





Abstract
The impact of cigarette smoke (CS), a major cause of lung diseases, on the composition and metabolism of lung lipids is incompletely understood. Here, we integrated quantitative lipidomics and proteomics to investigate exposure effects on lung lipid metabolism in a C57BL/6 and an Apolipoprotein E-deficient (Apoe−/−) mouse study. In these studies, mice were exposed to high concentrations of 3R4F reference CS, aerosol from potential modified risk tobacco products (MRTPs) or filtered air (Sham) for up to 8 months. The 2 assessed MRTPs, the prototypical MRTP for C57BL/6 mice and the Tobacco Heating System 2.2 for Apoe−/− mice, utilize “heat-not-burn” technologies and were each matched in nicotine concentrations to the 3R4F CS. After 2 months of CS exposure, some groups were either switched to the MRTP or underwent cessation. In both mouse strains, CS strongly affected several categories of lung lipids and lipid-related proteins. Candidate surfactant lipids, surfactant proteins, and surfactant metabolizing proteins were increased. Inflammatory eicosanoids, their metabolic enzymes, and several ceramide classes were elevated. Overall, CS induced a coordinated lipid response controlled by transcription regulators such as SREBP proteins and supported by other metabolic adaptations. In contrast, most of these changes were absent in the mice exposed to the potential MRTPs, in the cessation group, and the switching group. Our findings demonstrate the complex biological response of the lungs to CS exposure and support the benefits of cessation or switching to a heat-not-burn product using a design such as those employed in this study.
Smoking has been causally linked to many diseases, especially those of the respiratory and cardiovascular systems. In the developed world, tobacco smoke exposure is the major risk factor for chronic obstructive pulmonary disease (COPD), a major and increasing global health problem (Postma et al., 2015). COPD is characterized by persistent and progressive airflow limitation associated with chronic inflammation that is implicated in parenchymal tissue destruction and emphysema. Cigarette smoke (CS) provokes an array of biological responses in the respiratory tract, including apoptosis, oxidative stress, and endoplasmic reticulum stress which, with sustained exposure, may lead to nonresolving inflammation and emphysematous lesions (Phillips et al., 2015).
Smoking cessation is the most effective measure to reduce COPD morbidity and progression (Godtfredsen et al., 2008). As an alternative for smokers who otherwise would not quit, switching to less harmful products provides an alternative means to reduce the harm from smoking-related diseases, such as COPD. The US Family Smoking Prevention and Tobacco Control Act of 2009 describes this type of product as a modified risk tobacco product (MRTP), defined as “any tobacco product that is sold or distributed for use to reduce harm or the risk of tobacco-related disease associated with commercially marketed tobacco products.” Here, we conducted an integrated analysis of 2 comprehensive mouse exposure studies, in C57BL/6 and Apolipoprotein E-deficient (Apoe−/−) mice, in which 2 different potential MRTPs—developed by Philip Morris International—were assessed (Phillips et al., 2015 and Phillips et al., 2015b). Both tobacco products are based on the “heat-not-burn” principle, in which tobacco is heated in a controlled fashion to release nicotine and volatiles contributing to tobacco flavors but combustion is prevented. Thermal decomposition of organic tobacco compounds at elevated temperatures results in both pyrogenesis and pyrosynthesis of many harmful or potentially harmful constituents (HPHCs). Therefore, preventing combustion can produce, when compared with CS, a simpler aerosol with a strongly reduced content of these toxicants (Baker, 2006; Phillips et al., 2015).
In the prototypical MRTP (pMRTP), which was assessed in C57BL/6 mice, this goal is achieved with a carbon tip attached to a column of tobacco filler, serving as a heat source to generate an aerosol containing water, glycerin, nicotine, and tobacco flavors (Gladden et al., 2012; Maeder et al., 2011; Phillips et al., 2015). Of note, the pMRTP was an early (proof-of-concept) prototype of this carbon-tip based heat-not-burn platform. In contrast, the tobacco heating system (THS) 2.2, which was assessed in Apoe−/− mice, represents a more refined heat-not-burn product. It relies on an electrically controlled heat source with an improved design compared with the electrically heated smoking systems described previously (Schorp et al., 2012). The compositions of aerosols, including 56 HPHCs, from pMRTP and THS2.2 and smoke from 3R4F reference cigarettes are summarized in Supplementary Data. Although the levels of most of the measured HPHCs were strongly reduced—several by more than 90%—there were a few noteworthy exceptions. Acrylamide remained at the same level in THS2.2 as in 3R4F (at approximately 2 µg/mg nicotine) and arsenic and polycyclic aromatic hydrocarbons (PAHs) were only strongly reduced in THS2.2, but not in pMRTP aerosol. The more refined design and tighter temperature control of the THS2.2 system likely explains this further reduction of these latter important HPHCs in the aerosol.
Apoe−/− mice were chosen for the THS2.2 assessment study as they support the concomitant evaluation of effects on the respiratory and cardiovascular system. Apoe−/− mice are a commonly used model for atherogenesis (Veniant et al., 2001), especially to investigate smoking-related atherosclerosis (Boué et al., 2012; Chan et al., 2012; Lietz et al., 2013; von Holt et al., 2009) as well as CS-induced lung inflammation and emphysema (Arunachalam et al., 2010; Boue et al., 2013; Han et al., 2012). Although this manuscript is focused on the effects on the lung, our accompanying manuscript discusses the cardiovascular effects in more detail (Phillips et al., 2015b).
Though not all symptoms of COPD may be recapitulated in rodent lungs, CS-exposed mice are particularly well-suited to model the early phases of human COPD (GOLD stages 1 and 2) (Churg et al., 2008). High-throughput approaches, including genome-wide expression analyses, have highlighted a number of genes and biological pathways involved in the development of COPD (Phillips et al., 2015). In addition, a few studies have already demonstrated that specific lipid classes play key roles in lung pathobiology, and there is evidence that CS exposure can modulate lung lipid metabolism (Lundstrom et al., 2011; Telenga et al., 2014). Recently, Morissette et al. (2015) presented an intriguing model how CS-induced damage of surfactant lipids, with the involvement of foam-cell-like macrophages, can elicit lung inflammation. It is anticipated that the recent progress in mass spectrometry-based analyses of lipids (lipidomics) will facilitate further insights into the complex compositions and responses of lung lipidomes (t’Kindt et al., 2015).
The goals of our study were to gain deeper insights into the effects of CS exposure on the lung lipidome and lipid-related proteome (ie, the set of proteins with enzymatic or structural roles in lipid-related processes) and to compare these to effects of cessation, continuous exposure to aerosol from the potential MRTP, and switching to the MRTP. We conducted an integrated analysis of the lung lipidome and proteome response profiles in lungs from 2 comprehensive mouse exposure studies (Figure 1A). In the first study, we exposed C57BL/6 mice for up to 7 months to assess effects of pMRTP aerosol. In the second study, we exposed Apoe−/− mice for up to 8 months to assess effects of aerosol from the THS 2.2. Certainly, this manuscript, which is focused on the effects on lung lipid metabolism, does not constitute a full risk assessment of these heat-not-burn products. For additional endpoints of these 2 studies, the reader is referred to our overview publications (Phillips et al., 2015 and Phillips et al., 2015b); and importantly, these studies are part of a much more comprehensive assessment framework and results from additional studies will be reported elsewhere.
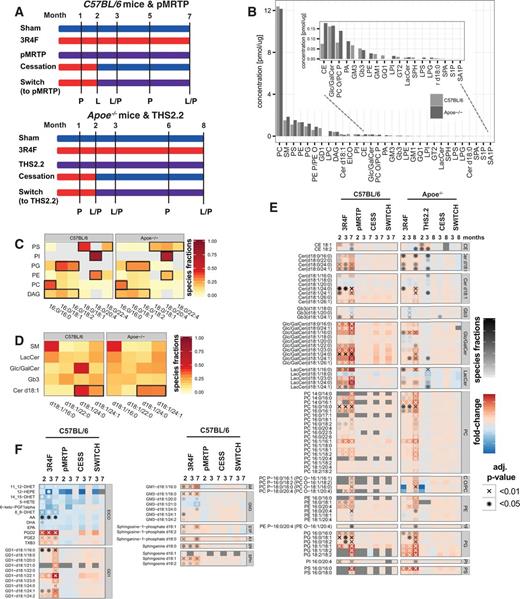
Effect of cigarette smoke exposure, MRTP aerosol exposure, cessation, and switching to a potential MRTP on the lung lipidomes of C57BL/6 and Apoe−/− mice. A, Experimental design for the 2 mouse inhalation studies, investigating exposure effects in C57BL/6 and Apoe−/− mice. Mice were exposed to cigarette smoke, a potential MRTP (THS2.2 or pMRTP) or fresh air (sham). Some mice were exposed to CS for 2 months and then either underwent cessation or were switched to the MRTP. Lung tissue samples for lipidomics and proteomics were collected at the indicated time points (L = lipidomics, P = proteomics). B, Lipid class concentrations for both studies. The sums of the median lipid species concentrations of a class across the whole study are shown. If a lipid class was quantified in both studies, only lipid species detected in both studies were considered in calculating the lipid class sum. Values were sorted based on the maximum values in both studies. The insert shows the lower abundance lipid classes. See Materials and Methods for the list of abbreviations. C, Detailed composition profiles for glycerophospholipids and DAG. Only lipid compositions contributing at least 20% to the total concentration of either lipid class were included. D, Composition profiles for d18:1 Cer, Glc/GalCer, LacCer, and Gb3. E, Differential abundance profiles for lipid species with significant differential abundance in both studies. Each row shows 1 lipid species, each column an exposure condition compared to the sham group at the respective time point. The fold-change compared with sham exposure is color-coded (see color key). Significantly abundant differences are indicated (*BH-adjusted P-value < .05, XBH-adjusted P-value < .01). In addition, the molar contribution of significantly abundant lipid species to their lipid class is indicated by the shade of the significance indicator (“species fractions”). (See Supplementary Data for the full set of quantified lipids). F, Differential abundance profiles for lipid species that were quantified only in the C57BL/6 study.
MATERIALS AND METHODS
For more details, see Supplementary Data.
Experimental design
The integrative analysis of the effects of CS on lung lipid metabolism, as reported here, was performed on samples from 2 larger systems toxicology studies (the C57BL/6 and the Apoe−/− studies, respectively). A more detailed description of these studies and results from multiple other endpoints have been reported in our overview publications (Phillips et al., 2015 and Phillips et al., 2015b). Of note, these studies were independently conducted—with the C57BL/6 study preceding the Apoe−/− study. For the rational of combining cardiovascular and respiratory endpoints and adjustments in the sampling time points in the Apoe−/− study (Figure 1A), the reader is referred to our overview publication (Phillips et al., 2015b).
Briefly, these studies tested effects of CS, potential MRTP aerosols and air on lung tissue of female C57BL/6 or Apoe−/− mice. In both studies we focused on the effects on female mice to limit the number of animals (and omics measurements), while selecting the possibly more sensitive gender. For example, clinical studies suggest that women might be more susceptible to emphysema development than men and a higher susceptibility of female mice to develop emphysema has been reported (Aryal et al., 2013; March et al., 2006;Sørheim et al., 2010).
Each study included 5 groups of mice, each exposed as follows: (1) sham (exposed to fresh air); (2) CS (exposed to mainstream smoke from 3R4F, a reference cigarette from the University of Kentucky); (3) MRTP (exposed to MRTP aerosol); (4) smoking cessation (CS followed by smoking cessation); and (5) switching to MRTP (CS followed by MRTP). Mice were exposed as indicated for up to 7 and 8 months in the C57BL/6 and Apoe−/− mouse studies, respectively. To model the effects of smoking cessation and switching to an MRTP, groups of mice were first exposed to CS for 2 months and exposure was changed to filtered air (cessation) or MRTP (switching) for an additional 5 or 6 months. C57BL/6 mice were exposed to aerosol from pMRTP and Apoe−/−mice to THS2.2.
Lung tissue samples for lipidomics analysis were collected after 2, 3, and 7 months exposure in the C57BL/6 study and 2, 3, and 8 months exposure in the Apoe−/− study. Each group analyzed for lipidomics included 8 mice, providing 8 biological replicates. Lung tissue samples for proteomics analysis were collected after 1, 3, 5, and 7 months exposure in the C57BL/6 study and after 1, 2, 3, 6, and 8 months exposure in the Apoe−/− study. For proteomics analysis, each C57BL/6 group included 6 mice and each Apoe−/− group included 8 mice, providing 6 and 8 biological replicates, respectively.
Animals
All procedures involving animals were approved by an Institutional Animal Care and Use Committee, in compliance with the National Advisory Committee for Laboratory Animal Research Guidelines on the Care and Use of Animals for Scientific Purposes. In accordance with the “3Rs” principles of animal research, Replacement, Reduction, and Refinement, we used the minimum number of animals needed to obtain valid results and the least invasive procedures to minimize pain and distress. Moreover, our laboratory technicians and veterinary specialists are trained in the latest techniques to most effectively and humanely manage and care for animals.
Female C57BL/6 mice bred under specific pathogen-free conditions were obtained from Charles River, Wilmington, Massachusetts. Female B6.129P2-Apoe (tm1Unc)N11 (Apoe−/−) mice bred under specific pathogen-free conditions were from Taconic Biosciences (Germantown, New York). The mice were 8- to 10-week old at the start of exposures.
Reference cigarettes and MRTPs
3R4F cigarettes were purchased from the University of Kentucky (Lexington, Kentucky, http://www.ca.uky.edu/refcig). The candidate pMRTP (Philip Morris Products S.A., Neuchâtel, Switzerland) has a carbon tip attached to a tobacco plug that serves as a fast-lighting heat source to generate an aerosol. The candidate MRTP, THS2.2, consists of a stick containing a tobacco plug inserted into a holder that electrically heats the tobacco in a controlled manner to create an aerosol. THS2.2 sticks were produced at Philip Morris International (PMI; Neuchâtel, Switzerland) and holders were provided by PMI. For both candidates the temperature applied to the tobacco plug is below the combustion temperature. This technology, by avoiding tobacco combustion, reduces formation of HPHCs. The resulting aerosols contain mainly water, glycerin, nicotine, and volatile substances contributing to the specific tobacco flavors.
Comparative analytical specifications of the pMRTP aerosol, THS2.2 aerosol, and 3R4F CS yields are given in Supplementary Data, which shows quantification of 56 HPHCs plus water and glycerol (as a humectant) for a total of 58 analytes from 3R4F CS and pMRTP and THS2.2 aerosols. In the table, the concentrations of these analytes are normalized to that of nicotine.
Smoke generation and animal exposure
Mainstream CS from 3R4F cigarettes was generated on 30-port rotary smoking machine (type PMRL-G, SM2000) as described previously (Phillips et al., 2015). Aerosols from THS2.2 sticks were generated on modified 30-port rotary smoking machines equipped with the appropriate holders. Two modified smoking machines per chamber were required to achieve the target THS2.2 aerosol concentration. pMRTP aerosol was generated in modified SM2000 machines. 3R4F cigarettes, THS2.2 sticks, and pMRTP sticks were smoked according to the Health Canada Intensive Smoking Protocol based on ISO standard 3308 (revised in 2000), with the exception of the puff volume (55 ml) and puff frequency (1 puff every 30 s), as described previously (Phillips et al., 2015). Several additional minor deviations from ISO standard 3308 were necessary for technical reasons (Phillips et al., 2015). Aerosols were further diluted with conditioned fresh air to achieve the target concentrations.
The C57BL/6 mice were whole body-exposed to diluted mainstream smoke from 3R4F (750 mg total particulate matter (TPM)/m3, equivalent to 34.4 mg nicotine/m3), pMRTP aerosol (nicotine-matched to 3R4F, 34.4 mg/m3) or filtered air for 4 h per day, 5 days per week, for up to 7 months, with intermittent daily exposure to fresh filtered air for 30 min after the first hour of smoke exposure and for 60 min after the second and third hour of exposure. The mice exposed only to fresh air served as the control (sham) group. The Apoe−/− mice were whole body-exposed to diluted mainstream smoke from 3R4F (600 mg TPM/m3, equivalent to 29.9 mg nicotine/m3), THS2.2 aerosol (nicotine-matched to 3R4F, 29.9 mg/m3) or filtered air for 3 h per day, 5 days per week, for up to 8 months, with intermittent daily exposure to fresh filtered air for 30 min after the first hour of smoke exposure and for 60 min after the second hour of exposure. This was done to avoid accumulation of excessive carboxyhemoglobin (COHb) in the 3R4F group. For the sham group, mice were exposed only to air. Reproducibility of smoke generation was ensured by periodic analysis of the test atmosphere during the study.
Proteomics analysis
Our general quantitative proteomics approach has been previously described (Phillips et al., 2015; Titz et al., 2014) and the reader is referred to our main study publications for a high-level summary of the proteomics results (Phillips et al., 2015 and Phillips et al., 2015b). For convenience, we have summarized the experimental details in the Supplementary Data. Briefly, samples were homogenized in tissue lysis buffer, proteins were precipitated, and 50 µg protein from each sample was used for further analysis. In the C57BL/6 study, samples were processed using iTRAQ 8-plex labeling (AB Sciex, Framingham, Massachusetts). For the Apoe−/− study, samples were processed using TMT 6-plex labeling (Thermo Scientific, Waltham, Massachusetts). Each procedure was performed according to the manufacturer’s instructions. Samples were analyzed by liquid chromatography coupled with tandem mass spectrometry using an Easy-nanoLC 1000 instrument connected online to a Q-Exactive MS mass-analyzer (Thermo Scientific). Data were normalized, quantified, and used to identify differentially abundant proteins in the R environment. The Benjamini–Hochberg FDR multiple test correction was employed and proteins with an adjusted P-value <.05 were considered to be differentially expressed.
Lipidomics analysis
Molecular lipids from lung tissue were extracted and quantified by Zora Biosciences (Espoo, Finland) using synthetic nonendogenous standards (Boué et al., 2012) and specific details are given in the Supplementary Data. Lipid levels were normalized to their respective internal standards and the sample volume or tissue weight. Concentrations of molecular lipids were presented as nmol/mg wet tissue. For differential abundance analysis, only those lipid species that were present in at least 50% of the samples of each compared group were included. The Benjamini–Hochberg FDR multiple test correction was employed for t-test derived P-values and lipid species with an adjusted P-value < .05 were considered to be differentially abundant.
Annotations of lipid species
Wherever possible, we report lipids following the Lipid Maps classification (Fahy et al., 2009). Briefly, the different lipid species of phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylserine (PS), phosphatidylinositol (PI), phosphatidic acid (PA), phosphatidylglycerol (PG), and diacylglycerol (DAG) are listed with the 2 fatty acyl groups separated with a hyphen, eg, PC 16:0-18:1, when positions are unknown and with a slash, eg, PC 16:0/16:0, when known. LysoPC and lysoPE are abbreviated as LPC and LPE, respectively, and cholesteryl esters are abbreviated CE. Ether-linked phospholipids are shown as PC O (alkyl), PC P (alkenyl), PE O (alkyl), and PE P (alkenyl). Fatty acyl groups of ether-linked lipids and N-amidated fatty acyl groups for sphingomyelin (SM), ceramide (Cer), sphingolipids, and gangliosides are shown after the slash. Sphingolipids are: sphingosine (SPH), sphingosine-1-phosphate (S1P), sphinganine-1-phosphate (SA1P), glucosylceramide (GlcCer), lactosylceramide (LacCer), and globotriaosylceramide (Gb3). Gangliosides are: GD1, GM1, GM3, GQ1, and GT2. Fatty acids and eicosanoids are: docosahexaenoic acid (DHA), arachidonic acid (AA), eicosapentaenoic acid (EPA), 12-hydroxyeicosatetraenoic acid (12-HETE), 11-hydroxyeicosatetraenoic acid (11-HETE), 15-hydroxyeicosatetraenoic acid (15-HETE), 8-hydroxyeicosatetraenoic acid (8-HETE), 5-hydroxyeicosatetraenoic acid (5-HETE), prostaglandin E2 (PGE2), prostaglandin D2 (PGD2), 13-hydroxyoctadecadienoic acid (13-HODE), 9-hydroxyoctadecadienoic acid (9-HODE), 6-keto prostaglandin F1α (6-keto-PGF1alpha), prostaglandin F2α (PGF2alpha), tromboxane B2 (TXB2), tromboxane B3 (TXB3), 12-hydroxyeicosapentaenoic acid (12-HEPE), 14,15-dihydroxyeicosatrienoic acid (14_15-DHET), 11,12-dihydroxyeicosatrienoic acid (11_12-DHET), 8,9-dihydroxyeicosatrienoic acid (8_9-DHET), 5,6-dihydroxyeicosatrienoic acid (5_6-DHET), hydroxyoctadecatrienoic acid (13-HOTrE), 12-oxo-eicosatetraenoic acid (12-oxoETE), 5-oxo-eicosatetraenoic acid (5-oxoETE), 5-hydroxyicosapentaenoic acid (5-HEPE), 15-hydroxyicosapentaenoic acid (15-HEPE).
Additional data analysis methods
Lipid pathway maps were obtained from the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (http://www.genome.jp/kegg/). The log2 fold-change was used as the protein statistic, the mean as the set statistic and sample permutation within the group comparisons and P-value adjustment with the Benjamini–Hochberg procedure were used to estimate statistical significances. The sparse partial least squares (sPLS) approach was conducted in the canonical mode. For the proteomics data, 100 variables were kept in the loadings and 50 variables were kept for the lipidomics data. The analysis of transcription factor activities and the network perturbation amplitude analysis for the Nfe2l2 signaling network were based on transcriptomics measurements (Phillips et al., 2015) and performed as described, previously (Martin et al., 2012, 2014).
Data availability
The mass spectrometry proteomics data are available from the ProteomeXchange Consortium via the PRIDE partner repository (http://www.ebi.ac.uk/pride/archive/). The database identifier for the C57BL/6 dataset is PXD001369 and the identifier for the Apoe−/− dataset is PXD002530. The lipidomics data are available in the supplement (Supplementary Data). In addition, the complete data for the C57BL/6 systems toxicology study has been summarized in a data descriptor article (manuscript in revision).
RESULTS
The Lung Lipidome Shows a Consistent Response to CS Exposure in C57BL/6 and Apoe−/− Mice
To investigate effects of CS exposure, exposure to aerosols from heat-not-burn tobacco products (ie, the potential MRTPs), cessation and switching on the mouse lung lipidome, we conducted an extensive LC-MS/MS-based lipidomics analysis for 3 exposure time points in 2 different mouse exposure studies (Figure 1A). Lung tissue samples in the C57BL/6 mouse study were collected after 2, 3, and 7 months and, in the Apoe−/− mouse study, after 2, 3, and 8 months of exposure. The potential MRTPs tested were pMRTP in the C57BL/6 study and THS2.2 in the Apoe−/− study. Smoking cessation and switching to the MRTP occurred, in the indicated groups, after 2 months CS exposure. Thus, the effects of cessation and switching were assessed at 1 month and, depending on which mouse strain, 5 or 6 months after the change of aerosol (3- and 7 or 8-month time points). The nicotine concentrations were matched between each MRTP and 3R4F and, importantly, we could demonstrate a similar uptake of nicotine for the 3R4F and MRTP groups and a reduction in COHb for the MRTP group (Supplementary Data).
The quantified lipid classes of the lung lipidome covered a wide range of concentrations (Figure 1B; see methods for the used lipid nomenclature and abbreviations). PC had by far the highest concentration in the lung lipidome, most likely because of its role as a surfactant lipid (see below). PC is followed in abundance by several lipid classes with important structural roles including SM, PS, PE, and PG. Lipids present at the lower end of the concentration range include sphingosine-1-P and sphinganine-1-P, important lipid signaling mediators. Overall, the C57BL/6 and Apoe−/− mouse studies showed generally good agreement in concentrations of these lipid classes, with some exceptions. For example, CE concentrations were greater in the Apoe−/− mouse study because of the general perturbation of cholesterol metabolism in this mouse strain (Nakashima et al., 1994).
Our LC-MS/MS approach enabled resolution of the exact composition of the analyzed lipid species. The glycerophospholipids and DAG showed an especially intriguing fatty acid composition pattern, with different composition profiles for the different lipid classes (Figure 1C). Dipalmitoylphosphatidylcholine (PC 16:0/16:0) dominated the class of PC lipids, which is most likely because of the dominant role of this lipid as a component of lung surfactant (Batenburg, 1992). The phosphatidyl-glycerine class was enriched for PG 16:0/18:1 and PG 16:0/18:2 lipid species, which, similarly, is likely to reflect their role as surfactant lipids (Postle et al., 2001). Both PE and PI had a relatively abundant 18:0/20:4 lipid species. The 20:4 fatty acid most likely represents AA and a particularly high abundance of this lipid in PI and PE has been noted previously (Balgoma et al., 2010). Therefore, PI 18:0/20:4 and PE 18:0/20:4 represent important substrates for the release of AA by phospholipases, for example, to initiate prostaglandin signaling in the lung. In contrast, ceramide lipids had a less distinct pattern of composition (Figure 1D). However, as discussed further below, an enrichment of longer-chain ceramide species (Cer d18:1/24:0 and Cer d18:1/24:1) was apparent.
