012). These findings indicate that SIRT1 is critical to maintain mitochondrial function and reduce oxidative stress status. Interestingly, we found that EX 527 also abolished the protection of STS against CSE-induced mitochondrial dysfunction and oxidative stress. These above data further suggest that SIRT1 activation mediates the STS-induced effects on CSE-induced mitochondrial damage, oxidative stress and apoptosis in AECs.
We acknowledge there are some limitations in this study. Although lots of studies have demonstrated that A549 cells shows the similar response to that of AECs following CSE treatment, and that A549 cells are also used for in vitro experimental models in emphysema/COPD (Eurlings et al., 2014; Guan et al., 2019; Somborac-Bačura et al., 2018; Wang et al., 2019). This cell line may include constitutive overexpression of Nrf-2 driven gene products such as HO-1, which may promote the resistance of cells to apoptosis (Kweon et al., 2006). Further experiments are warranted to verify whether STS is protective against mitochondrial damage and apoptosis in primary human AECs. Second, mitophagy-dependent necroptosis is also involved in epithelial cell death and contributes to the pathogenesis of COPD (Mizumura et al., 2014), which was not investigated in this study. Third, we did not evaluate whether STS affects lung senescence, apoptosis, and mitochondrial dysfunction including mtROS, OXPHOS, and mitophagy in CS-exposed mice. This remains to be investigated in future study.
In conclusion, STS protects against CSE-induced apoptosis of AECs. The protective role of STS in AECs involves the inhibition of mitochondrial dysfunction and oxidative stress through enhancing SIRT1 pathway. Enhancing SIRT1 pathway by STS provides novel mechanisms and promising approaches for halting the development of emphysema/COPD.
AUTHOR CONTRIBUTIONS
W.L. and J.W. conceived the project and designed experiments. R.G. and H.Y. wrote the manuscript. W.L., H.Y., Z.L. and D.S. edited the manuscript. R.G., Z.L., Z.C., L.Y., W.L., M.D., J.X., and J.Q. performed experiments. R.G., Y.L., and J.Q. conducted data analysis. All authors have read and approved the final manuscript.
FUNDING
National Natural Science Foundation of China (81770043, 81800072, and 81520108001); National Key R&D Project (2016YFC0903700); Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (2017BT01S155); Natural Science Foundation of Guangdong Province (2018A030310291, 2016A030311020, 2018A030310290, and 2016A030313606); Guangzhou Municipal Science and Technology (201607020030 and 201804010052); Changjiang Scholars and Innovative Research Team in University (IRT0961).
DECLARATION OF CONFLICTING INTERESTS
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
REFERENCES
Author notes
Ruijuan Guan, Hongwei Yao, and Ziying Li contributed equally to this study.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments