





Abstract
A fundamental aspect of symbiotic relationships is host specificity, ranging from extreme specialists associated with only a single host species to generalists associated with many different species. Although symbionts with limited dispersal capabilities are expected to be host specialists, some are able to associate with multiple hosts. Understanding the micro- and macro-evolutionary causes of variations in host specificity is often hindered by sampling biases and the limited power of traditional evolutionary markers. Here, we studied feather mites to address the barriers associated with estimates of host specificity for dispersal-limited symbionts. We sampled feather mites (Proctophyllodidae) from a nearly comprehensive set of North American breeding warblers (Parulidae) to study mite phylogenetic relationships and host–symbiont codiversification. We used pooled-sequencing (Pool-Seq) and short-read Illumina technology to interpret results derived from a traditional barcoding gene (cytochrome c oxidase subunit 1) versus 11 protein-coding mitochondrial genes using concatenated and multispecies coalescent approaches. Despite the statistically significant congruence between mite and host phylogenies, mite–host specificity varies widely, and host switching is common regardless of the genetic marker resolution (i.e., barcode vs. multilocus). However, the multilocus approach was more effective than the single barcode in detecting the presence of a heterogeneous Pool-Seq sample. These results suggest that presumed symbiont dispersal capabilities are not always strong indicators of host specificity or of historical host–symbiont coevolutionary events. A comprehensive sampling at fine phylogenetic scales may help to better elucidate the microevolutionary filters that impact macroevolutionary processes regulating symbioses, particularly for dispersal-limited symbionts. [Codiversification; cophylogenetics; feather mites; host switching; pooled sequencing; species delineation; symbiosis, warblers.]
Symbionts are organisms that form close, persistent associations with hosts and comprise the majority of biodiversity on Earth (Larsen et al. 2017; Okamura et al. 2018; Carlson et al. 2020). One fundamental feature of symbiotic relationships is host specificity, which in the simplest sense is a measure of the breadth of associated taxa with a symbiont (Lymbery 1989). Symbionts can range from host specialists that are associated with only one single host species to broad host generalists that are associated with many host species (Futuyma and Moreno 1988; Poulin et al. 2011). Variation in host specificity is typically related to the degree that a specific symbiont can disperse, survive, reproduce, and maintain a population on different hosts (Agosta et al. 2010; Araujo et al. 2015; Mestre et al. 2020). In parasitology, and symbioses more generally, these are known as encounter and compatibility filters (Combes 2001); the encounter filter is related to the potential of a parasite (symbiont) to disperse to a new host, whereas the compatibility filter is related to parasite survival on that new host (Timms and Read 1999). Understanding the mechanisms that regulate these filters requires accurate estimates of host specificity.
Host specificity is presumably straightforward to estimate for symbionts with limited dispersal abilities because these symbionts are thought to be excluded from reaching novel hosts during the initial encounter filter. Thus, dispersal-limited symbionts are expected to be highly host-specific (i.e., specialists; Poulin and Keeney 2008; Hayward et al. 2021). Accordingly, high host specificity has been observed in dispersal-limited symbionts such as pocket gopher lice (Psocodea: Trichodectidae; Hafner et al. 1994; Reed and Hafner 1997) and in mutualistic fungi grown by attine ants (Basidiomycota: Agaricaceae; Beigel et al. 2021). However, when dispersal barriers are removed because the encounter filter is eliminated, the high degree of host specificity can collapse, as has been demonstrated experimentally in dispersal-limited bat bugs (Hemiptera: Polyctenidae; Dick et al. 2009) and ectomycorrhizal fungi (Basidiomycota: Suillaceae; Pérez-Pazos et al. 2021). Relaxed host specificity despite symbiont dispersal limitations has also been observed naturally in the fungus–termite mutualism (van de Peppel and Aanen 2020) and in avian quill mites (Acariformes: Syringophilidae; Hromada et al. 2016). However, estimates of host specificity for many symbionts (including seemingly specialist and dispersal-limited symbionts) are biased due to incomplete sampling efforts within and across hosts, presence-only host–symbiont association records, and methodological limitations in detecting cryptic species (Wells and Clark 2019; Ellis et al. 2020). These same obstacles can consequently hinder our understanding of macroevolutionary processes such as cospeciation and host switching. Thus, it is necessary to establish complete and reliable patterns of host–symbiont associations (i.e., host specificity) in order to more thoroughly assess the causes and consequences of coevolutionary processes.
Given this context, vane-dwelling avian feather mites (Acari: Acariformes: Astigmata: Analgoidea, Pterolichoidea; hereinafter “feather mites” or “mites”) represent an ideal system to address the weaknesses in our understanding of host specificity and host–symbiont codiversification. These mites are a large group of symbionts highly adapted for their obligate, permanent life on feathers and, as such, are presumed to have limited dispersal abilities. For example, feather mites have specialized morphological features such as pretarsi ambulacral discs (“feet”), complex leg spines and spurs, and modified body setae (“hairs”) that allow them to remain tightly secure on and move amongst feathers (Dabert and Mironov 1999; Proctor 2003). Their diet consists of host-specific waxy uropygial secretions (Proctor 2003; Soini et al. 2013) and microbes (Doña et al. 2019b) found on feathers that they eat at night when it is presumably safer to move across feathers (Labrador et al. 2022). They are primarily transmitted vertically from parent to offspring, most often during the brooding and nestling period in which parents are in direct contact with offspring (Doña et al. 2017a; Peet et al. 2022). Furthermore, they are nonphoretic, so they do not “hitchhike” on other species for transport/dispersal to new hosts (Jovani et al. 2001). As such, feather mites are dispersal-limited and often exhibit a high degree of host specificity toward one or a few closely related hosts (Doña et al. 2018a). However, evidence for clade-limited host switching in this group has been accumulating (Doña et al. 2017b; Matthews et al. 2018a; Dabert et al. 2022), and host specificity does indeed vary among feather mites (Stefan et al. 2018; Doña et al. 2019a; Mironov and Galloway 2021). Some feather mite species are highly host specific (specialized to a single host species), whereas others are host-general (i.e., multi host or polyxenous symbionts). Thus, despite their dispersal limitations and encounter filter barriers, some feather mite species are able to reach and persist across multiple host species. Nevertheless, the true host specificity of nearly all feather mites is currently uncertain because of two main limitations—an incomplete sampling of host species and the limited power of traditional evolutionary markers—which we address in this study.
First, incomplete sampling (e.g., taxonomically, geographically, and temporally) of host species is known to cause biases and uncertain estimates of host specificity across many symbiotic systems (Novotny et al. 2002; Bell et al. 2015; Ruiz-Aravena et al. 2022). Sampling hosts at broad taxonomic scales poses the risk of underestimating symbiont host specificity and overlooking signatures of (co)evolutionary processes that occur at lower taxonomic levels, as has been documented in the fig–fig wasp mutualism (Machado et al. 2005) and avian lice parasitism (Sweet et al. 2016). For feather mites, previous studies have broadly sampled representative host species across a wide range of passerine families (Doña et al. 2017b; Klimov et al. 2017; Dabert et al. 2022). For example, Klimov et al. (2017) sampled mites across 140 host species of 100 genera from 56 families, and Dabert et al. (2022) sampled mites across 47 host species of 33 genera from 16 families. Although this is a practical sampling strategy to study mite evolution at a broad scale, it neither thoroughly represents the range of hosts with which feather mites are associated (e.g., “stragglers”; Doña et al. 2018b) nor accurately captures signatures of evolution occurring at a finer taxonomic scale (e.g., cryptic divergence).
The second reason for uncertain estimates of host specificity is that research on feather mite systematics, evolution, and biodiversity has mostly been based on mite morphology (e.g., Constantinescu et al. 2021; Mironov et al. 2021; Hernandes 2022) or a few barcoding markers (Matthews et al. 2018a; Stefan et al. 2018; Doña et al. 2019a; Pedroso et al. 2021; Dabert et al. 2022). Most commonly, the cytochrome c oxidase subunit 1 (cox1) mitochondrial gene has been used. Cox1 is an effective barcode for feather mite species delineation (Doña et al. 2015a) and can be useful for high-throughput biodiversity surveys (Vizcaíno et al. 2018). However, it potentially has limited functionality to detect cryptic variation between closely related taxa or species undergoing incipient speciation, including symbionts like avian malaria parasites (Apicomplexa: Haemosporida; Galen et al. 2018) and mutualistic attine ants (Hymenoptera: Formicidae; Beigel et al. 2021). Thus, cox1 may misinform estimates of host specificity and biodiversity. Multilocus genetic studies on feather mites are rarer (Doña et al. 2017b; Klimov et al. 2017) and have been constrained for these small-bodied organisms due to the minimal yield of high-quality DNA usable for multilocus sequencing. An alternative solution is to conduct pooled sequencing (Pool-Seq). With Pool-Seq, DNA from multiple individuals (e.g., multiple mites from a single host feather) is pooled together, which can increase DNA concentrations and the likelihood of successful sequencing in a cost-effective and time-efficient manner. This strategy is similar to metabarcoding in that multiple individuals from a bulk extract can be sequenced together, as has previously been conducted with feather mites (Vizcaíno et al. 2018) and other invertebrates (Hajibabaei et al. 2011; Bittleston et al. 2016; Watts et al. 2019). However, metabarcoding is generally focused on a single locus and mistagging (or “index hopping”) events can cause errors in sample assignment (Carlsen et al. 2012; Schnell et al. 2015). Alternatively, Pool-Seq can be a highly efficient strategy for shotgun genomic (e.g., multilocus) sequencing, and studies of small-bodied organisms such as soil mites (Acari: Acariformes and Parasitiformes; Arribas et al. 2020) have successfully applied this technique. Although shotgun sequencing of pooled samples carries a risk for chimeric sequence assemblies if heterospecifics are present, it helps to alleviate library preparation and sequencing issues related to low DNA concentrations. Overall, the combination of incomplete sampling, limited genetic markers, and sequencing obstacles may lead to significant underestimates of feather mite biodiversity and host specificity (Gaud and Atyeo 1996; Mironov 2003; Doña et al. 2016). Ultimately, these barriers restrict our ability to untangle the factors contributing to the variation in host specificity in these dispersal-limited symbionts.
In this study, we focus on the feather mites of New World warblers in the family Parulidae (“wood-warblers”). Previous molecular (cox1; Matthews et al. 2018a) and morphological (Mironov and Chandler 2017, 2020; Hernandes et al. 2018; Mironov and Galloway 2021) efforts have shown that parulid feather mites, despite their limited dispersal abilities, exhibit unexpected degrees of host specificity, from highly host-specific to very general. However, our current understanding of parulid mite host specificity could be influenced in either direction if the cryptic variation or additional host associations have gone undetected due to indistinguishable mite morphology, limited genetic resolution, or incomplete sampling of hosts. The relatively recent and rapid diversification of the parulid warblers (Lovette and Bermingham 1999; Rabosky and Lovette 2008; Lovette et al. 2010) and the close evolutionary relationships between the mites (Matthews et al. 2018a) make this system useful to address the sampling and genetic limitations of previous studies. Moreover, our system allows us to more accurately identify variation in host specificity, explore diversification at lower taxonomic levels, and elucidate fine-scale signatures of historical (co)evolutionary processes between dispersal-limited symbionts and their hosts.
Here, we use Pool-Seq technology to compare a single traditional barcoding gene (cox1) to a multilocus data set of protein-coding mitochondrial genes to understand how taxonomic or evolutionary inferences might change depending on the composition of genetic markers used. Our specific aims are to: (i) estimate a phylogeny from feather mites found on nearly all species of parulids that breed in North America; (ii) delineate feather mite lineages based on molecular and morphological data to inform taxonomy and host specificity; and (iii) test for signals of phylogenetic congruence between feather mites and their parulid hosts. Our novel approach and comprehensive results will help clarify patterns of diversification and host specificity of avian feather mites more broadly and are also useful for studies of other small-bodied and/or dispersal-limited symbionts.
Materials and Methods
Field Collections
We captured live birds and examined both live and dead birds for feather mites from 92% of parulid species that breed in the United States and Canada (45/49 species across 13 genera and two hybrids) from 2014 to 2021. For each potential host species, we examined at least two individuals (range: 2–306; average per host species: 33 individuals; median per host species: 16 individuals; Supplementary Table S1) in an attempt to identify the presence of feather mites. To obtain mites from live birds, we captured birds (n = 1376) during the migratory or breeding seasons using mist nets. We either used passive netting or lured adults into nets using playback of recorded vocalizations such as alarm calls or territorial songs. Once captured, we closely inspected hosts for feather mites across their primary, secondary, and rectrix feather tracts. Mites were collected from each individual by removing one feather from each of the three feather tracts where mites were present and placing each feather in an individually labeled glassine envelope to prevent cross-contamination. Birds were then safely released. Mites from dead birds were typically acquired from carcasses that had been collected from window-collision surveys (n = 136); in these cases, mites were collected from all feathers. Birds were captured and handled under the United States Geological Survey Bird Banding Laboratory permit #23877 and under the auspices of an IACUC protocol from Arkansas State University #638636.
In total, mites were found on at least one individual for all but seven species of parulids (Cardellina rubrifrons, Leiothlypis luciae, L. virginiae, Myioborus pictus, Setophaga graciae, S. nigrescens, and S. occidentalis). Of the 38 host species with mites, we found that 31 species (82%) harbored Pterodectinae (Analgoidea: Proctophyllodidae) mites of the genera Amerodectes and Tyrannidectes (Supplementary Table S1). These two genera cannot be distinguished morphologically without compound microscopy (Mironov and Galloway 2021) and the arrangement of their close phylogenetic relationships is still uncertain. Likewise, we found 31 species (82%) which harbored Proctophyllodes (Analgoidea: Proctophyllodidae: Proctophyllodinae), whereas only 28 (74%) harbored Trouessartia (Analgoidea: Trouessartidae; Supplementary Table S1). We focused the present study on Amerodectes and Tyrannidectes as they were one of the most prevalent groups from our sampling. We used Proctophyllodes and Trouessartia as close and distant outgroups, respectively.
Mites were later isolated from individual feathers using fine forceps under a stereomicroscope, identified to genus or subfamily (e.g., in the case of Amerodectes vs. Tyrannidectes), and preserved in vials of 95% ethyl alcohol at −20 °C until DNA extraction. Field collection data and associated host data for each sample used in this study are listed in Supplementary Table S2.
DNA Extraction
We extracted mite DNA from 30 out of the 31 host species that harbored Pterodectinae mites (i.e., genera Amerodectes and Tyrannidectes–ingroup taxa). We excluded Setophaga palmarum as we only collected a single individual Pterodectinae mite from this host species. We included two Setophaga virens samples: one individual of the nominate S. virens race and one individual of S. v. waynei, a disjunct and distinct subspecies (Carpenter et al. 2022). Each DNA extract pool consisted of male and female mites from a single feather collected from the 30 Pterodectinae-harboring host species. Because Pterodectinae mites are morphologically very similar under a standard dissecting microscope, it is possible to pool closely related, heterospecific (or heterogeneric in the case of Amerodectes and Tyrannidectes) individuals together (i.e., those living in synhospitality) for extraction. However, the risk of synhospitality on a single feather is low, as synhospitality is most common with heterospecific mites inhabiting different microhabitats on a host or a feather (Mestre et al. 2011) or heterospecific mites inhabiting conspecific hosts that are allopatric/geographically separated (Bochkov and Mironov 2008). We prioritized samples that had at least 50 mites to ensure enough yield for Pool-Seq. Some host species have naturally low mite abundances (Matthews et al. 2018b); from these host species, we chose the feather sample that had the most mites and extracted DNA from as many individual mites that were present. We extracted Proctophyllodes and Trouessartia DNA from an individual bird that harbored both outgroup genera on a single feather (host: Setophaga petechia). The number of mites in each extraction pool (mean number of mites ± SE = 45.7 ± 5.2; range = 2–160 mites) is listed in Supplementary Table S2.
DNA was isolated from pools of mites using the QIAamp DNA Micro Kit (Qiagen) following the manufacturer’s protocol for tissue samples with several modifications: (i) whereas in 180 µl of buffer ATL, we used a small sterile pestle to crush mites; (ii) proteinase K was added and mixed by pipetting; (iii) samples were incubated (lysed) for 24 h and gently vortexed by pipetting every 3 h; (iv) after adding buffer AL, samples were inverted instead of pulse-vortexed and were placed on a heat block (70 °C) for 5 min; (v) no carrier RNA was added; (vi) chilled ethanol was used; (vii) buffer AE (35 µl) was heated to 70 °C before eluting DNA bound to the membrane; (viii) eluted DNA was incubated at room temperature for 5 min; and (ix) the final centrifugation step was extended to 2 min at 14,000 rpm. After extraction, 4 µl of DNA was used for quantification using a Qubit 4.0 Fluorometer (Invitrogen, Carlsbad, CA, USA) using the manufacturer’s recommended protocols and reagents. The remaining DNA was then stored at −20 °C prior to library preparation. Mite exoskeletons were recovered from the elution membranes, stored in 70% ethanol, and mounted on slides in PVA as primary vouchers after DNA extraction. An average of nine mites per extraction pool were intact enough for individual slide-mounting. Additional voucher mites were slide-mounted from the original collection vials (i.e., those that were not subjected to DNA extraction) if available. Slide-mounted feather mite vouchers are deposited in the Arkansas Center for Biodiversity Collections at Arkansas State University (ASUMZ) in Jonesboro, AR, USA and in the Department of Ecology and Zoology at the Universidade Federal de Santa Catarina (ECZ-UFSC), in Florianópolis, Santa Catarina, Brazil.
Library Preparation and Sequencing
Construction of all but one (see below) of the whole-genome shotgun genomic libraries was carried out at the Roy J. Carver Biotechnology Center at the University of Illinois at Urbana-Champaign (UIUC) and sequenced on an Illumina NovaSeq 6000 instrument. The dual-indexed DNA libraries were constructed from up to 10 ng of DNA after sonication with a Covaris ME220 (Covaris, MA) to an average fragment size of 400bp with either the UltraLow Input DNA Library Construction kit (Tecan, CA) or the Hyper Library construction kit from Kapa Biosystems (Roche), depending on starting input. Libraries were amplified with 8 cycles of PCR and run on a Fragment Analyzer (Agilent, CA) to confirm the absence of free primers and primer dimers and to confirm the presence of DNA of the expected size range. Libraries were pooled in equimolar concentration and further quantitated by qPCR on a BioRad CFX Connect Real-Time System (Bio-Rad Laboratories, Inc. CA). The pooled barcoded shotgun libraries were loaded on a NovaSeq 6000 SP lane for cluster generation and paired-end sequencing for 151 cycles. Base calling and demultiplexing were performed with the bcl2fastq v2.20 Conversion Software (Illumina, San Diego, CA). One library (MOWA611R) was constructed after sonication with a QSonica to an average fragment size of 400 bp using New England Biolab’s NEBNext® Ultra™ II DNA Library Prep kit. This library was sequenced at Novogene on an Illumina HiSeq 2500 sequencer generating 150 bp paired-end reads. Raw reads are deposited in the NCBI Short Read Archive (SRA) database (Supplementary Table S2).
Sequence Quality Control
Short reads were trimmed to remove Illumina sequencing adapters, and poor quality bases and sequences using Cutadapt version 3.0 (Martin 2011) using a Phred score quality threshold of 30. The resulting reads were then analyzed using FastQC version 0.11.5 (Babraham Bioinformatics). We further trimmed the 3′ end of resulting reads to 140 bases using BBDuk (https://sourceforge.net/projects/bbmap/) to remove-poor quality bases. One library (MOWA611R) was sequenced at higher coverage, so 35% of its reads were randomly sampled to match the average coverage of the other samples using BBDuk (reformat.sh). We conducted read deduplication using BBDuk (dedupe.sh) for three libraries (MOWA611R, CAWA366A, and BPWA337C) because the number of duplicated reads exceeded 10% for these three libraries. We then reanalyzed the cleaned reads using FastQC to check for any remaining errors.
Mitochondrial Gene Assembly
Quality-trimmed fastq files were converted to “localized” Basic Local Alignment Search Tool (BLAST; Altschul et al. 1990) libraries using the automated Target Restricted Assembly Method (aTRAM; Allen et al. 2018). We targeted 13 mitochondrial protein-coding genes for assembly using aTRAM version 2.3.4. Specifically, we used translated amino acid sequences from Proctophyllodes miliariae (family Proctophyllodidae) as reference loci for aTRAM, which was the most closely related species to our collected mites with all 13 mitochondrial genes present in a reference data set (Doña et al. 2017b). Although a published feather mite reference mitogenome exists (Esteban et al. 2018), it is in a different family (Trouessartidae: Trouessartia rubecula) than our ingroup. We ran five aTRAM iterations and used SPAdes (Bankevich et al. 2012) for de novo assembly. We ran an additional postprocessing script within aTRAM that uses Exonerate version 2.2.0 (Slater and Birney 2005) to identify and stitch together exons for each of the genes assembled, as well as measure assembly completeness by each gene. Of the 13 mitochondrial genes, aTRAM assembled 11 with over 95% completeness. aTRAM was not able to assemble atp8 or nad4l, so these genes were excluded from analyses.
Phylogenetic Analyses
Coding sequences were aligned separately with MAFFT version 7.481 (Katoh et al. 2002) using the default parameters (—auto, which implemented an “L-INS-i” accurate alignment strategy). Alignments were checked manually and were translated to amino acids using the invertebrate mitochondrial genetic code (5) to check for the presence of stop codons. We rooted trees on two species of Proctophyllodes: P. miliariae collected from Miliaria calandra (see Doña et al. 2017b), and Proctophyllodes quadratus collected from S. petechia (this study), as well as an undescribed Trouessartia sp., also collected from S. petechia (this study).
For the cox1-only phylogeny, we ran the cox1 alignment (1506bp) in IQ-Tree version 1.6.12 (Nguyen et al. 2015) to estimate a maximum likelihood (ML) phylogeny and PhyloBayes-MPI version 1.9 (Lartillot et al. 2013) to estimate a Bayesian inference (BI) phylogeny. For the ML analyses, we used ModelFinder (Kalyaanamoorthy et al. 2017) bundled in IQ-Tree to test for the appropriate nucleotide substitution model and used the corrected Akaike Information Criterion (AICc) to determine the best-fitting model. We used 1000 ultrafast bootstrap replicates as measures of branch support. For the BI analysis, we ran two independent Markov chain Monte Carlo (MCMC) chains in parallel for a minimum of 30,000 generations under a GTR-CAT model. We analyzed chain convergence and stationarity using bpcomp and tracecomp. A 50% majority-rule consensus tree was obtained using bpcomp by combining trees from both MCMC chains and discarding the first 5000 generations as burn-in.
We used a concatenation-based approach for our phylogenetic estimation of the alignment that included all 11 mitochondrial genes (9150bp). Mitochondrial genes were concatenated using FASconCAT (Kück and Meusemann 2010), and then IQ-Tree was used to estimate an ML phylogeny. We used ModelFinder to test for the appropriate nucleotide substitution models given the different gene partitions of our multilocus concatenated data set. We used AICc to determine the best-fitting models. We used 1000 ultrafast bootstrap replicates as measures of branch support. We followed the same BI methods for the multilocus data set as we did for the cox1-only data set described above.
Our Pool-Seq approach affords us a novel, suitable, and unique situation with which to apply the multispecies coalescent (MSC) model as an auxiliary analysis of our data for two reasons. First, evidence from an unpublished feather mite Pool-Seq study indicates that there is high population genetic diversity on an individual feather. Second, data from the mitochondrial barcoding gene (cox1) supports a positive correlation between genetic diversity and feather mite abundance (Doña et al. 2015b). Although the use of organelle data with MSC models remains contentious (Doyle 2022), these two reasons suggest that coalescent-based approaches may help to capture species relationships from a pooled sample with high genetic diversity by accounting for potential gene tree heterogeneity. Furthermore, the MSC model has been shown to reliably resolve lepidopteran (Kim et al. 2020) and coccinellid (Li et al. 2021) relationships using mitogenomic data. Thus, for the coalescent-based analyses, we estimated ML phylogenies for each gene separately using IQ-Tree, which implemented the best model of nucleotide substitution for each gene and 1000 ultrafast bootstrap replicates. We then summarized the best gene trees with ASTRAL version 5.7.7 (Zhang et al. 2018), which has been shown as an appropriate method to summarize small numbers of both mitochondrial and nuclear genes (Hundsdoerfer and Kitching 2020; Kergoat et al. 2021; Lebedev et al. 2022). We used the local posterior probability (Sayyari and Mirarab 2016) as measure of branch support.
Species Delineation and Morphological Analyses
Most of the specimens we used for these analyses were not able to be identified as species prior to sequencing (i.e., by morphology or well-known host associations), and some are likely new species. Thus, we used multiple approaches to delimit feather mite lineages as species (if others in the clade are already described) or operational taxonomic units (OTUs) for putatively new species. We first used the Automatic Barcode Gap Discovery (ABGD) method (Puillandre et al. 2012) with the cox1-only alignment as input. ABGD was run using Jukes–Cantor, Kimura-2P, and simple raw distance models with the default minimum (0.001) and maximum (0.1) a priori intraspecific divergence values. We also calculated pairwise genetic distances using these same three distance models using dist.dna() implemented in the R package “ape” version 5.5 (Paradis and Schliep 2019). A previous study of feather mites suggests a conservative and reliable 3.4% (K2P) threshold value for barcoding genes (Doña et al. 2015a). We used the best partitioned multilocus tree from IQ-Tree as input into the online version of bPTP (Zhang et al. 2013). For bPTP, we removed outgroup taxa and ran MCMC for 100,000 generations with thinning set to 100 and a 10% burn-in, which was confirmed as an appropriate burn-in value based on model convergence.
Specimens were subjected to morphological examination to evaluate species delineations. We slide-mounted specimens (males and females, one mite per slide) from the primary vouchers (i.e., samples which underwent DNA extraction) using standard protocols (Krantz and Walter 2009; Lutz et al. 2017). We then took approximately 4000 high-resolution images of slides using an Olympus BX51 optical microscope equipped with differential interference contrast (DIC) lenses. Morphological examination of slides and images was conducted by F.A.H. and A.E.M. Mite lineages that were genetically distinct from a previously described sister lineage, but were not morphologically distinct, were given affinis (“aff.”) status.
Cophylogenetic Analyses
To conduct cophylogenetic analyses, we obtained a rooted host tree directly from the Open Tree of Life (Hinchliff et al. 2015) as deposited by Lovette et al. (2010). We then removed outgroup host taxa (and other nonfocal taxa) prior to converting host and feather mite trees to patristic distance matrices for cophylogenetic analyses. We explored alternate trees of our focal host taxa through the BirdTree online database (Jetz et al. 2012; http://birdtree.org) and included three relatively closely related outgroups, which range in relatedness to the core Parulidae clade (Xenoligea montana, Zeledonia coronata, and Icteria virens; Lovette et al. 2010). We downloaded 1000 trees from the Hackett “All Species” backbone tree (Hackett et al. 2008). We then summarized these 1000 trees by computing a single 95% majority-rule consensus tree, a 50% majority-rule consensus tree, and a maximum clade credibility tree using SumTrees version 4.5.2 (https://github.com/jeetsukumaran/DendroPy) in DendroPy version 4.5.2 (Sukumaran and Holder 2010), dropping the first 200 trees as burn-in. However, some species relationships in these trees were not concordant with the taxonomy from Lovette et al. (2010), particularly between Oporornis and Geothlypis, and low branch support led us to select the Open Tree of Life phylogeny for our hosts.
For cophylogenetic analyses, we collapsed the feather mite ML phylogenies to represent only a single tip (randomly chosen) for each species/OTU. We used the distance-based approaches, ParaFit (Legendre et al. 2002) and PACo (Balbuena et al. 2013), as well as the event-based reconciliation approach, JANE version 4.0.1 (Conow et al. 2010) to assess phylogenetic congruence. Distance-based methods are highly computationally efficient and generate summary (i.e., “global-fit”) statistics to measure congruence between host and symbiont phylogenies, whereas event-based methods infer specific evolutionary events (e.g., cospeciation and host-switching) that may have occurred between hosts and symbionts (often through a parsimony-based cost structure) and can thus help contextualize global-fit statistics (reviewed in Dismukes et al. 2022).
Distance-based methods were implemented in R version 3.6.3, with ParaFit implemented with the package “ape” and PACo with the package “paco” version 0.4.2 (Hutchinson et al. 2017). Briefly, these distance-based methods convert phylogenies to patristic distance matrices and generate global-fit statistics to assess the overall congruence of the two phylogenies along with how each individual host–symbiont association (“link”) contributes to the global fit. ParaFit specifically tests whether hosts and symbionts have evolved independently of one another by analyzing whether host–symbiont associations are random (α = 0.05). We ran ParaFit 100 times to account for variation in the host–symbiont randomization procedure and assess the consistency of results across runs. We used 999 permutations within each run with a “cailliez” correction for negative eigenvalues. We corrected the ParaFitLink1 (F1) individual link P-values using the Benjamini–Hochberg correction for false discovery rate (Benjamini and Hochberg 1995), which allows us to assess the contribution of each link to the overall congruence and correct for multiple tests. We ran PACo for 999 permutations using the r0 method, which permutes the host–symbiont associations explicitly following the assumption that the symbiont phylogeny tracks the evolution of the host (α = 0.05) and calculates the relative “contribution weight” of each individual host–symbiont link to the global fit. To test the statistical significance of each link’s contribution to the global fit, we additionally implemented the jackknife procedure in “ape,” “phangorn” version 2.7.0 (Schliep 2011), and “vegan” version 2.5-7 (Oksanen et al. 2020). If the 95% confidence intervals did not overlap with the median square residual (i.e., were lower than m2XY), we considered the link significant.
We ran the event-based reconciliation program, JANE, with default event costs (cospeciation = 0, duplication = 1, host switch = 2, loss = 1, failure to diverge = 1) and set the Genetic Algorithm parameters to 100 generations with a population size of 250. We then tested whether the optimal solution (i.e., observed score) was lower than expected by chance by running randomized tip mapping (i.e., randomizing the host–symbiont associations whereas maintaining the same number of symbionts with which a host is originally associated) 999 times. A significant result from the randomization procedure indicates significant congruence between the host and symbiont phylogenies. As has been discussed before (Matthews et al. 2018a; Dabert et al. 2022), multi host symbionts are problematic for event-based cophylogenetic methods, but JANE is a computationally efficient, commonly used reconciliation tool for systems with multi host symbionts (Libeskind-Hadas 2022). eMPRess, JANE’s successor, cannot yet handle multi host symbionts (Santichaivekin et al. 2021). However, once a tip has been assigned as a failure to diverge in JANE, the tip cannot be reassigned as a host switch (Charleston and Libeskind-Hadas 2014), so computational limitations still exist.
Sequence quality control and mitochondrial gene assembly were conducted using the Arkansas High Performance Computing Center (AHPCC) resources. Phylogenetic, species delineation, and cophylogenetic analyses were conducted on the Jetstream cloud server provided by Indiana University through XSEDE allocations and the AHPCC. All bioinformatics pipelines and scripts are available on GitHub (https://github.com/alixmatthews/parulidae_proctophyllodidae_cophylo).
Results
Parulid Feather Mite Diversity, Host Specificity, and Species Delineation Comparisons
Genetic data and morphological analyses confirmed the presence of two feather mite genera, Amerodectes and Tyrannidectes (Mironov and Galloway 2021) collected for the ingroup, as well as two other genera, Proctophyllodes and Trouessartia, collected for the outgroups. We uncovered eight Amerodectes lineages across 27 host species (Table 1 and Supplementary Table S2). Six of these lineages have previously been described: Amerodectes helmitheros, A. hribari, A. ischyros, A. jonesborensis, A. protonotaria, and A. seiurus. One of the two new lineages is very similar morphologically to A. ischyros and shares some of the same hosts as A. ischyros and is herein referred to as A. aff. ischyros. The other new lineage is currently undescribed and is herein called Amerodectes sp. n. 1. We found two Tyrannidectes lineages across five host species: Tyrannidectes charitomenos and T. aff. charitomenos (Table 1; Supplementary Table S2). The former was recently transferred to the genus Tyrannidectes from the genus Amerodectes by Mironov and Galloway (2021). Tyrannidectes aff. charitomenos is a new lineage that is very similar in morphology to T. charitomenos and shares with it some of the same hosts. The Proctophyllodes outgroup is P. quadratus (cox1 pairwise identity of 99% over 1189bp), and the Trouessartia sp. outgroup is currently undescribed. We determined the Trouessartia genus first by morphology and then by its cox1 gene, which showed an 84% pairwise identity to the T. rubecula reference over 1497bp. We conducted an additional BLASTn of the Trouessartia sp. outgroup cox1 sequence to a small database of ten other Trouessartia sp. cox1 sequences (Doña et al. 2017b) and measured 84–86% pairwise identity over 1413–1501bp.
Summary of the number of parulid host species with which each Amerodectes and Tyrannidectes feather mite lineage was associated in this study
| Mite lineage | Number of host species |
|---|---|
| Amerodectes helmitheros | 2 |
| Amerodectes hribari | 4 |
| Amerodectes ischyros | 12 |
| Amerodectes aff. ischyros | 2 |
| Amerodectes jonesborensis | 2 |
| Amerodectes protonotaria | 1 |
| Amerodectes seiurus | 3 |
| Amerodectes sp. n. 1 | 1 |
| Tyrannidectes charitomenos | 4 |
| Tyrannidectes aff. charitomenos | 1 |
| Mite lineage | Number of host species |
|---|---|
| Amerodectes helmitheros | 2 |
| Amerodectes hribari | 4 |
| Amerodectes ischyros | 12 |
| Amerodectes aff. ischyros | 2 |
| Amerodectes jonesborensis | 2 |
| Amerodectes protonotaria | 1 |
| Amerodectes seiurus | 3 |
| Amerodectes sp. n. 1 | 1 |
| Tyrannidectes charitomenos | 4 |
| Tyrannidectes aff. charitomenos | 1 |
Summary of the number of parulid host species with which each Amerodectes and Tyrannidectes feather mite lineage was associated in this study
| Mite lineage | Number of host species |
|---|---|
| Amerodectes helmitheros | 2 |
| Amerodectes hribari | 4 |
| Amerodectes ischyros | 12 |
| Amerodectes aff. ischyros | 2 |
| Amerodectes jonesborensis | 2 |
| Amerodectes protonotaria | 1 |
| Amerodectes seiurus | 3 |
| Amerodectes sp. n. 1 | 1 |
| Tyrannidectes charitomenos | 4 |
| Tyrannidectes aff. charitomenos | 1 |
| Mite lineage | Number of host species |
|---|---|
| Amerodectes helmitheros | 2 |
| Amerodectes hribari | 4 |
| Amerodectes ischyros | 12 |
| Amerodectes aff. ischyros | 2 |
| Amerodectes jonesborensis | 2 |
| Amerodectes protonotaria | 1 |
| Amerodectes seiurus | 3 |
| Amerodectes sp. n. 1 | 1 |
| Tyrannidectes charitomenos | 4 |
| Tyrannidectes aff. charitomenos | 1 |
The ABGD analyses (all models) of cox1 data recovered the presence of ten lineages, and the bPTP analysis of the same cox1 data recovered eleven lineages. The only difference between the two analyses was that bPTP separated mites collected on Geothlypis trichas from the other hosts harboring A. hribari. However, in an analysis using all eleven mitochondrial genes, bPTP lumped mites collected on G. trichas with the other A. hribari-harboring hosts and also split the mites collected on Setophaga cerulea and S. tigrina as two separate lineages from A. ischyros. Morphological analyses were in line with the ABGD species delineations. Given morphological analyses and the inconsistency of bPTP results, we delineated lineages based on results from the ABGD cox1-only method (ten lineages; interspecific and intraspecific genetic distances are in Table 2).
Pairwise cox1 distances of Amerodectes and Tyrannidectes feather mites. Pairwise genetic distances for Amerodectes and Tyrannidectes feather mites based on the cox1 barcode gene. The bold values along the diagonal are the intraspecific K2P distances. The values below the bold diagonal are the interspecific K2P distances
| A. hribari | A. ischyros | A. jonesborensis | A. protonotaria | A. seiurus | A. aff. ischyros | A. helmitheros | A. sp. n. 1 | T. charitomenos | T. aff. charitomenos | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| A. hribari | Min | 0.001 | |||||||||
| Max | 0.020 | ||||||||||
| A. ischyros | Min | 0.137 | 0.001 | ||||||||
| Max | 0.145 | 0.014 | |||||||||
| A. jonesborensis | Min | 0.093 | 0.131 | 0.009 | |||||||
| Max | 0.097 | 0.136 | 0.009 | ||||||||
| A. protonotaria | Min | 0.096 | 0.127 | 0.092 | – | ||||||
| Max | 0.098 | 0.134 | 0.094 | – | |||||||
| A. seiurus | Min | 0.091 | 0.123 | 0.074 | 0.090 | 0.001 | |||||
| Max | 0.097 | 0.131 | 0.077 | 0.091 | 0.002 | ||||||
| A aff. ischyros | Min | 0.128 | 0.101 | 0.132 | 0.134 | 0.128 | – | ||||
| Max | 0.132 | 0.107 | 0.135 | 0.134 | 0.129 | – | |||||
| A. helmitheros | Min | 0.103 | 0.134 | 0.101 | 0.090 | 0.092 | 0.130 | 0.005 | |||
| Max | 0.110 | 0.141 | 0.102 | 0.094 | 0.094 | 0.133 | 0.005 | ||||
| Amerodectes sp. n. 1 | Min | 0.104 | 0.149 | 0.106 | 0.091 | 0.101 | 0.145 | 0.080 | – | ||
| Max | 0.110 | 0.154 | 0.107 | 0.091 | 0.102 | 0.145 | 0.082 | – | |||
| T. charitomenos | Min | 0.163 | 0.170 | 0.156 | 0.163 | 0.153 | 0.168 | 0.165 | 0.170 | 0.005 | |
| Max | 0.170 | 0.179 | 0.158 | 0.165 | 0.155 | 0.169 | 0.172 | 0.172 | 0.005 | ||
| T. aff. charitomenos | Min | 0.158 | 0.172 | 0.144 | 0.167 | 0.148 | 0.169 | 0.164 | 0.170 | 0.046 | – |
| Max | 0.164 | 0.177 | 0.148 | 0.167 | 0.148 | 0.169 | 0.164 | 0.170 | 0.049 | – |
| A. hribari | A. ischyros | A. jonesborensis | A. protonotaria | A. seiurus | A. aff. ischyros | A. helmitheros | A. sp. n. 1 | T. charitomenos | T. aff. charitomenos | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| A. hribari | Min | 0.001 | |||||||||
| Max | 0.020 | ||||||||||
| A. ischyros | Min | 0.137 | 0.001 | ||||||||
| Max | 0.145 | 0.014 | |||||||||
| A. jonesborensis | Min | 0.093 | 0.131 | 0.009 | |||||||
| Max | 0.097 | 0.136 | 0.009 | ||||||||
| A. protonotaria | Min | 0.096 | 0.127 | 0.092 | – | ||||||
| Max | 0.098 | 0.134 | 0.094 | – | |||||||
| A. seiurus | Min | 0.091 | 0.123 | 0.074 | 0.090 | 0.001 | |||||
| Max | 0.097 | 0.131 | 0.077 | 0.091 | 0.002 | ||||||
| A aff. ischyros | Min | 0.128 | 0.101 | 0.132 | 0.134 | 0.128 | – | ||||
| Max | 0.132 | 0.107 | 0.135 | 0.134 | 0.129 | – | |||||
| A. helmitheros | Min | 0.103 | 0.134 | 0.101 | 0.090 | 0.092 | 0.130 | 0.005 | |||
| Max | 0.110 | 0.141 | 0.102 | 0.094 | 0.094 | 0.133 | 0.005 | ||||
| Amerodectes sp. n. 1 | Min | 0.104 | 0.149 | 0.106 | 0.091 | 0.101 | 0.145 | 0.080 | – | ||
| Max | 0.110 | 0.154 | 0.107 | 0.091 | 0.102 | 0.145 | 0.082 | – | |||
| T. charitomenos | Min | 0.163 | 0.170 | 0.156 | 0.163 | 0.153 | 0.168 | 0.165 | 0.170 | 0.005 | |
| Max | 0.170 | 0.179 | 0.158 | 0.165 | 0.155 | 0.169 | 0.172 | 0.172 | 0.005 | ||
| T. aff. charitomenos | Min | 0.158 | 0.172 | 0.144 | 0.167 | 0.148 | 0.169 | 0.164 | 0.170 | 0.046 | – |
| Max | 0.164 | 0.177 | 0.148 | 0.167 | 0.148 | 0.169 | 0.164 | 0.170 | 0.049 | – |
Pairwise cox1 distances of Amerodectes and Tyrannidectes feather mites. Pairwise genetic distances for Amerodectes and Tyrannidectes feather mites based on the cox1 barcode gene. The bold values along the diagonal are the intraspecific K2P distances. The values below the bold diagonal are the interspecific K2P distances
| A. hribari | A. ischyros | A. jonesborensis | A. protonotaria | A. seiurus | A. aff. ischyros | A. helmitheros | A. sp. n. 1 | T. charitomenos | T. aff. charitomenos | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| A. hribari | Min | 0.001 | |||||||||
| Max | 0.020 | ||||||||||
| A. ischyros | Min | 0.137 | 0.001 | ||||||||
| Max | 0.145 | 0.014 | |||||||||
| A. jonesborensis | Min | 0.093 | 0.131 | 0.009 | |||||||
| Max | 0.097 | 0.136 | 0.009 | ||||||||
| A. protonotaria | Min | 0.096 | 0.127 | 0.092 | – | ||||||
| Max | 0.098 | 0.134 | 0.094 | – | |||||||
| A. seiurus | Min | 0.091 | 0.123 | 0.074 | 0.090 | 0.001 | |||||
| Max | 0.097 | 0.131 | 0.077 | 0.091 | 0.002 | ||||||
| A aff. ischyros | Min | 0.128 | 0.101 | 0.132 | 0.134 | 0.128 | – | ||||
| Max | 0.132 | 0.107 | 0.135 | 0.134 | 0.129 | – | |||||
| A. helmitheros | Min | 0.103 | 0.134 | 0.101 | 0.090 | 0.092 | 0.130 | 0.005 | |||
| Max | 0.110 | 0.141 | 0.102 | 0.094 | 0.094 | 0.133 | 0.005 | ||||
| Amerodectes sp. n. 1 | Min | 0.104 | 0.149 | 0.106 | 0.091 | 0.101 | 0.145 | 0.080 | – | ||
| Max | 0.110 | 0.154 | 0.107 | 0.091 | 0.102 | 0.145 | 0.082 | – | |||
| T. charitomenos | Min | 0.163 | 0.170 | 0.156 | 0.163 | 0.153 | 0.168 | 0.165 | 0.170 | 0.005 | |
| Max | 0.170 | 0.179 | 0.158 | 0.165 | 0.155 | 0.169 | 0.172 | 0.172 | 0.005 | ||
| T. aff. charitomenos | Min | 0.158 | 0.172 | 0.144 | 0.167 | 0.148 | 0.169 | 0.164 | 0.170 | 0.046 | – |
| Max | 0.164 | 0.177 | 0.148 | 0.167 | 0.148 | 0.169 | 0.164 | 0.170 | 0.049 | – |
| A. hribari | A. ischyros | A. jonesborensis | A. protonotaria | A. seiurus | A. aff. ischyros | A. helmitheros | A. sp. n. 1 | T. charitomenos | T. aff. charitomenos | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| A. hribari | Min | 0.001 | |||||||||
| Max | 0.020 | ||||||||||
| A. ischyros | Min | 0.137 | 0.001 | ||||||||
| Max | 0.145 | 0.014 | |||||||||
| A. jonesborensis | Min | 0.093 | 0.131 | 0.009 | |||||||
| Max | 0.097 | 0.136 | 0.009 | ||||||||
| A. protonotaria | Min | 0.096 | 0.127 | 0.092 | – | ||||||
| Max | 0.098 | 0.134 | 0.094 | – | |||||||
| A. seiurus | Min | 0.091 | 0.123 | 0.074 | 0.090 | 0.001 | |||||
| Max | 0.097 | 0.131 | 0.077 | 0.091 | 0.002 | ||||||
| A aff. ischyros | Min | 0.128 | 0.101 | 0.132 | 0.134 | 0.128 | – | ||||
| Max | 0.132 | 0.107 | 0.135 | 0.134 | 0.129 | – | |||||
| A. helmitheros | Min | 0.103 | 0.134 | 0.101 | 0.090 | 0.092 | 0.130 | 0.005 | |||
| Max | 0.110 | 0.141 | 0.102 | 0.094 | 0.094 | 0.133 | 0.005 | ||||
| Amerodectes sp. n. 1 | Min | 0.104 | 0.149 | 0.106 | 0.091 | 0.101 | 0.145 | 0.080 | – | ||
| Max | 0.110 | 0.154 | 0.107 | 0.091 | 0.102 | 0.145 | 0.082 | – | |||
| T. charitomenos | Min | 0.163 | 0.170 | 0.156 | 0.163 | 0.153 | 0.168 | 0.165 | 0.170 | 0.005 | |
| Max | 0.170 | 0.179 | 0.158 | 0.165 | 0.155 | 0.169 | 0.172 | 0.172 | 0.005 | ||
| T. aff. charitomenos | Min | 0.158 | 0.172 | 0.144 | 0.167 | 0.148 | 0.169 | 0.164 | 0.170 | 0.046 | – |
| Max | 0.164 | 0.177 | 0.148 | 0.167 | 0.148 | 0.169 | 0.164 | 0.170 | 0.049 | – |
Comparison of Phylogenetic Analyses
The ML and BI analyses estimated similar feather mite phylogeny topologies with high support. The MCMC chains from the Bayesian analyses converged to stationarity (<0.1 maximum discrepancy clade support scores in bpcomp; <0.1 relative difference scores and effective sample size for all parameters > 200 in tracecomp) for both the multilocus concatenated and cox1 data sets. Phylogenies estimated from both data sets were largely congruent with one another in both topology and support, as was the MSC phylogeny. The multilocus concatenated tree (Fig. 1) had the highest support overall, followed by the MSC tree (Supplementary Fig. S1), and then the cox1 tree (Fig. 2). There were eight partitions with six best models of nucleotide substitution as estimated by AICc for the ML multilocus concatenated tree (atp6: TPM3u+F+R2; cob: TIM+F+I+G4; cox1: GTR+F+I+G4; cox2 and nad5: GTR+F+I+G4; cox3: HKY+F+I+G4; nad1: TIM3+F+I+G4; nad2 and nad6: TIM3+F+R3; nad3 and nad4: GTR+F+I+G4) and TIM3+F+I+G4 was the best-fit model for the cox1-only ML tree.
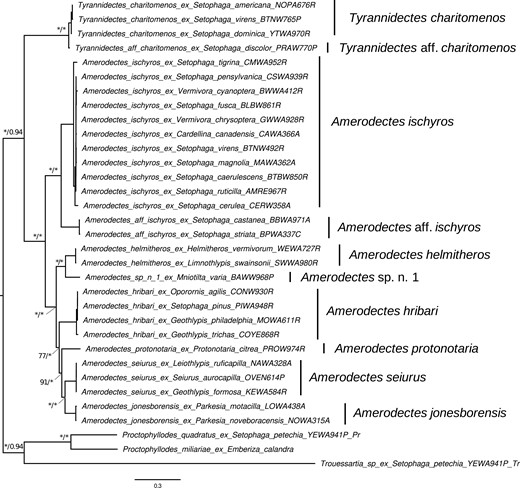
Maximum likelihood (ML) topology of parulid Amerodectes and Tyrannidectes feather mites estimated from a partitioned analysis of 11 concatenated mitochondrial genes. Values at nodes between lineages indicate ML ultrafast bootstrap (BS) support values followed by Bayesian posterior probabilities (PP); nodes with >99 BS or >0.95 PP are indicated by asterisks (*). The scale bar indicates nucleotide substitutions per site. Lineages as recovered by the species delineation analyses are labeled to the right of tip labels.
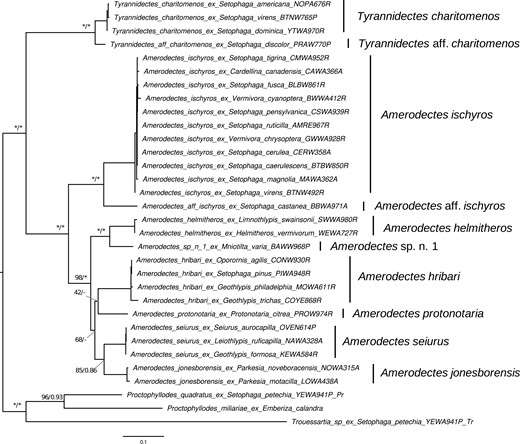
Maximum likelihood (ML) topology of parulid Amerodectes and Tyrannidectes feather mites estimated from the cox1 gene. Values at nodes between lineages indicate ML ultrafast bootstrap (BS) support values followed by Bayesian posterior probabilities (PP); nodes with >99 BS or >0.95 PP are indicated by asterisks (*). The scale bar indicates nucleotide substitutions per site. Lineages as recovered by the species delineation analyses are labeled to the right of tip labels.
Monophyletic relationships between lineages were recovered with the multilocus concatenated data set and all nodes were supported by greater than 75% bootstrap support values (ML) and >0.8 posterior probability (PP) support values (BI), with 78% of nodes (excluding outgroups) having very high bootstrap support (≥99%) and 89% of nodes having very high (≥0.95) PP support. Comparable results were found in the MSC tree, with 89% of nodes with >0.8 local PP support and 67% with ≥0.95 local PP support (Supplementary Fig. S1). With the cox1 tree, 78% of monophyletic lineage nodes were supported by greater than 75% bootstrap support values and 100% with >0.8 PP support, with 56% of nodes having very high bootstrap support (≥99%) and 86% of nodes having very high (≥0.95) PP support. In the ML analyses, the relationship between A. hribari and A. protonotaria differed in the cox1 tree (sister to one another) compared with the concatenated and coalescent-based approaches (A. protonotaria is sister to the most recent common ancestor of A. seiurus and A. jonesborensis). In the multilocus BI consensus tree, the relationships between these lineages were consistent with the ML multilocus tree but were not well resolved and contained polytomies in the cox1 BI consensus tree. Although we conducted downstream analyses using the multilocus concatenated and cox1-based data sets, a notable use of the MSC phylogeny is outlined below.
We initially included samples from all 31 host species (including outgroup hosts) in our phylogenetic analyses. However, our initial multilocus concatenated ML analysis assigned T. charitomenos and A. ischyros as sister taxa (albeit with low bootstrap support; 40–42%) and we observed inconsistent patterns (e.g., sister vs. nonsister, T. charitomenos was occasionally nested within A. ischyros) across various reconstruction attempts (e.g., implementing different models of nucleotide evolution and parsing models to different codon positions). Instead, the cox1-based phylogeny (Fig. 2) and MSC phylogeny (Supplementary Fig. S1) both correctly separated these two genera with high support. By investigating individual gene trees, we detected the presence of both genera in a single sample (host: Setophaga citrina) using several lines of evidence. First, branch support for the split between the two genera was low (≤76%) in six of the 11 gene trees. Second, the S. citrina mite sample clustered within T. charitomenos in 6/11 gene trees and A. ischyros in 5/11 gene trees (Supplementary Table S2). Morphological analyses using high-powered DIC microscopy on voucher specimens recovered from this sample resulted in ambiguous conclusions as to whether both A. ischyros and T. charitomenos were present and pooled together during DNA extraction; males appear to all be consistent with A. ischyros, but females are very difficult to distinguish between these two genera (Mironov and Galloway 2021). Based on molecular results, we removed this individual and re-ran phylogenetic analyses. The resulting tree using the multilocus concatenated data set separated the two genera with much higher bootstrap support (≥99%; Fig. 1). Although this individual sample was removed from the phylogenetic and species delineation analyses, the host (S. citrina) was included as associated with both mite species in cophylogenetic analyses.
Cophylogenetic Analyses Yielded Overall Phylogenetic Congruence
Using the multilocus data set for the mite phylogeny, distance-based cophylogenetic analyses between feather mites and their parulid hosts suggest there is phylogenetic congruence in this system. Both ParaFit (ParaFitGlobal = 0.044; P = 0.001) and PACo (PACo m2XY = 0.12; global P = 0.001) global tests suggest there is significant phylogenetic congruence between feather mites and their parulid hosts despite the tangled web of interactions (Fig. 3a). With respect to the specific hypotheses that each method is testing, the significant global ParaFit test statistic indicates nonrandom associations between feather mites and hosts, suggesting that the evolution of these two groups has not been independent of one another. In contrast, the significant global value from PACo indicates that host speciation is driving mite speciation in this system. However, the individual link tests between methods are not in agreement. The adjusted individual link tests from ParaFit (ParaFitLink1) suggest that there are 22 (out of 32) significant host-mite associations that contribute to the overall congruence of this system, plus an additional (one) link fluctuates between significant and nonsignificant across runs (Fig. 3a and Supplementary Table S2). Conversely, the Procrustes residuals from PACo show that only two out of 32 links, which are both unambiguously nonsignificant links recovered from ParaFit, contribute the most to the overall congruence of these phylogenies (i.e., the m2XY; Fig. 4a and Supplementary Table S2). Using the event-based/reconciliation analysis, JANE, we detected a significant signal of phylogenetic congruence in this system (total observed cost = 75; P < 0.001). Despite the significant congruence detected by JANE, the specific event reconstruction recovered only one cospeciation event, along with four duplication events (i.e., intrahost speciation), four duplications + host switches, 41 losses (i.e., extinction or sporting events), and 22 failures to diverge (Supplementary Fig. S2).
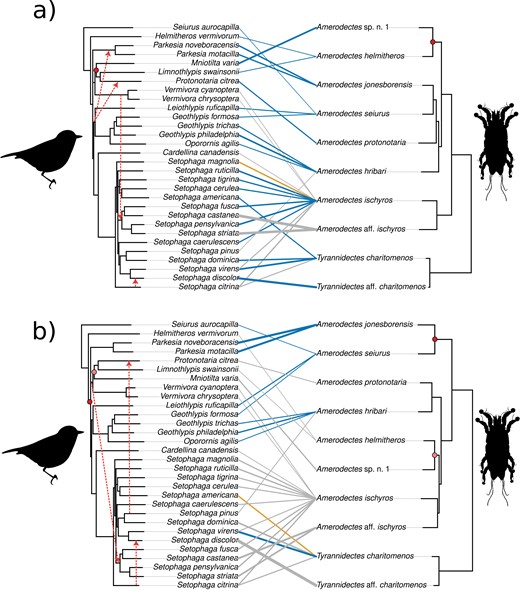
Tanglegrams illustrating the evolutionary history between New World warbler (parulid) hosts (left) and their Amerodectes and Tyrannidectes feather mites (right). The mite phylogenies were estimated from (a) eleven concatenated mitochondrial genes and (b) cox1-only using maximum likelihood. The host phylogeny is from Lovette et al. (2010) in both (a) and (b). Line colors indicate the level of the significance of each host–mite link estimated by ParaFitLink1 after correction (blue: significant; orange: fluctuation between 100 ParaFit runs; gray: nonsignificant). Line thickness is inversely proportional to the residuals as estimated by PACo, multiplied by a factor of two for illustrative purposes (thicker lines indicate higher contribution to the overall congruence). Red shaded circles indicate cospeciation events recovered from JANE reconciliation analyses, with codivergences filled with the same level of shading. Red dotted arrows indicate host switches recovered from JANE reconciliation analyses.

PACo jackknife test results of individual host-mite links (x-axis) of the (a) multilocus data set and (b) cox1-only data set. Each bar represents the jackknife goodness-of-fit statistics with 95% confidence intervals (CIs; y-axis) and the blue horizontal dashed line represents the median global square residual (). Links in which the 95% CIs do not overlap the m2XY (dark blue) are considered to contribute significantly to the overall congruence between the host and mite phylogenies.
Using the cox1 data set, distance-based ParaFit (ParaFitGlobal = 0.015; P = 0.003) and PACo (PACo m2XY = 0.12; global P = 0.001) both suggest there is significant phylogenetic congruence between feather mites and their parulid hosts. However, the number of significant links differed from the multilocus data set. ParaFit recovered nine out of 32 significant host-mite associations that contribute to the overall congruence of this system, plus one additional link that fluctuates between significant and nonsignificant in the permutations (Fig. 3b and Supplementary Table S2). PACo recovered seven out of 32 links as significantly contributing to the overall congruence (Fig. 4b and Supplementary Table S2). Using JANE, we detected a significant cophylogenetic signal in this system (total observed cost = 73; P < 0.001), although again only recovering a small number of cospeciation events (two), along with four duplications, three duplications + host switches, 41 losses, and 22 failures to diverge (Supplementary Fig. S3). Thus, despite the slightly different topologies, global results from both distance-based and event-based cophylogenetic analyses using the cox1 data set remained generally the same as the multilocus data set.
Discussion
Dispersal-limited symbionts, especially those that are permanent and vertically transmitted, are hypothesized to be host specialists because symbiont populations likely transmit within a particular host species and are ultimately sorted (i.e., speciate) according to that particular host species (Clayton et al. 2016). However, from our comprehensive sampling of 92% of parulid species that breed in North America, Pool-Seq approach, and thorough phylogenetic analyses, we uncovered unexpected variations in the degree of host specificity for dispersal-limited avian feather mites. These data indicate that host specificity varies from highly specialist mites found on only a single host species (e.g., A. protonotaria) to broadly generalist mites found on ≥12 host species (e.g., A. ischyros). This variation in host specificity suggests that contemporary processes such as dispersal, even if limited or rare, and ongoing cross-host gene flow can differ across closely related species. In addition to the signatures of microevolutionary processes underlying host–symbiont associations, our results lend support for significant statistical phylogenetic congruence between feather mites and their parulid hosts. This congruence suggests that, at some level, historical macroevolutionary processes such as host switching and cospeciation also play a role in shaping symbioses. In general, these results are consistent across data sets (multilocus and barcode). However, the multilocus data set was more effective in detecting the presence of a heterogeneous sample and ultimately impacting our understanding of symbiont–host specificity.
Microevolutionary filters (e.g., ecological, geographical, and behavioral) can either disrupt or strengthen the degree of host specificity within symbiotic systems, consistent with the concepts of ecological fitting and niche or host expansion (Agosta et al. 2010; Araujo et al. 2015; Mestre et al. 2020). For symbionts with dispersal stages or that use vectors, host switching by ecological fitting is common (Fecchio et al. 2019; McKee et al. 2019; Pérez et al. 2019). For dispersal-limited symbionts, host encounter/switching opportunities are less well understood, but likely increase when hosts occur in sympatry (e.g., chipmunk pinworms, Nematoda: Oxyuridae; Bell et al. 2016; passerine lice, Psocodea: Menoponidae; Martinů et al. 2015). Specifically related to feather mites, several nonmutually exclusive host-centric factors can widen encounter filters with novel hosts in breeding sympatry, such as hosts sharing similar nesting ecologies (Matthews et al. 2018a), exchanging nest material (Dabert et al. 2022), and behaving antagonistically (Jones et al. 2007, 2016; Wynia and Bednarz 2021). Additionally, the parulid hosts surveyed in this study are all Neotropical-Nearctic migrants, so ecological, geographical, or behavioral overlap occurring during the nonbreeding seasons (e.g., migration and “winter”) could alter the opportunities for ecological fitting (Matthews et al. 2018a). An overlap of any one of these factors can theoretically promote host generalism, which is likely the case for the most generalist (polyxenous) mite here, A. ischyros.
Conversely, barriers that limit encounter filters related to these same factors can promote host specialization, such as for the highly specific (monoxenous) mite found here, A. protonotaria. In addition to host-centric factors, variation in host specificity may be driven by symbiont-centric traits such as differential abilities to disperse phoretically (e.g., feather lice, Psocodea: Ischnocera; Harbison et al. 2009) or contrasting reproductive strategies (e.g., fish and coral copepods, Copepoda: Podoplea; Doherty et al. 2022). In feather mites, the probability of survival in the absence of hosts is greater for A. ischyros (generalist), but A. protonotaria (specialist) has a greater chance of dispersal (Matthews et al. 2022a), suggesting that a selective trade-off between life history traits may lead to differential host specificities. Ultimately, a combination of statistical and network modeling techniques (e.g., Satler et al. 2019) should be implemented to measure the directionality and relative importance of these factors. Doing so may help determine the microevolutionary factors (e.g., encounter and/or compatibility filters) that relax or restrict host–symbiont specificity and modulate macroevolutionary (e.g., coevolutionary) processes over time.
Despite the variation in host specificity, all cophylogenetic analyses (distance- and event-based; multilocus concatenated and cox1-only data sets) uncovered significantly more phylogenetic congruence between mites and parulids than would be expected by chance alone. Phylogenetic congruence despite variable host specificity may be the result of preferential host switching (Charleston and Robertson 2002) or pseudocospeciation (de Vienne et al. 2007). Pseudocospeciation, or the adaptive radiation of symbionts on multiple closely related hosts (Hafner and Nadler 1988), is especially likely in our system given that the hosts and symbionts are all very closely related and there are multiple multi host mite species. Disagreements with respect to the composition and number of significant host–symbiont links between our data sets in our distance-based tests (Figs. 3 and 4) may also be signatures of pseudocospeciation (de Vienne et al. 2013), results of the differences in the underlying hypotheses between the two distance-based tests (Legendre et al. 2002; Balbuena et al. 2013), or consequences of variable phylogenetic resolution of input data sets (Sweet et al. 2016; Beigel et al. 2021). Our event-based analysis found that cospeciation between mites and their hosts is unlikely to be the dominant evolutionary mechanism underlying the phylogenetic congruence between these two taxa. Instead, host switching, losses, and failures to diverge drive the statistical congruence. This conclusion is consistent with previous feather mite studies (Doña et al. 2017b; Matthews et al. 2018a; Dabert et al. 2022) and those of other dispersal-limited symbionts such as two-toed sloth fur algae (Fountain et al. 2017) and bat mites (Mesostigmata: Spinturnicidae; Bruyndonckx et al. 2009). Collectively, our distance- and event-based results underscore the concept that phylogenetic congruence does not always result from cospeciation. Host switching and cospeciation are nonmutually exclusive processes, particularly in cases involving multi host symbionts found on closely related hosts in which statistical congruence may be caused by pseudocospeciation (de Vienne et al. 2013).
For this symbiotic system and others, the ability to detect pseudocospeciation and accurately measure host specificity for a group of symbionts is ultimately reliant on sampling coverage of possible host species and intensity (e.g., how many individuals of each potential host species are sampled). In this study and with our increased sampling efforts (despite only sequencing ~1 sample per host species), we uncovered a series of new mite–host associations that were previously unknown. Specifically, we found one new host of A. jonesborensis (Parkesia motacilla), one new host of A. seiurus (Leiothlypis ruficapilla), two new hosts of A. hribari (Oporornis agilis and Setophaga pinus), five new hosts of A. ischyros (Cardellina canadensis, S. citrina, S. magnolia, S. virens, and Vermivora cyanoptera) and three new hosts of T. charitomenos (S. americana, S. citrina, and S. virens). Additionally, we discovered a potentially new species of Amerodectes (A. sp. n. 1), which was associated with Mniotilta varia (a host species that has only otherwise been associated with A. seiurus; Mironov and Galloway 2021). Although we sampled a nearly comprehensive set of closely related host species, sequencing was limited to only ~1 sample per host species. It would thus be beneficial to sequence additional mites from multiple individuals of each host species across their known geographic ranges to evaluate the consistency of the host-mite associations found in the present study. Preliminary data from expanded sampling efforts (i.e., multiple individuals of conspecific hosts across their entire range) indicate that host specificity in this system still varies from highly host-specific mites (e.g., A. protonotaria) to highly host generalist mites (e.g., A. ischyros; Matthews et al. 2022b). Sequencing additional individuals per host species would also help to identify if there are uncommon or ephemeral host-mite associations (e.g., “stragglers”). Although these uncommon associations are often overlooked as contaminations in dispersal-limited symbionts like feather mites (Doña et al. 2018b) and chewing lice (Psocodea: Menoponidae; Martinů et al. 2015), they may represent established associations in certain host populations.
Our additional sampling coupled with higher resolution (i.e., multilocus) phylogenetic analyses also uncovered that two previously recorded hosts associated with A. ischyros (S. castanea and S. striata; Mironov and Galloway 2021) may in fact be associated with a separate but closely related lineage (herein A. aff. ischyros). As suggested by Mironov and Galloway (2021), A. ischyros may actually be a species complex in statu nascendi. Our results here support that possibility as there were no clear morphological differences, species delimitation analyses showed these separate from core A. ischyros clade, and pairwise cox1 distances between A. ischyros and A. aff. ischyros is 10.1–10.7% (Table 2). These genetic distance values are approximately three times greater than the suggested species threshold for feather mites of 3.42% (Doña et al. 2015a). Additional collection and sequencing of A. ischyros from more host individuals may help to further understand the breadth of this species’ genetic diversity.
Intraspecifically, parulids can harbor multiple closely related feather mite species. For example, Oporornis agilis harbors both A. hribari (this study) and A. tiffanyluiae (Mironov and Galloway 2021), and S. discolor harbors A. ischyros (this study) and T. charitomenos (Matthews et al. 2018a). This phenomenon, also known as synhospitality (Eichler 1966), has been documented previously in other host families (Doña et al. 2016). Synhospitality may be the result of geographically isolated host populations (Bochkov and Mironov 2008), as we detected in this study with the host S. virens; the sample of the nominate S. virens race harbored A. ischyros, whereas the sample of the disjunct subspecies, S. v. waynei, harbored T. charitomenos. Whether this heterogeneric synhospitality pattern is consistent across individuals of these genetically and geographically distinct populations (Carpenter et al. 2022) remains to be tested, but could give further insight into host population dynamics. In contrast to allopatric hosts, synhospitality can even occur on the same individual bird (Hernandes and OConnor 2017). Heterogeneric mites found on the same individual oftentimes physically separate themselves across different feather microhabitats (e.g., dorsal vs. the ventral side of a feather), likely as a result of competition for space or resources (Mestre et al. 2011; Fernández-González et al. 2015; Stefan et al. 2015). Typically, heterogeneric feather mites are distinguishable by their microhabitat or by morphology using a standard dissecting microscope and can be separated prior to sequencing. However, based on the genetic data gathered from our Pool-Seq approach in this study, we found one instance in which heterogeneric mites coexisted in the same feather microhabitat (T. charitomenos and A. ischyros on the ventral surface of a single S. citrina feather; see Results). Had we only sequenced one individual mite from this S. citrina individual (as opposed to pooling), the additional mite association would have been overlooked and the composition of host-mite associations would have been underestimated. It is also possible that congeneric mites of different species exist on the same individual host or even the same feather. As a result, they may be unknowingly pooled together and thus could potentially lead to chimeric assemblies. Besides applying coalescent-based approaches (see below) and/or slide-mounting exoskeletons postextraction as done here, an alternate strategy to minimize this risk would be to conduct metabarcoding on the pooled samples to confirm that the composition of species is homogenous within samples (Vizcaíno et al. 2018).
Methodologically, this study provided a test case for our Pool-Seq approach, which also offered a unique situation to apply a coalescent-based approach to phylogenetic estimation from mitochondrial data. Feather mites can have high genetic diversity (Doña et al. 2015b) and can have high speciation rates (Dabert et al. 2010; Knowles and Klimov 2011), and thus Pool-Seq of individuals from a single feather represents a situation similar to what would be observed under incomplete lineage sorting or hybridization with nuclear loci (Kim et al. 2020). Consequently, applying a coalescent-based model to pooled mitochondrial data can help to uncover or otherwise support species relationships using different approaches (e.g., multilocus or barcoding genes). This coalescent-based strategy can also help identify chimeric sequence assemblies from pooled data (e.g., if mites of different species were pooled together). Our results also support the robustness of mitochondrial data for phylogenetic analyses, including multilocus concatenated data sets (Doña et al. 2017b) and cox1-only data sets (Matthews et al. 2018a). However, phylogenies estimated from nuclear data may also help to inform species delineations that were recovered by mitochondrial data alone, particularly if any species have high effective population sizes (Klimov et al. 2019). Nuclear data may also help us to explore the potential for sex-biased (Matthews et al. 2022a) gene flow in feather mites, which has not yet been explicitly studied, but could help explain patterns of dispersal, population structure, and other mechanisms (e.g., behavioral, ecological) underlying host specificity.
Conclusion
Our comprehensive sampling and novel Pool-Seq approach allowed us to demonstrate that the presumed dispersal abilities of symbionts may not be strong indicators of their host specificity. We stress that host specificity can vary significantly for dispersal-limited symbionts, even at fine taxonomic scales. We also emphasize that phylogenetic congruence does not always imply high degrees of cospeciation and may instead represent pseudocospeciation. These findings highlight the potential for microevolutionary processes related to encounter and/or compatibility filters as being key drivers of macroevolutionary patterns in host–symbiont systems. Future studies should focus on generalizing these filters to better understand the ecological and evolutionary processes mediating host specificity.
Supplementary Data
Data available from the Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.n02v6wx15.
Funding
This work was supported by the Kays Foundation to T.J.B.; the Society of Systematic Biologists Graduate Student Research Award to A.E.M.; an Ernst Mayr Travel Grant from the Museum of Comparative Zoology–Harvard University to A.E.M.; the Arkansas Audubon Society Trust to A.E.M.; Arkansas State University Student Research and Creativity Grant to A.E.M.; the SUPERB Scholarship Program (National Science Foundation; DUE-1564954); the National Council for Scientific and Technological Development—CNPq (304479/2019-5) to. F.A.H.; Pennsylvania State University to D.P.L.T.; and startup funds from the Eberly College of Science and the Huck Institutes of the Life Sciences to D.P.L.T.
Acknowledgements
We thank Lee Bryant, Lesley Bulluck, Jeff Larkin, Eliot Miller, Morgan Slevin, Doug Raybuck, and Alex Worm for collecting field samples; Laura Porturas and the Penn State University Frost Entomological Museum for coordinating and contributing museum samples; Jennifer Mandel and Ramhari Thapa (University of Memphis) for allowing us to use their QSonica for preliminary samples; Kyle Gustafson (Arkansas State University) for access to the microscope and imaging station; Jeff Pummill, David Chaffin, and Pawel Wolinski at the AHPCC (University of Arkansas) and Sandaruwan Ratnayake (Arkansas State University) for their computational assistance; Alvaro Hernandez and Chris Wright at the Roy J. Carver Biotechnology Center at the University of Illinois for their assistance in library preparation and sequencing; Arkansas State University undergraduate assistants, Tryniti Garred and Kendall Lee, for their collections and curatorial help; and we thank Mary DuBose (University of Memphis), Travis Marsico (Arkansas State University), as well as Isabel Sanmartín (Real Jardín Botánico, CSIC), Lauren Esposito (California Academy of Sciences), and two anonymous reviewers for conducting constructive reviews that improved the manuscript.
Author Contributions
A.E.M. and T.J.B. conceived the study, secured funding, and designed the methodology with contributions from A.J.W. and A.D.S.; T.J.B. secured permits; A.E.M., T.J.B., and D.P.L.T. conducted fieldwork and collected samples; A.E.M. performed molecular work and analyzed the molecular data with input from A.J.W. and A.D.S.; A.E.M. and F.A.H. analyzed morphological data; A.E.M. wrote initial manuscript drafts, and all authors were involved in writing, editing, and/or constructively revising the final manuscript. All authors gave approval for publication. The authors declare no conflict of interest.
DATA AVAILABILITY
All raw sequencing data are available from NCBI’s Short Read Archive under the project number PRJNA817584 at https://www.ncbi.nlm.nih.gov/bioproject/PRJNA817584. All bioinformatic pipelines, sequence alignments, and analytical scripts are available on the Dryad Digital Repository (https://doi.org/10.5061/dryad.n02v6wx15) and on GitHub (https://github.com/alixmatthews/parulidae_proctophyllodidae_cophylo). Sampling efforts for all host species and prevalence data are in Supplementary Table S1 and sample metadata and accession numbers are listed for individual samples in Supplementary Table S2 of the Supplementary Material; both tables are on Dryad (https://doi.org/10.5061/dryad.n02v6wx15).

{kind=link}
{kind=link}
{kind=link}
{kind=link}