Having established general lipid patterns and the overall agreement between the 2 studies, we next investigated how the quantified lipid species were affected in both mouse strains by the various exposure conditions. These data are expressed as changes in abundance of each analyte, when compared with its levels in lungs from sham-treated mice at the same time point. A broad range of lipids was significantly affected by CS throughout the exposure time in both studies (Figure 1E, Supplementary Data). These included several major lipid classes such as glycerophospholipids (PC, PE, PI, PG, PS; and lipid ethers of PC and PE), sphingolipids (Cer, Glc/GalCer, LacCer, and Gb3), and CE. The majority of these significantly affected lipid species were upregulated by CS exposure. In contrast to the effects of CS exposure, in both mouse strains, there was a much more limited response of these lipids in mice from the MRTP exposure, cessation and switching groups. In the most prominent such effect, after 3 months exposure to THS2.2, levels of only several ceramides (Cer), lactosylceramide (LacCer), and one Glc/Gal ceramide species were changed in Apoe−/− mice. Furthermore, the changes in these lipids were primarily in the opposite direction as the changes occurring with CS exposure and there was no consistent time-dependence.
Additional lipid classes measured only in the C57BL/6 study also showed significant differential abundance with CS exposure (Figure 1F). Notably, these included several eicosanoids (eg, PGD2, PGE2, and TXB3), sphingosine and sphingosine-1-phosphate, and gangliosides (GD1, GM1, and GM3). For these lipid species, significant changes in abundance were detected only in the CS-exposed groups, with some time-dependent trends detectable when CS exposure was discontinued. For example, PGD2 demonstrated a similar trend as Glc/Gal ceramides in the C57BL/6 study: Levels of these lipids were strongly upregulated at all 3 time points of 3R4F exposure, some upregulation remained 1 month after cessation or switching, but levels were close to those in sham mice at the last time point, 7 months.
In summary, lipidomics profiling of lung tissues in both exposure studies demonstrated a strong and consistent effect of CS exposure on many lipid classes. Upon cessation or switching to THS2.2 or pMRTP, these increases had mostly returned to sham levels by the end of each study, whereas THS2.2 and pMRTP exposure by itself had only limited effects without consistent time-dependences.
CS Exposure Induces Wide-Ranging Changes in the Lipid-Related Lung Proteome
We next investigated if the global alterations induced by CS exposure in the lung lipidome are reflected by changes in proteins involved in lipid-related processes. For this, we leveraged quantitative lung proteomics data that we had generated at 4 time points (1, 3, 5, and 7 months) in the C57BL/6 study and 5 time points (1, 2, 3, 6, 8 months) in the Apoe−/− study. In both mouse strains, 3R4F (CS) exposure induced strong changes in the lung proteome, whereas both potential MRTPs (pMRTP and THS2.2) exposed groups showed limited effects and the protein levels in the cessation and switching groups return to sham levels over the course of the study (Figure 2A). Interestingly, the subset of proteins involved in lipid-related processes (lipid metabolic process [GO:0006629] and lipid homeostasis [GO:0055088]) demonstrated a response similar to this global profile, with a robust differential expression response for all CS exposure time points in both studies.
![Effects of exposure conditions on the lipid-related lung proteome of C57BL/6 and Apoe−/− mice. A, Effect on the whole and lipid-related lung proteome. Volcano plots show the amplitude (log2 fold-change, x-axis) and significance (−log10 BH-adjusted p-value, y-axis) for the exposure groups as compared to the appropriate sham exposed group. Lipid-related proteins (lipid metabolic process [GO:0006629] and lipid homeostasis [GO:0055088]) are indicated with red/blue dots and remaining proteins with gray dots. Proteins with statistically significant differential regulation are above the BH-adjusted P-value threshold line of .05. B, Protein set enrichment for lipid-related pathways from the KEGG database. The fold-change was used as the protein-level metric and the mean as the protein set statistic (Väremo et al., 2013); protein pathways were obtained from the KEGG database (http://www.genome.jp/kegg/). Only lipid-related pathways with a BH-adjusted P-value < .05 in any of the comparisons are shown. The enrichment score is color coded (see key) and the statistical significance of an enrichment is indicated with “*” (BH-adjusted P-value < .05). C, Protein expression profiles for the KEGG fatty acid biosynthesis pathway. The fold-changes and statistical significance of differential expression are color-coded and marked with a symbol, respectively (see key, BH-adjusted p-value, *<0.05, x< 0.01). Missing values are gray. D, As in C, but for fatty acid degradation pathway.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/toxsci/149/2/10.1093_toxsci_kfv244/2/m_kfv244f2p.jpeg?Expires=1750191899&Signature=lq0aZ64GecQaUbR-mgjvjkS1vcatEGGIvGpcTOmbWi6KC8N4h0g55AxOd~MVTu8DNOz94GCuju4bqRxg7KizUitLxw1jDWonBvBdEPqLhQ0V3RmBRiWZLxTS7jy-hEhbDAkNSxFRE~txCLyNsbcWGVJYnckrQgoT4~ka~L6PRTs4F9sQke2y81c1gAkrqjH9VtWBpoQJvY06IiKVPSSHxoe0Kivnq6vXDXQIscsV66tiswT2K7t-ZTitrlCeUayXAcKOsCjecOfug62b9lARvGZuI7zDAR-03ZtWMCePot3jY2wpBs5o~mBypiHdUCCptfz4sj2GVvmrDAlP4iQPLQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Effects of exposure conditions on the lipid-related lung proteome of C57BL/6 and Apoe−/− mice. A, Effect on the whole and lipid-related lung proteome. Volcano plots show the amplitude (log2 fold-change, x-axis) and significance (−log10 BH-adjusted p-value, y-axis) for the exposure groups as compared to the appropriate sham exposed group. Lipid-related proteins (lipid metabolic process [GO:0006629] and lipid homeostasis [GO:0055088]) are indicated with red/blue dots and remaining proteins with gray dots. Proteins with statistically significant differential regulation are above the BH-adjusted P-value threshold line of .05. B, Protein set enrichment for lipid-related pathways from the KEGG database. The fold-change was used as the protein-level metric and the mean as the protein set statistic (Väremo et al., 2013); protein pathways were obtained from the KEGG database (http://www.genome.jp/kegg/). Only lipid-related pathways with a BH-adjusted P-value < .05 in any of the comparisons are shown. The enrichment score is color coded (see key) and the statistical significance of an enrichment is indicated with “*” (BH-adjusted P-value < .05). C, Protein expression profiles for the KEGG fatty acid biosynthesis pathway. The fold-changes and statistical significance of differential expression are color-coded and marked with a symbol, respectively (see key, BH-adjusted p-value, *<0.05, x< 0.01). Missing values are gray. D, As in C, but for fatty acid degradation pathway.
To gain an initial view on which functional categories are involved in the overall CS-induced response of lipid-related proteins, we conducted a set enrichment analysis focused on lipid metabolism pathways of the KEGG database (http://www.genome.jp/kegg/). Many lipid metabolism pathways were significantly affected by CS exposure in the C57BL/6 and the Apoe−/− studies. These included several pathways with links to the lipidome alterations discussed in the previous section. Examples of these pathways include glycerophospholipid metabolism, sphingolipid metabolism, and AA metabolism, all showing consistent upregulation with CS exposure. In contrast, effects observed for the other exposure groups, MRTP-exposed, cessation and switching, were more limited or showed inconsistent time dependence (Figure 2B).
The broad effects of CS on the lung lipidome likely require de novo synthesis of lipids. The fatty acid biosynthesis pathway was significantly upregulated in both mouse strains with differential regulation of several key enzymes (Figure 2C). In both studies, CS exposure resulted in significant upregulation of the core fatty acid synthase (Fasn) as well as of the acetyl-CoA carboxylase alpha (Acaca), which catalyzes the rate-limiting step in the biogenesis of long-chain fatty acids. In addition, the long-chain-fatty-acid-CoA ligases 4/5 (Acsl4/5) were upregulated, whereas isoform 1 (Acsl1) was downregulated. Acsl enzymes conjugate fatty acids to coenzyme-A, facilitating the incorporation of these fatty acids into glycerolipids and glycerophospholipids. This is important, for example, in the synthesis of surfactant lipids or synthesis of AA-conjugated glycerophospholipids. In this context, it would be interesting to further investigate the intriguing finding that CS-induced opposing effects on Acsl1 and Acsl4/5. This observation might be related to mechanisms for channeling individual fatty acids to specific synthetic pathways (Robichaud and Surette, 2015). Simultaneously several fatty acid degradation enzymes were upregulated (eg, Acaa1a, Acads, Acox1, and Hadha) (Figure 2D), which could reflect the need to dispose of lipids that have, for example, been damaged by CS-linked reactive oxygen species (Morissette et al., 2015).
The proteome alterations observed upon CS exposure are consistent with a major shift in the lipid metabolism of the lung, which likely forms the basis for our observed CS-induced alterations in the lung lipidome. Again, the MRTP-exposed and the cessation and switching groups exhibited much reduced effects, when compared with effects of CS exposure, on these lipid metabolism pathways.
CS Exposure Represents the Main Correlated Trend in the Lung Lipidome and Proteome
The lung lipidome and proteome both showed a broad response to CS exposure with corresponding alterations in lipid classes and lipid metabolism pathways. To further relate the exposure effects on the lung lipidome to those in the proteome, we applied a multivariate sPLS canonical analysis approach (sPLS-can). Conceptually, sPLS-can is related to principal component analyses (PCA). However, rather than identifying the major new data components that best explain the variance of individual datasets, sPLS-can identifies those new components in the data that best correlate between 2 datasets. Note that, for this analysis, only samples shared between the lipidomics and proteomics datasets could be considered, that is only lung samples obtained from the same mouse and analyzed by both procedures. As for PCA, mapping of samples onto the new components is visualized in score plots. Strikingly, for both the C57BL/6 (Figure 3A) and Apoe−/− (Figure 3B) studies the first component, that is the component best correlated between the data modalities, clearly distinguished the exposure groups. Primarily, the CS-exposed mice are clearly separated from the remaining, highly overlapping exposure groups. In addition, in the CS-exposed groups, time-dependent trends with increased scores for the latest time points were apparent.
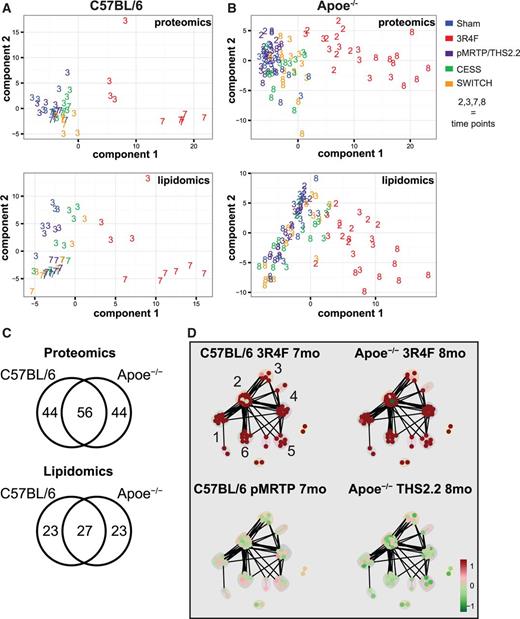
Integration of lipidomics and proteomics data show 1 major shared effect component for C57BL/6 and Apoe−/− mice. A, Lipidomics and proteomics data from the C57BL/7 study were integrated using sparse PLS-can to identify the major shared trends among data types. PLS-can is conceptually similar to PCA, but is designed to identify shared correlated components among datasets. These score plots show the projection of each sample shared between the data modalities onto the first (x-axis) and second (y-axis) PLS-can component for the lipidomics (top) and proteomics (bottom) data. Exposure groups are color-coded and the numbers indicate the time point in months. B, As described in A, but with data from the Apoe−/− study. C, Venn diagrams showing overlap between the 2 studies, with respect to the molecules contributing to the first component. D, Clustered functional association network for proteins positively contributing to component 1 (see Supplementary Data for details). The identified clusters were manually annotated guided by ToppGene (Chen et al., 2009): 1) lipid-related, 2) pentose-phosphate pathway, unfolded-protein response, and oxidative stress response (mixed), 3) ATPase complex, 4) immune-related / lysosomal, 5) surfactant-related, and 6) xenobiotic response.
To gain deeper insights into this major sPLS-can component correlated between the proteomics and lipidomics data, we first evaluated the overlap between the C57BL/6 and the Apoe−/− studies. Interestingly, more than half the contributing proteins and lipids were shared between studies with the 2 strains (Figure 3C), suggesting that this main effect has a common basis. We employed a functional clustering approach to identify the functional protein categories contributing to this separation (Figure 3D and Supplementary Data). The clusters with a positive contribution to the first component, that is, proteins tending toward having higher expression in CS-exposed mice, involved a broad range of functions. These included those involved in lipid-related functions (cluster 1, eg, Fasn, Acsl5, and Abcd3), components of the pentose-phosphate pathway (cluster 2), immune-related proteins (cluster 4), surfactants (cluster 5), and xenobiotic response proteins (cluster 6). Overall, this analysis clearly illustrates the complexity of the major and correlated responses to CS exposure in these data sets. Lipid metabolism and the other functional categories are tightly connected components of the overall exposure response. These connections, as they apply to surfactant proteins, immune-related proteins, and the pentose-phosphate pathway, will be discussed below.
CS Induces Surfactant Lipid–Protein Complexes
Having established that CS demonstrates a global effect on lung lipid and lipid-protein profiles, we next investigated its effects on selected functional categories in more detail. Pulmonary surfactant is an especially important lipid–protein complex for lung homeostasis. It stabilizes the alveoli by reducing surface tension and functions as an innate component of the lung immune-system (Whitsett et al., 2010).
Quantitative proteomics analysis showed that CS exposure had statistically significant effects on several surfactant and surfactant metabolism proteins (Figure 4A). These effects were substantially lower in the MRTP-exposed, cessation, and switching groups. Specifically, Sftpa1 and Sftpd, 2 surfactant proteins involved in immune defense and surfactant homeostasis (Whitsett et al., 2015), were strongly induced by CS exposure. However, Sftpc and Sftpb, the main structural protein components of surfactant (Whitsett et al., 2015), showed slightly less consistent changes with Sftpb down-regulation in the C57BL/6 mice. Dipalmitoyl-phosphatidyl choline (PC 16:0/16:0) is the dominant lipid component of surfactant. Lpcat1, a critical enzyme for synthesis of this surfactant lipid in mice (Bridges et al., 2010; Nakanishi et al., 2006), was significantly upregulated by CS exposure in both mouse strains. In contrast, Prdx6, which acts as a lysosomal-type phospholipase A2 for surfactant lipid, was significantly downregulated (Fisher et al., 2005). In addition, ABCA3, a transporter critically important for surfactant lipid secretion, and Slc34a2, a marker of alveolar type II cells that secrete surfactant (Whitsett et al., 2015), were upregulated by CS exposure in both mouse strains.
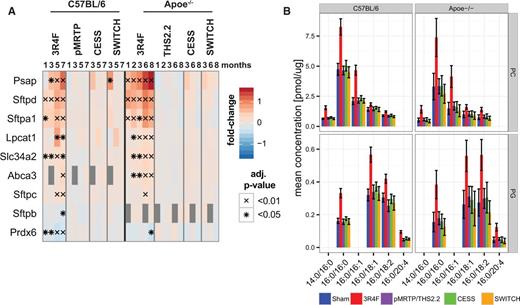
Exposure effects on surfactant metabolism. A, Protein expression profiles of surfactant-related proteins in the lung from the C57BL/6 and Apoe−/− studies. The fold-change is color-coded and statistical significance is marked (BH-adjusted P-value, *< 0.05, x< 0.01). Missing values are gray. B, Comparison of lipid concentration profiles for the surfactant-associated PC and PG lipid classes. The mean and 95% confidence intervals of the measured lipid concentration are shown for the last time point of each study (7 months for the C57BL/6 and 8 months for the Apoe−/− study). Only those lipid species with fatty acid chain compositions contributing at least 5% to the total lipid class concentration of either PC or PG are included. Missing bars indicate missing values, eg, for the C57BL/6 study no values were obtained for PG 16:0/20:4.
These alterations in surfactant metabolism likely contribute to the CS-induced changes in lipidome profiles of surfactant lipids (Figure 4B). Consistent with its dominant effects on surfactant, CS exposure resulted in the highest measured concentrations within the PC class for PC 16:0/16:0 at the latest time points of both studies. A strong CS-induced increase was also observed for PC 16:0/16:1 and PC 16:0/14:0, both known to contribute to the surfactant (Goss et al., 2013). In addition to phosphatidyl-choline (PC), phosphatidyl-glycerine (PG) plays an important role in the surfactant (Postle et al., 2001; Whitsett et al., 2015). Those PG species most significantly changed by CS exposure were PG 16:0/18:1, PG 16:0/18:2 and PG 16:0/16:0. This observation is consistent with previous analyses of mammalian surfactant lipids (Postle et al., 2001).
Overall, this integrative analysis of surfactant changes revealed consistent and aligned surfactant protein and surfactant lipid responses in both C57BL/6 and Apoe−/− mice. CS exposure strongly affected both protein and lipid components of surfactant, while pMRTP and THS2.2 exposure did not induce such changes and the cessation and switching groups rapidly returned to sham levels of these proteins and lipids.
CS Exposure Affects Regulation of the Immune Response Through Eicosanoid and Sphingolipid Metabolism
Eicosanoids and sphingolipids are key signaling mediators regulating the immune response and cellular processes such as differentiation, survival, and apoptosis (Gilroy, 2010; Lee et al., 2015). Several sphingolipids also play structural roles in the cell. We measured eicosanoid lipid response profiles in only the C57BL/7 mouse study, though several enzymes of the AA metabolism pathway were quantified in both studies (Figure 5A). Mapping of the protein and lipid alterations onto the AA metabolism pathway revealed an intriguing pattern (Figure 5B). The metabolic branch leading to prostaglandins D2 and E2 (PGD2, PGE2), including the enzyme Ptgs1 (COX-1), was strongly induced by CS exposure, whereas the branch leading to dihydroxylated-eicosatrienoic acids (8,9-DHET, 11,12-DHET, and 14,15-DHET), including the enzyme Cyp2b10, was downregulated. PGD2 showed particularly high upregulation with CS exposure (Figure 1F). This is in line with Strebbins et al. (2010) who reported that pharmacological blockade of DP2, 1 of the 2 G-protein coupled PGD2 receptors, strongly suppressed the CS-induced inflammatory response in a mouse exposure model. Considering this, our observed increased levels of PGD2 and PDE2 likely contributed to the strong inflammatory response in the lungs of these mice, specifically those exposed to CS (Figure 5C) (Phillips et al., 2015). Downregulation of DHET molecules is likely indicative of downregulation of their more active epoxide precursor lipids, epoxyeicosatrienoic acids (EETs), not measured in our lipidomics experiment because of their instability. Among other functions, EETs have been reported to reduce the lung immune response (Morin et al., 2008). This indicates that suppression of this branch of eicosanoid synthesis might further amplify the CS-induced inflammatory response in the lungs.
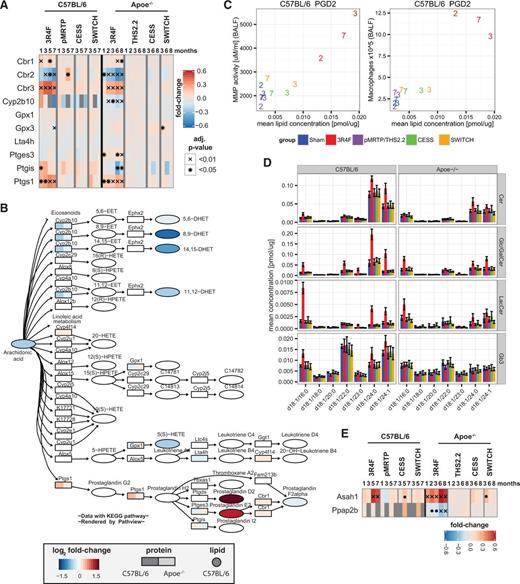
Exposure effects on eicosanoid and sphingomyelin metabolism. A, Expression profiles of quantified proteins of “arachidonic acid metabolism” pathway of the KEGG database. Heatmap representation as in Figure 2C. B, Arachidonic acid metabolism pathway map. For detected enzymes, the maximum absolute fold-change in the 3R4F exposure groups is color-coded for each study (left: C57BL/6, right: Apoe−/−). Eicosanoids were measured only in the Apoe−/− study and the measured fold-changes are color-coded. C, Comparison of measured matrix-metalloproteinase (MMP) activity and the number of macrophages in BALF with measured PGD2 concentrations. The values for the different exposure groups (see color key in panel B) and time points (numbers in months) are compared. D, Comparison of lipid concentration profiles for sphingolipids: ceramides (Cer), Glc/Gal ceramides (Glc/GalCer), Lac ceramides (LacCer), and globotriaosylceramide (GB3). The mean and 95% confidence intervals of the measured lipid concentrations are shown for the last time point from each study (7 months for C57BL/6 and 8 months for Apoe−/− study). Only those lipid species with compositions contributing at least 5% to the total concentration of either lipid class are included. E, As in A, but for expression profiles of Asah1 and Ppap2b.
Several sphingolipids had increased abundance following CS exposure (Figure 1F) and showed a chain length-specific pattern (Figure 5D). That is, long chain (24:0/24:1) and 16:0 fatty acid-containing species were significantly upregulated for the Cer, Glc/GalCer, and LacCer classes, whereas Gb3 demonstrated a more limited response that was less consistent between the 2 studies. It is notable that, while the 24:0 and 24:1 species contributed most to the Cer and Glc/GalCer classes under CS exposure, the profile for LacCer differed, showing the largest contribution from LacCer d18:1/16:0. The increases in levels of these lipids were strongly attenuated in the pMRTP- and THS2.2-exposed groups and these levels returned to those of the sham groups only 1 month after either smoking cessation or switching to a potential MRTP.
Other bioactive sphingolipids such as sphingosine (Sph) and sphingosine -1-phosphate (S1P) play key roles in determining the fate, survival, proliferation, differentiation, and migration of cells. Importantly, the balance between pro-apoptotic Cer and Sph and pro-survival S1P, the “sphingolipid rheostat”, was demonstrated as being vital for lung homeostasis and maintenance of alveolar septal integrity (Lee et al., 2015). Sph and S1P, measured only in the C57BL/6 study, were both specifically and significantly induced by CS exposure (Figure 1F). Sph is generated from Cer through the activity of ceramidase enzymes and its phosphorylation to S1P is subsequently controlled through antagonistic effects of sphingosine kinases and phosphatases. We identified the acid ceramidase Asah1 as a likely candidate enzyme causing the CS-mediated increase in Sph synthesis, finding that it was strongly and consistently upregulated upon CS exposure in both the C57BL/6 and Apoe−/− studies. Interestingly, this enzyme has been implicated in CS-mediated inhibition of apoptotic cell clearance, or efferocytosis, by alveolar macrophages (Petrusca et al., 2010). In addition, Ppap2b, a lipid phosphate phosphatase acting on S1P and other substrates, was strongly downregulated by CS exposure in the Apoe−/− study (Figure 5E), though was not reliably measured in the C57BL/6 mice. It has been proposed that this lipid phosphatase, through its effect on S1P levels, is involved in changing vascular permeability (Ren et al., 2013). This suggests that Asah1 upregulation and Ppap2b downregulation are likely to contribute to the complex response of sphingolipids to CS-exposure in our study, and to be implicated in disease-related effects of CS exposure on the lung, including suppressed apoptotic cell clearance and vascular disturbances.
Although the specific effects of these extensive changes in eicosanoid and sphingolipid metabolism will be difficult to unravel, it is likely that, overall, they contribute to disease development in CS-exposed mice, including the emphysematous effects observed in our studies (Phillips et al., 2015).
CS Exposure Affects Lipid Metabolism Regulators and Induces Associated Metabolic Alterations
Having demonstrated that CS induces extensive alterations in lung lipid metabolism in both mouse strains, we investigated whether a subset of these alterations could be attributed to effects on transcriptional regulators of lipid metabolism. Activity of transcriptional regulators can be deduced though expression of their downstream genes. We applied an established scoring approach to quantitatively deduce activities of transcriptional regulators related to lipid metabolism (Martin et al., 2012) from the transcriptomics datasets of the C57BL/6 and the Apoe−/− studies (Phillips et al., 2015) (Phillips et al., 2015b). Specifically, we employed this approach to evaluate activities of transcriptional regulators of the SREBP, C/EBP, and PPAR families, all implicated in lipid metabolic processes (Jeon and Osborne, 2012; Varga et al., 2011). We also included Ahr and Nfe2l2 (Nrf2), key regulators of the xenobiotic and oxidative stress responses, respectively. We found that, among these, only the activities of Srebf1a/ Srebf1c and Cebpb/Cebpd/Cebpe were predicted to be significantly upregulated in both C57BL/6 and the Apoe−/− mice (Figure 6A). Sterol responsive element binding proteins (SREB) including Srebf1a/c, are central hubs in the regulation of lipid metabolism (Bommer and MacDougald, 2011). Interestingly, Srebf1c and Cebpb were recently linked to a control mechanism of early adipocyte differentiation (Ayala-Sumuano et al., 2011). In lung tissue, SREBP and C/EBP proteins have been implicated in regulation of surfactant-related lipogenesis (Zhang et al., 2004) and as components of a key regulatory module of surfactant lipid homeostasis (Xu et al., 2010). Taken together, these results suggest that upregulation of surfactant synthesis by CS, which appears to represent the largest quantitative effect on lipid biosynthesis identified in our data, is controlled by activation of SREBP and C/EBP transcriptional complexes.
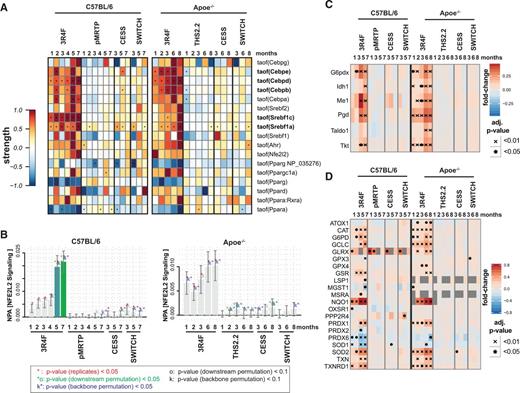
Transcriptional regulation of lipid metabolism alterations and metabolic adaptation toward production of NADPH. A, Transcription regulator activity analysis indicates upregulation of SREBP and C/EBP transcriptional activities with CS exposure. Each row of the heatmap represents the predicted activity of a transcriptional regulator, each column represents an exposure group compared to sham exposure at the same time point. The perturbation strength is color-coded and statistical significance is indicated (*BH-adjusted P-value < .05). B, Quantitative scores of exposure effects on the entire NFE2L2 signaling pathway. The bars show the overall network perturbation amplitude (NPA), the error bars show the 95% confidence interval. Three statistics are included: the red star indicates statistical significance with respect to the biological replicates, the green star (o statistics) indicates significance with respect to permutation of the downstream genes of the network nodes, and the blue star (k statistics) indicates significance with respect to permutation of the NFEL2 network topology. C, Upregulation of proteins involved in NADPH generation by CS exposure. Heat map representation is as described in Figure 2C. D, Proteins implicated in reactive oxygen species response. The human orthologs of differentially expressed proteins of the “Hallmark Reactive Oxygen Species Pathway” are shown (www.broadinstitute.org/gsea/msigdb).
Nfe2l2 (Nrf2) is a key regulator of the oxidative stress response (Hayes and Dinkova-Kostova, 2014). Nfe2l2 transcriptional activity only showed a trend indicating induction by CS exposure in both studies. For a more comprehensive assessment of effects on Nfe2l2 signaling, we employed the network perturbation amplitude approach as previously reported for the Nfe2l2 signaling network (Figure 6B) (Boué et al., 2015; Martin et al., 2014). Using this analysis, CS exposure induced a significant perturbation of Nfe2l2 signaling after 5 and 7 months exposure in C57BL/6 mice. Nfe2l2 signaling was also induced by CS exposure in Apoe−/− mice with the downstream node permutation showing marginal significance (P < .1).
Besides inducing expression of proteins to directly attenuate oxidative stress, Nfe2l2 signaling also reprograms cellular metabolism toward NADPH production (Figure 6C) (Hayes and Dinkova-Kostova, 2014). For example, upregulated proteins include the rate-limiting enzymes of the pentose phosphate (PP) pathway (G6pdx, Pgd) and NADPH-generating malic enzyme (Me1). Increasing reduction power in the cell, in the form of NADPH, by the PP pathway is an essential component of both the cellular response to oxidative stress and of fatty acid biosynthesis. Moreover, SREBP regulators have also been reported to facilitate lipid biosynthesis through upregulation of NADPH generating enzymes including Me1, glucose-6-phosphate dehydrogenase (G6pd), and Pgd (Shimomura et al., 1998).
In further support of a strong oxidative stress response of the lung tissue upon CS exposure, several oxidative stress enzymes were significantly and specifically upregulated by CS exposure in both studies (Figure 6D). These proteins, which as a whole likely mitigate the detrimental effects of CS-induced oxidative damage by their anti-oxidative effects, included NAD(P)H dehydrogenase, quinone 1 (Nqo1), superoxide dismutase 2 (Sod2), thioredoxin reductase 1 (Txnrd1), and glutamate-cysteine ligase (Gclc).
Taken together, the extensive alterations in pulmonary lipid metabolism induced by CS exposure are controlled by a complex regulatory program, which, concomitantly, may also mediate other adaptive and destructive tissue responses. Thus, changes in lipid metabolism are intrinsic to the broad response of the tissue to CS exposure, including oxidative stress and xenobiotic stress responses, immune response, and other metabolic alterations.
DISCUSSION
This integrative study addressed how lung lipid metabolism is affected by exposures to CS or aerosols of heat-not-burn tobacco products. We integrated comprehensive lung lipidomics and proteomics measurements from 2 exposure studies, in C57BL/6 and Apoe−/− mice, respectively (Phillips et al., 2015 and Phillips et al., 2015b). These studies were part of our systems toxicology assessment framework (Sturla et al., 2014) under which we complement the classical toxicological endpoints, such as histopathology and lung function measurements, with extensive molecular measurements, such as transcriptomics, proteomics, and lipidomics, and computational analysis approaches. The goal is to identify causal molecular events linking initial exposure effects with adverse functional and other outcomes. Such detailed analyses enable better understanding of observed toxicological responses, building confidence in risk-assessment. At the same time, by maximizing the number of measurements per animal, this approach is effective under the 3Rs strategy (Flecknell, 2002).
Proteomics technology is rapidly evolving with growing evidence that analysis of proteome alterations can help unravel mechanisms of toxicity, including smoke-induced effects (Titz et al., 2014). Similarly, improved mass-spectrometry based assays have enabled rapid progress in lipidomics (Blanksby and Mitchell, 2010). Lipid alterations have been increasingly implicated in toxicological mechanisms and improved lipidomics methods have been already demonstrated to support the elucidation of complex toxicological responses (Fernando et al., 2011;Soriano et al., 2005). Previously, applying lipidomics to characterize effects of CS, we reported that, in Apoe−/− mice, smoking cessation can halt accumulation of proatherogenic lipids in the vessel wall (Boué et al., 2012; Lietz et al., 2013) and Telenga et al. (2014) investigated the sputum lipidome in cigarette smokers, COPD patients, and subjects after smoking cessation.
In the present report, we integrated lung lipidome and proteome profiles to comprehensively assess the complex effects of CS exposure on lipid-related processes. Previously, Wilmes et al. (2013) have, for example, demonstrated the benefits of integrating proteomics, transcriptomics, metabolomics, and pharmacokinetics to elucidate mechanisms of drug-induced cell stress. Our integrative analysis revealed a complex and consistent response of the lung lipidome and lipid-related proteome to CS exposure in the 2 mouse strains, C57BL/6 and Apoe−/− (Figure 7). Apoe−/− mice are more sensitive than C57BL/6 mice to the effects of CS exposure (Boue et al., 2013). With this, the consistency of the response between the two strains was likely supported by the correspondingly adjusted nicotine concentrations and daily exposure durations of 34.4 mg/m3 for 4 hours (C57BL/6) and 29.9 mg/m3 for 3 hours (Apoe−/−) that would correspond to a roughly estimated daily human consumption of approximately 49 and 32 cigarettes, respectively (FDA Guidance for Industry, UCM078932, http://www.fda.gov/cder/guidance/index.htm).

Working model—interplay among lipids and proteins in CS-induced lung damage. Schematic summary of some relevant relationships among molecular entities and biological functions relevant to lung disease. The analyzed lipids (green text) and related proteins (yellow text) are emphasized along with their putative roles in CS-induced lung pathobiology.
Importantly, CS-induced effects on lipid metabolism were part of a broader exposure response in the lung, including xenobiotic and oxidative stress responses and other metabolic adaptations. Among these lipid-related effects, we studied those on surfactant, eicosanoid, and sphingolipid metabolism in more detail.
In our study, CS exposure led to concerted increase in surfactant lipids and proteins in both mouse strains (Figure 4). The upregulated surfactant proteins included major structural components of the surfactant (Sftpc), surfactant proteins involved in pulmonary immune defenses and surfactant homeostasis (Sftpa and Sftpd), and proteins involved in surfactant metabolism (eg, Lpcat1 and Abca3) (Whitsett et al., 2015). Surfactant alterations were reported in several lung diseases including COPD and cigarette smoking directly affects surfactant levels (Mazur et al., 2011;Winkler et al., 2011). Recently, Morissette et al. (2015) presented an intriguing model how oxidative damage of surfactant lipids that are taken up by alveolar macrophages can contribute to the induction of an inflammatory response. Consistent with this model, we have observed a strongly upregulated oxidative stress response and observed the induction of general lipid oxidation markers for CS exposure (4-HNE and MDA) (Phillips et al., 2015b). With this, increased surfactant synthesis is likely required to compensate for damaged surfactant that is taken up by macrophages in the CS-exposed mice.
The CS response included prominent changes in tissue levels of eicosanoids, which are important in pathophysiological processes such as development of COPD (Lundstrom et al., 2011). The metabolic branch, including the enzyme Cyp2b10, leading from AA, via EETs, to dihydroxylated-eicosatrienoic acids (DHETs) was downregulated. Conversely, the branch leading to prostaglandin D2 and E2 (PGD2, PGE2) including the key enzyme Ptgs1 (COX-1), was strongly induced (Figure 5). For example, PGE2 can stimulate inflammation and induce lung fibroblast senescence, effects associated with COPD (Dagouassat et al., 2013). Pharmacological blockade of the PGD2 G protein-coupled receptor DP2 inhibited smoke-induced inflammation, mucus cell metaplasia and epithelial hyperplasia in mouse lung (Stebbins et al., 2010). EETs, in contrast, reduce the lung immune response (Morin et al., 2008). Taken together, the opposing effects of CS on PGD2/PGE2 and EETs/DHETs synthesis may amplify lung inflammation and associated development of emphysema in the CS-exposed mice, which was observed in the same studies (Phillips et al., 2015) (Phillips et al., 2015b).
Several ceramide lipids and related sphingolipids, including sphingosine (Sph) and sphingosine-1-phosphate (S1P), were more abundant in CS-exposed mice (Figs. 1E and 5D). Inflammatory cell recruitment is an early event leading to emphysema (Sharma et al., 2012) and involves ceramides. Ceramides have also been implicated as crucial mediators of alveolar destruction. Inhibition of de novo ceramide synthesis prevented alveolar cell apoptosis and oxidative stress in a rodent model of emphysema (Petrache et al., 2005). In contrast to the pro-apoptotic effects of Cer, S1P suppresses apoptosis (Maceyka et al., 2002) and is regarded as vital for lung homeostasis and maintenance of alveolar septal integrity. Based on such reports, imbalance of these regulatory systems in CS-exposed mice might further amplify lung inflammation and emphysema development.
Overall, CS exposure resulted in extensive, consistent lipid metabolism changes, potentially associated with adverse effects on the lung, including the observed emphysematous changes and increased inflammation in the lungs of these mice (Figure 7) (Phillips et al., 2015 and Phillips et al., 2015b). Certainly, a key goal of our study was to address whether these lipid-related changes are affected by smoking cessation or by potential MRTPs. The two MRTPs tested here, pMRTP and THS2.2, are both based on the same principle: Tobacco is heated in a controlled fashion rather than burned, producing an aerosol containing nicotine, glycerin, water, and volatiles contributing to the specific tobacco flavor but generating much lower levels of HPHCs (Supplementary Data). However, there were a few exceptions, most notably arsenic and PAHs that were only reduced in THS2.2, but not in pMRTP aerosol. This further reduction of the released HPHCs in THS2.2 is likely explained by the more refined design and tighter temperature control of this product compared with the pMRTP.
In this investigation, pMRTP or THS2.2 aerosol exposures induced only limited effects on the lung lipid metabolism. For example, the assessed MRTPs did not change levels of surfactant lipids and surfactant-related proteins (Figure 4) nor induce the PGD2 and PGE2 branch of the eicosanoid pathway (Figs. 5A and 1F). Apoe−/− mice exposed to THS2.2 for 3 months showed some downregulation of several sphingolipids, a response opposite to that with 3R4F exposure and not consistently observed. In the cessation and switching groups effects of CS exposure appeared rapidly reversed, mainly within 1 month. Finally, further supporting these trends, a multivariate canonical PLS analysis revealed a major correlated effect in the datasets of both studies clearly separating the 3R4F-exposed groups from the remaining, highly overlapping, exposure groups (Figure 3).
Undoubtedly, CS has been associated with a multitude of adverse biological effects that go far beyond the ones assessed in this study, ie, the effects on the lung lipid metabolism in these mouse models. Therefore, this study can only represent a cornerstone within our much more comprehensive assessment framework toward the overall evaluation of the risk reduction potential of MRTPs. For example, we have already leveraged the Apoe−/− mouse study to concomitantly evaluate the reduction potential of THS2.2 aerosol for cardiovascular endpoints (Phillips et al., 2015b). In another example, to evaluate a relevant mechanism of the CS-mediated cardiovascular effects (ie, atherosclerosis), we have compared the effects of THS2.2 and 3R4F on chemotaxis and transendothelial migration of a monocytic cell line (van der Toorn et al., 2015).
CONCLUSION
CS, with over 5000 constituents (Talhout et al., 2011), triggers a complex biological response in lung tissue. Several biological stress-responses maintain lung homeostasis but, when overwhelmed, permanent damage such as emphysema and chronic bronchitis, hallmarks of COPD, can result. Initiation and progression of these diseases involve an intricate interplay among different classes of biological molecules, such as the cooperation of lipids and proteins to regulate inflammation. To explore how CS exposure affects this complex biology, we performed an integrative systems toxicology analysis of alterations in levels of lung lipids and lipid-metabolism related proteins in the lungs of CS-exposed mice. In 2 mouse strains, we found an extensive, highly consistent response of the lung lipidome and lipid-related proteome to CS exposure. These lipid-related alterations are intrinsic to the overall response “program” of the lungs and we found several direct links between the protein and lipid alterations induced by CS exposure, for example, in surfactant and eicosanoid metabolism. Importantly, we also evaluated effects of smoking cessation, exposure to potential MRTPs (THS2.2 and pMRTP), and switching from CS to MRTP exposure. Exposure to THS2.2 or pMRTP showed little to no effects on the lipid-related processes. Both the cessation and switching groups showed rapidly normalized lipid metabolism related profiles with respect to sham-exposed mice. Therefore, our systems toxicology analysis supports an integrated product risk assessment of MRTPs, including the biological benefits of cessation or switching, when compared with continued smoking. Moreover, this work could form the foundation for the identification of translational molecular markers for human risk assessment in the future.
SUPPLEMENTARY DATA
Supplementary data are available online at Supplementary Data.
ACKNOWLEDGMENTS
The authors would like to thank the PMI R&D COPD II program team, especially acknowledging the technical assistance of Didier Goedertier, Elena Seow, Teo Hutt Wang, Woon Ching Qing, Clement Foong, Eric Yew, and Yeo Chor Kiat and their teams. We thank Hector de Leon for his expertise and guidance on this project. This study was funded by Philip Morris International, and all authors except for T.V., K.E., and R.L. are employees of Philip Morris International. T.V., K.E., and R.L. are employees of Zora Biosciences. An international patent application relating to the use of lipid biomarkers for the diagnosis of lung injury was filed (WO 2015/085225 A1).
REFERENCES
Author notes
1S.B. and B.T. contributed equally to this work.
2Present address: Novartis Pharma AG, Novartis. Institutes for Biomedical Research (NIBR), 4002, Basel, Switzerland.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments