






Abstract
Pseudocataplexy is a rare functional neurological disorder that mimics cataplexy, pathognomonic for narcolepsy type 1 (NT1). We describe the psychiatric comorbidity and personality traits of patients with pseudocataplexy versus NT1 cases.
The case–control observational study enrolled consecutive patients with pseudocataplexy and a control group of age-matched consecutive NT1 patients. The diagnostic work-up included a structured interview, 48-hour polysomnography, multiple sleep latency test, cataplexy provoking test, and hypocretin-1 measurement in cerebrospinal fluid. All participants were administered Beck Depression Inventory, State-Trait Anxiety Inventory, Patient Health Questionnaire-15 (PHQ-15), Personality Inventory for DSM-5 brief form, and quality-of-life (QoL) measurement by 36-item Short Form health survey (SF-36).
Fifteen patients with pseudocataplexy and 30 with NT1 were included. Despite the suspicion of possible cataplexy, none of the pseudocataplexy participants fulfilled international diagnostic criteria for NT1. Pseudocataplexy patients presented higher rates of moderate state anxiety (40% vs. 10%, p = 0.018), medium level of somatic symptoms, defined by PHQ-15 score > 10 (66.7% vs. 16.7%, p = 0.003), and a trend towards moderate-to-severe depressive symptoms (33.3% vs. 10%, p = 0.054) compared to NT1. No significant differences in personality traits emerged. Pseudocataplexy patients had worse QoL profiles in almost all SF-36 domains including physical (mean ± SD: 37.7 ± 9.88 vs. 51.13 ± 7.81, p < 0.001) and mental (mean ± SD: 33.36 ± 12.69 vs.42.76 ± 11.34, p = 0.02) summary scores.
Patients with pseudocataplexy present more severe psychiatric symptoms and a lower QoL profile in comparison with patients with NT1. The severe somatoform and affection impairment in pseudocataplexy may explain the poorer QoL and should require a tailored therapeutic approach.
This research is a case–control observational study addressing the clinical features and psychiatric profile of patients with a functional neurological disorder, namely pseudocataplexy, in comparison with patients with narcolepsy with cataplexy. Our results show that subjects with pseudocataplexy present more psychiatric symptoms (i.e. in somatoform and affection domains) and worse quality of life than patients affected by narcolepsy with cataplexy, despite the latter already repeatedly showing a severe psychological impacts impairing the quality of life. Moreover, this study provides some clues about features that allow the clinical distinction between cataplexy and pseudocataplexy, despite mentioning some elements that can overlap between the two entities.
Introduction
Cataplexy is a transient and sudden loss of muscle tone with preserved consciousness, typically provoked by strong emotions, particularly laughter. Cataplexy is the pathognomonic symptom of narcolepsy type 1 (NT1, formerly defined as narcolepsy with cataplexy), a rare neurological disorder also characterized by excessive daytime sleepiness (EDS) and by impaired hypocretin neurotransmission, demonstrated by hypocretin-1 (hcrt-1) deficiency in the cerebrospinal fluid (CSF) [1]. The differential diagnosis of cataplexy includes several disorders such as epilepsy, syncope, drop attacks, and psychogenic attacks the latter defined as “pseudocataplexy” [2, 3].
Pseudocataplexy typically presents some features unusual for cataplexy: it is often evoked by negative emotions, instead of the typical positive ones; weakness in pseudocataplexy is mostly generalized but focal attacks can rarely occur; deep tendon reflexes are preserved, while transiently abolished during generalized genuine cataplexy; the duration of the pseudocataplectic attack is mostly longer than the few seconds-one minutes durations that characterizes cataplexy, and can last minutes or even hours [4]. When video recorded, cataplexy is often heralded by hypotonia involving at first the facial district, facial jerks and grimaces, head drop, and trunk fall, leading to falls to the ground, objective findings that may be useful for differential diagnosis [2], despite deep tendon reflexes can be preserved in partial cataplexy [5].
According to the current psychiatric nosography, pseudocataplexy falls in the spectrum of somatic symptoms disorders, namely functional neurologic disorder (FND), which has replaced conversion disorder in the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5) [6]. In particular, pseudocataplexy can be included among functional movement disorders (FMD), that, together with psychogenic non-epileptic seizures (PNES), represent one of the subtypes of FND [7]. Very few reports addressed pseudocataplexy phenomenology, its psychopathological features, and its psychiatric comorbidity.
Pseudocataplexy can be also included among transient functional paralysis, a broader term encompassing all reversible weaknesses of psychogenic origin that can be in differential diagnosis with neurological conditions beyond cataplexy (i.e. seizures, syncope, and transient ischemic attacks) [8].
In literature, the term “atypical” cataplexy can also be found, identifying spells described by the patients with unusual features such as unilateral weakness, absence of clear triggers, only negative emotions as triggers, hyperacute generalized weakness, only generalized attacks, prolonged recovery [9]. It is to be noted that “atypical” cataplexy is a definition applied to the medical history, and it includes cases of pseudocataplexy (as the two entities share several features), that can be defined when the episode is documented as functional by an experienced physician, and a diagnosis of narcolepsy is ruled out, as well as of cataplexy with peculiar features. Indeed, several “atypical” cataplexy features are now well-recognized features of cataplexy itself in children [10, 11], or may be documented in late-onset cases [12], pointing to the need to better investigate, document, and understand these intriguing phenomena that may vary across the life span [13]. Instead of the a priori distinction between “typical” and “atypical” cataplexy, which can be misleading, some authors suggest classifying the patient’s report of attacks based on different levels of certainty of genuine cataplexy [14].
To the best of our knowledge, the first cases of pseudocataplexy were described by Schenck and Mahowald (1993) in eight patients evaluated as conversion disorder [15]. Krahn et al. [16] reported a female middle-aged patient presenting with sleep paralyzes and spells lasting up to 10 min and triggered by anger, diagnosed with “dependent and narcissistic personality traits” without axis-I psychiatric disorder. In another case of pseudocataplexy, a 42-year-old male was found comorbid with major depressive disorder [8]. In a case series of cataplexy mimics, two out of six were classified as “conversion disorder” and “paranoid schizophrenia” [17]. With further complication of differential diagnosis, patients with confirmed NT1 and both cataplexy and functional spells have been reported [3], in one case mimicking status cataplecticus [18] In these patients, functional generalized episodes were distinguished by preserved deep tendon reflexes, in the context of unexplained exacerbation of symptoms. Meinen et al. suggested a diagnosis of somatoform disorder and focused on several factors predisposing to stress such as personality traits such as perfectionism and health worries in NT1 patients presenting also pseudocataplexy [18].
Overall, several psychiatric disorders—including somatoform, mood, and personality disorders—have been associated with pseudocataplexy through few isolated case reports. The objective of the present study is to evaluate psychiatric comorbidity and personality traits in a consecutive sample of patients with pseudocataplexy attending a national referral center for narcolepsy and to compare them with a pathological “control” group of NT1 patients.
Methods
Study design
Case–control observational study of patients with pseudocataplexy with a pathological “control” group of patients with NT1.
Participants
Consecutive patients with final diagnosis of FND with pseudocataplexy, evaluated between 2019 and 2021 at Narcolepsy Center of IRCCS Istituto delle Scienze Neurologiche di Bologna, were recruited. The pathological “control” group consisted of consecutive patients with NT1, attending follow-up visits at Narcolepsy Center, matched for age and presenting cataplexy at the time of the diagnosis. Two “controls” were included for each case with cataplexy.
Participants from both groups had been referred to the Narcolepsy Center for suspected NT1 after triage based on the presence of subjective complaints of EDS and episodes of transient generalized or focal weakness associated with emotions suggesting possible cataplexy.
Diagnostic work-up included a clinical evaluation with a standardized interview performed by a neurologist trained in sleep disorders (G.P., F.P.), a standardized test to document cataplexy including a semi-structured interview on episodes phenomenology [19], 48-hour continuous polysomnography (PSG) followed by a fixed 5-naps multiple sleep latency test (MSLT) [20, 21], a blood drawn to search for HLA-DQB1*0601 positivity, and, whenever possible, lumbar puncture to measure CSF-hcrt-1 levels. All participants underwent brain magnetic resonance imaging to rule out lesions accountable for a secondary form of narcolepsy.
At the time of the diagnostic work-up, all participants were either drug-naïve or withdrawn from stimulants, anti-cataplectic, and antidepressant medications for at least 14 days.
The diagnosis of NT1 was based on the International Classification of Sleep Disorders, third edition (ICSD-3) [1], including CSF-hcrt-1 levels below 110 pg/mL and documented cataplexy at the in-laboratory test [1, 19]. The diagnosis of FND with pseudocataplexy relied on the description of repeated sudden transient falls or focal loss of strength, with preserved consciousness, and exclusion of a neurological disorder by means of the following criteria: (1) ruling out narcolepsy by polysomnographic findings, (2) normal CSF-hcrt-1 levels, (3) normal muscle tone on chin-EMG during documented focal and generalized attacks, (4) persistence of deep tendon reflexes when tested during generalized attacks, and (5) normal neurological examination. All patients with FND were subsequently referred to psychiatric consultation for a clinical assessment after the diagnostic work-up for suspected narcolepsy.
Sleep and cataplexy assessment
From the diagnostic work-up, the following data of PSG recording (second 24 hours after adaptation) and MSLT were collected: nocturnal Sleep Latency (nSL, min), nocturnal rapid eye movement (REM) sleep latency (nREML, min), nocturnal sleep efficiency (nSE, %), nocturnal total sleep time (nTST, min), nocturnal time in bed (nTIB), percent of TST spent in Non-REM sleep stage 1 (nN1TST, %), in Non-REM sleep stage 2 (nN2TST, %), in Non-REM sleep stage 3 (nN3TST, %), and in REM sleep (nREMTST, %), daytime number of naps (dNAP), number of spontaneous sleep onset REM periods (d-SOREMPs), daytime TST (d-TST), mean MSLT sleep latency (MSLT-SL), and number of MSLT-SOREMPs (MSLT-SOREMPs). We also collected results of CSF-hcrt-1 assay and HLA-DQB1*0602 allele positivity.
Clinical assessment of cataplexy included standardized information on relation of episodes with emotional triggers, type of triggers, frequency of spells, duration of spells, and topographical distribution of perceived weakness.
For each of the included patients, cataplexy and pseudocataplexy spells were documented by video-PSG standard test, including the evidence of chin-EMG atonia for cataplexy and persistence of muscle tone for pseudocataplexy as reported in the methods of previous works [2, 19]. None of the patients showed at diagnosis the co-existence of both cataplexy and pseudocataplexy at the test.
Psychiatric assessment
Participants were administered the following set of psychometric tools along with diagnostic hospitalization or outpatient clinic follow-up visits.
Beck Depression Inventory (BDI) is a 21-question multiple-choice self-report inventory that measures the presence and severity of depressive symptoms [22]. The current version is adapted to the diagnostic criteria of a major depressive episode according to the fourth version of the DSM. The cutoffs used for the total score are: 0–13 minimal depression, 14–19 mild depression, 20–28 moderate, and 29–63 severe depression. We applied the cutoff of > 19 to define patients with clinically relevant depressive symptoms.
State-Trait Anxiety Inventory (STAI) evaluates state anxiety (STAI-Y1 scale) and trait anxiety (STAI-Y2 scale), each scale consisting of 20 items [23]. Total score is set between 20 and 80. Predictive threshold for symptomatic anxiety is set above 40. Severity scales with score: 40–50 minor anxiety, 51–60 moderate anxiety, >61 high level anxiety.
Patient Health Questionnaire-15 (PHQ-15) is a brief self-administered instrument developed for screening for somatization [24]. It encompasses 15 somatic symptoms from the PHQ, each symptom scored 0= “not bothered at all,” 1= “bothered a little,” or 2= “bothered a lot.” PHQ-15 scores of 5, 10, and 15 represent cutoff points for low, medium, and high somatic symptom severity, respectively.
Personality Inventory for DSM-5 Brief Form (PID-5-BF) [25], derived from the original 220-item PID-5 version, consists of 25 items for the assessment of maladaptive personality traits, according to the Alternative Model of Personality Disorder developed in the DSM-5 [6]. These traits are described through 5 domains: negative affectivity, detachment, antagonism, disinhibition, and psychoticism. Each item can be assigned a Likert score from 0 = “always false/ often false” to 3 = “always true/ often true.” The mean scores were converted into normalized T points using the appropriate conversion tables [25].
Medical Outcome Study 36-item Short Form health survey (SF-36) measures health-related quality of life through 8 scales: physical activity, role and physical health, health in general, pain, vitality, role and emotional state, mental health, social activities. The questions and subscales of the SF-36 are organized in such a way that the higher the score, the better the health of the participant. Synthetic indices (Physical Component Summary, PCS, Mental Component Summary, MCS) that globally describe the state of physical and mental health were obtained from the weighted aggregation of the different subscales [26].
Statistical analysis
Descriptive statistics are presented as N (%) or mean ± SD for categorical or continuous variables respectively. Clinical, neurophysiological, and psychiatric items were compared between patients with pseudocataplexy and with NT1. Comparisons were performed with non-parametrical approaches using Chi-Square or Mann–Whitney U test for categorical and continuous variables, respectively. A P-value of ≤ 0.05 was considered statistically significant. Analyses were performed with SPSS, 23rd version.
Standard protocol approvals, registrations, and patient consent
The study was approved by the local ethical committee (number 17009). All patients signed written informed consent. Participants with writing, reading or text comprehension inability were excluded.
Reporting guideline statement
Study methods and results are reported following the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE).
Results
Patients
Fifteen patients with pseudocataplexy and 30 patients with NT1 were enrolled. There was a greater female and lower male prevalence in the pseudocataplexy group (males 13.3% vs. 50%, p = 0.017), but no differences in age or education.
Clinical and polysomnographic features
Reported age at onset of self-reported episodes of suspected cataplexy did not differ, while the onset of EDS occurred earlier in NT1 patients. At the time of diagnostic evaluation, the severity of subjective sleepiness (measured with the Epworth Sleepiness Scale) did not differ between groups.
As expected from diagnostic features, compared with the pseudocataplexy group, in NT1 group the HLA DQB1*0602 allele was more frequent, and CSF-hcrt-1 values were lower (CSF was available for 13/15 and 28/30 patients in the pseudocataplexy and NT1 group, respectively). NT1 patients also showed lower MSLT-SL compared to pseudocataplexy participants. In the pseudocataplexy group, five participants had MSLT-SL ≤ 10 minutes (4 below 8 minutes, and one between 8 and 10 minutes), one of them carried the HLA-DQB1*06:02 haplotype, while two other participants (both HLA-DQB1*06:02 negative) presented one spontaneous daytime nap with a SOREMP, but normal MSLT. These seven participants, and the other two with positive HLA-DQB1*0602, presented normal levels of CSF-hcrt-1. All these cases were evaluated by neurologists experienced in the field of narcolepsy (F.P., G.P.) and NT1 was ruled out. In only one case of the four patients with MSLT-SL < 8 minutes the shortened sleep latency was confirmed at follow-up reevaluation.
At PSG monitoring, NT1 patients showed a higher d-TST with more d-SOREMPs than participants with pseudocataplexy, despite comparable d-nNAP. During nighttime NT1 patients showed shorter REML and higher representation of nN1TST compared to participants with pseudocataplexy, without any further significant difference. Clinical and polysomnographic data are shown in Table 1.
Clinical, Laboratory, and Polysomnographic Features
| Pseudocataplexy (n = 15) | NT1 (n = 30) | ||||
|---|---|---|---|---|---|
| Clinical Features | Mean or % | SD | Mean or % | SD | P-Value |
| Male Gender, % | 13.30 | 50.00 | 0.017 | ||
| Age, years | 39.60 | 14.53 | 35.40 | 12.96 | 0.460 |
| Education, years | 13.13 | 2.64 | 13.25 | 2.65 | 0.805 |
| Cataplexy onset, years | 27.40 | 11.64 | 17.73 | 8.57 | 0.113 |
| EDS onset, years | 28.46 | 15.91 | 17.04 | 10.11 | 0.034 |
| Disease duration*, years | 15.25 | 16.70 | 17.38 | 9.07 | 0.135 |
| Epworth sleepiness scale, score | 12.47 | 5.21 | 15.22 | 4.52 | 0.089 |
| HLA DQB1*0602, % | 20.00 | 100 | <0.0001 | ||
| Lumbar puncture % | 86.70 | 93.30 | 0.459 | ||
| CSF-hcrt-1, pg/mL | 350.54 | 52.07 | 22.99 | 41.79 | <0.0001 |
| CSF-hcrt-1 ≤ 110 pg/mL % | 0.00 | 96.40 | <0.0001 | ||
| Polysomnographic Data | Mean | SD | Mean | SD | P-Value |
| MSLT-SL, min | 11.43 | 5.33 | 3.87 | 3.43 | 0.001 |
| MSLT-SL ≤ 8 min, % | 33.33 | 90.00 | <0.0001 | ||
| MSLT-SOREMPs, n | 0.00 | 0.00 | 3.60 | 1.52 | <0.0001 |
| nSL, min | 12.67 | 13.54 | 4.63 | 4.64 | 0.136 |
| nREML, min | 74.40 | 19.68 | 34.10 | 60.28 | <0.0001 |
| nSE, % | 84.34 | 9.63 | 84.90 | 11.13 | 0.525 |
| nTST, min | 411.23 | 65.83 | 430.19 | 85.96 | 1.00 |
| nTIB, min | 489.73 | 73.37 | 506.94 | 75.24 | 1.00 |
| nN1TST, % | 3.55 | 1.46 | 10.06 | 5.23 | <0.0001 |
| nN2TST, % | 37.95 | 9.62 | 41.04 | 9.04 | 0.520 |
| nN3TST, % | 34.72 | 9.38 | 26.30 | 8.73 | 0.198 |
| nREMTST, % | 23.76 | 6.59 | 22.61 | 6.17 | 1.00 |
| dNAP, n | 1.87 | 1.41 | 3.14 | 1.98 | 0.112 |
| d-SOREMPs, n | 0.20 | 0.56 | 2.11 | 1.59 | 0.004 |
| d-TST, min | 68.27 | 58.38 | 128.89 | 49.39 | 0.014 |
| Pseudocataplexy (n = 15) | NT1 (n = 30) | ||||
|---|---|---|---|---|---|
| Clinical Features | Mean or % | SD | Mean or % | SD | P-Value |
| Male Gender, % | 13.30 | 50.00 | 0.017 | ||
| Age, years | 39.60 | 14.53 | 35.40 | 12.96 | 0.460 |
| Education, years | 13.13 | 2.64 | 13.25 | 2.65 | 0.805 |
| Cataplexy onset, years | 27.40 | 11.64 | 17.73 | 8.57 | 0.113 |
| EDS onset, years | 28.46 | 15.91 | 17.04 | 10.11 | 0.034 |
| Disease duration*, years | 15.25 | 16.70 | 17.38 | 9.07 | 0.135 |
| Epworth sleepiness scale, score | 12.47 | 5.21 | 15.22 | 4.52 | 0.089 |
| HLA DQB1*0602, % | 20.00 | 100 | <0.0001 | ||
| Lumbar puncture % | 86.70 | 93.30 | 0.459 | ||
| CSF-hcrt-1, pg/mL | 350.54 | 52.07 | 22.99 | 41.79 | <0.0001 |
| CSF-hcrt-1 ≤ 110 pg/mL % | 0.00 | 96.40 | <0.0001 | ||
| Polysomnographic Data | Mean | SD | Mean | SD | P-Value |
| MSLT-SL, min | 11.43 | 5.33 | 3.87 | 3.43 | 0.001 |
| MSLT-SL ≤ 8 min, % | 33.33 | 90.00 | <0.0001 | ||
| MSLT-SOREMPs, n | 0.00 | 0.00 | 3.60 | 1.52 | <0.0001 |
| nSL, min | 12.67 | 13.54 | 4.63 | 4.64 | 0.136 |
| nREML, min | 74.40 | 19.68 | 34.10 | 60.28 | <0.0001 |
| nSE, % | 84.34 | 9.63 | 84.90 | 11.13 | 0.525 |
| nTST, min | 411.23 | 65.83 | 430.19 | 85.96 | 1.00 |
| nTIB, min | 489.73 | 73.37 | 506.94 | 75.24 | 1.00 |
| nN1TST, % | 3.55 | 1.46 | 10.06 | 5.23 | <0.0001 |
| nN2TST, % | 37.95 | 9.62 | 41.04 | 9.04 | 0.520 |
| nN3TST, % | 34.72 | 9.38 | 26.30 | 8.73 | 0.198 |
| nREMTST, % | 23.76 | 6.59 | 22.61 | 6.17 | 1.00 |
| dNAP, n | 1.87 | 1.41 | 3.14 | 1.98 | 0.112 |
| d-SOREMPs, n | 0.20 | 0.56 | 2.11 | 1.59 | 0.004 |
| d-TST, min | 68.27 | 58.38 | 128.89 | 49.39 | 0.014 |
* Duration from the reported age of onset of cataplexy/pseudocataplexy attacks.
EDS, Excessive daytime sleepiness; CSF-hcrt-1, Hypocretin-1 level in cerebral-spinal fluid; nSL, nocturnal Sleep Latency; nocturnal REM, rapid eye movement; nREML, sleep latency, nSE, nocturnal sleep efficiency;, nTST, nocturnal total sleep time; nTIB, nocturnal time in bed; nN1TST, nocturnal Non-REM sleep stage 1; nN2TST, Non-REM sleep stage 2; nN3TST, Non-REM sleep stage 3; nREMTST, nocturnal REM sleep; dNAP, daytime number of naps; d-SOREMPs, daytime spontaneous sleep onset REM periods; d-TST, daytime TST, MSLT, mean multiple sleep latency test; MSLT-SL, sleep latency; MSLT-SOREMPs, SOREMP during MSLT. In bold: P-Value < 0.05.
Clinical, Laboratory, and Polysomnographic Features
| Pseudocataplexy (n = 15) | NT1 (n = 30) | ||||
|---|---|---|---|---|---|
| Clinical Features | Mean or % | SD | Mean or % | SD | P-Value |
| Male Gender, % | 13.30 | 50.00 | 0.017 | ||
| Age, years | 39.60 | 14.53 | 35.40 | 12.96 | 0.460 |
| Education, years | 13.13 | 2.64 | 13.25 | 2.65 | 0.805 |
| Cataplexy onset, years | 27.40 | 11.64 | 17.73 | 8.57 | 0.113 |
| EDS onset, years | 28.46 | 15.91 | 17.04 | 10.11 | 0.034 |
| Disease duration*, years | 15.25 | 16.70 | 17.38 | 9.07 | 0.135 |
| Epworth sleepiness scale, score | 12.47 | 5.21 | 15.22 | 4.52 | 0.089 |
| HLA DQB1*0602, % | 20.00 | 100 | <0.0001 | ||
| Lumbar puncture % | 86.70 | 93.30 | 0.459 | ||
| CSF-hcrt-1, pg/mL | 350.54 | 52.07 | 22.99 | 41.79 | <0.0001 |
| CSF-hcrt-1 ≤ 110 pg/mL % | 0.00 | 96.40 | <0.0001 | ||
| Polysomnographic Data | Mean | SD | Mean | SD | P-Value |
| MSLT-SL, min | 11.43 | 5.33 | 3.87 | 3.43 | 0.001 |
| MSLT-SL ≤ 8 min, % | 33.33 | 90.00 | <0.0001 | ||
| MSLT-SOREMPs, n | 0.00 | 0.00 | 3.60 | 1.52 | <0.0001 |
| nSL, min | 12.67 | 13.54 | 4.63 | 4.64 | 0.136 |
| nREML, min | 74.40 | 19.68 | 34.10 | 60.28 | <0.0001 |
| nSE, % | 84.34 | 9.63 | 84.90 | 11.13 | 0.525 |
| nTST, min | 411.23 | 65.83 | 430.19 | 85.96 | 1.00 |
| nTIB, min | 489.73 | 73.37 | 506.94 | 75.24 | 1.00 |
| nN1TST, % | 3.55 | 1.46 | 10.06 | 5.23 | <0.0001 |
| nN2TST, % | 37.95 | 9.62 | 41.04 | 9.04 | 0.520 |
| nN3TST, % | 34.72 | 9.38 | 26.30 | 8.73 | 0.198 |
| nREMTST, % | 23.76 | 6.59 | 22.61 | 6.17 | 1.00 |
| dNAP, n | 1.87 | 1.41 | 3.14 | 1.98 | 0.112 |
| d-SOREMPs, n | 0.20 | 0.56 | 2.11 | 1.59 | 0.004 |
| d-TST, min | 68.27 | 58.38 | 128.89 | 49.39 | 0.014 |
| Pseudocataplexy (n = 15) | NT1 (n = 30) | ||||
|---|---|---|---|---|---|
| Clinical Features | Mean or % | SD | Mean or % | SD | P-Value |
| Male Gender, % | 13.30 | 50.00 | 0.017 | ||
| Age, years | 39.60 | 14.53 | 35.40 | 12.96 | 0.460 |
| Education, years | 13.13 | 2.64 | 13.25 | 2.65 | 0.805 |
| Cataplexy onset, years | 27.40 | 11.64 | 17.73 | 8.57 | 0.113 |
| EDS onset, years | 28.46 | 15.91 | 17.04 | 10.11 | 0.034 |
| Disease duration*, years | 15.25 | 16.70 | 17.38 | 9.07 | 0.135 |
| Epworth sleepiness scale, score | 12.47 | 5.21 | 15.22 | 4.52 | 0.089 |
| HLA DQB1*0602, % | 20.00 | 100 | <0.0001 | ||
| Lumbar puncture % | 86.70 | 93.30 | 0.459 | ||
| CSF-hcrt-1, pg/mL | 350.54 | 52.07 | 22.99 | 41.79 | <0.0001 |
| CSF-hcrt-1 ≤ 110 pg/mL % | 0.00 | 96.40 | <0.0001 | ||
| Polysomnographic Data | Mean | SD | Mean | SD | P-Value |
| MSLT-SL, min | 11.43 | 5.33 | 3.87 | 3.43 | 0.001 |
| MSLT-SL ≤ 8 min, % | 33.33 | 90.00 | <0.0001 | ||
| MSLT-SOREMPs, n | 0.00 | 0.00 | 3.60 | 1.52 | <0.0001 |
| nSL, min | 12.67 | 13.54 | 4.63 | 4.64 | 0.136 |
| nREML, min | 74.40 | 19.68 | 34.10 | 60.28 | <0.0001 |
| nSE, % | 84.34 | 9.63 | 84.90 | 11.13 | 0.525 |
| nTST, min | 411.23 | 65.83 | 430.19 | 85.96 | 1.00 |
| nTIB, min | 489.73 | 73.37 | 506.94 | 75.24 | 1.00 |
| nN1TST, % | 3.55 | 1.46 | 10.06 | 5.23 | <0.0001 |
| nN2TST, % | 37.95 | 9.62 | 41.04 | 9.04 | 0.520 |
| nN3TST, % | 34.72 | 9.38 | 26.30 | 8.73 | 0.198 |
| nREMTST, % | 23.76 | 6.59 | 22.61 | 6.17 | 1.00 |
| dNAP, n | 1.87 | 1.41 | 3.14 | 1.98 | 0.112 |
| d-SOREMPs, n | 0.20 | 0.56 | 2.11 | 1.59 | 0.004 |
| d-TST, min | 68.27 | 58.38 | 128.89 | 49.39 | 0.014 |
* Duration from the reported age of onset of cataplexy/pseudocataplexy attacks.
EDS, Excessive daytime sleepiness; CSF-hcrt-1, Hypocretin-1 level in cerebral-spinal fluid; nSL, nocturnal Sleep Latency; nocturnal REM, rapid eye movement; nREML, sleep latency, nSE, nocturnal sleep efficiency;, nTST, nocturnal total sleep time; nTIB, nocturnal time in bed; nN1TST, nocturnal Non-REM sleep stage 1; nN2TST, Non-REM sleep stage 2; nN3TST, Non-REM sleep stage 3; nREMTST, nocturnal REM sleep; dNAP, daytime number of naps; d-SOREMPs, daytime spontaneous sleep onset REM periods; d-TST, daytime TST, MSLT, mean multiple sleep latency test; MSLT-SL, sleep latency; MSLT-SOREMPs, SOREMP during MSLT. In bold: P-Value < 0.05.
Cataplectic symptoms phenomenology
The characteristics of self-reported episodes of suspected cataplexy are presented in Table 2. The relation of spells with emotions differed between groups, with NT1 patients more frequently reporting the attacks constantly triggered by emotions(70.0%), while participants with pseudocataplexy described spells triggered by emotions most of the time (60.0%) or never (13.3%). Despite the strong significance (p = 0.008), several patients overlapped in this item, with 26.7% of the pseudocataplexy group reporting a constant dependence on spells from emotional triggers, and 30.0% of NT1 patients claiming that most of the time emotions played a triggering role.
Self-Reported Features of Cataplexy. In bold: P-Value < 0.05.
| Pseudocataplexy (n = 15) | NT1 (n = 30) | ||
|---|---|---|---|
| Relation with emotional triggers | % | % | P-Value |
| Cataplexy triggered by emotions—Never | 13.30 | 0.00 | 0.008 |
| Cataplexy triggered by emotions—Most of times | 60.00 | 30.00 | |
| Cataplexy triggered by emotions—Always | 26.70 | 70.00 | |
| Triggers | % | % | P-Value |
| Cataplexy—Laughter | 33.30 | 100 | <0.0001 |
| Cataplexy—Telling Jokes | 13.30 | 46.70 | 0.028 |
| Cataplexy—Anger | 66.70 | 50.00 | 0.289 |
| Cataplexy—Surprise | 60.00 | 50.00 | 0.526 |
| Cataplexy—Meeting someone unexpectedly | 40.00 | 30.00 | 0.502 |
| Frequency | % | % | P-Value |
| Cataplexy Frequency: <1/y | 0.00 | 3.30 | 0.597 |
| Cataplexy Frequency: 1/y-1/m | 13.30 | 13.30 | |
| Cataplexy Frequency: 1/m-1/w | 26.70 | 16.70 | |
| Cataplexy Frequency: 1/w-1/d | 40.00 | 26.70 | |
| Cataplexy Frequency: >1/d | 20.00 | 40.00 | |
| Duration | % | % | P-Value |
| Cataplexy Duration: <10 s | 13.30 | 53.30 | 0.008 |
| Cataplexy Duration: 10s–2min | 33.30 | 36.70 | |
| Cataplexy Duration: 2–10 min | 26.70 | 3.30 | |
| Cataplexy Duration: >10 min | 26.70 | 6.70 | |
| Distribution | % | % | P-Value |
| Cataplexy—Generalized | 73.30 | 60.00 | 0.378 |
| Cataplexy—Face/Jaw | 40.00 | 86.70 | 0.001 |
| Cataplexy—Head | 26.70 | 46.70 | 0.197 |
| Cataplexy—Upper Limbs | 66.70 | 76.70 | 0.475 |
| Cataplexy—Lower Limbs | 60.00 | 53.30 | 0.671 |
| Cataplexy—Unilateral | 6.60 | 0.00 | 0.153 |
| Pseudocataplexy (n = 15) | NT1 (n = 30) | ||
|---|---|---|---|
| Relation with emotional triggers | % | % | P-Value |
| Cataplexy triggered by emotions—Never | 13.30 | 0.00 | 0.008 |
| Cataplexy triggered by emotions—Most of times | 60.00 | 30.00 | |
| Cataplexy triggered by emotions—Always | 26.70 | 70.00 | |
| Triggers | % | % | P-Value |
| Cataplexy—Laughter | 33.30 | 100 | <0.0001 |
| Cataplexy—Telling Jokes | 13.30 | 46.70 | 0.028 |
| Cataplexy—Anger | 66.70 | 50.00 | 0.289 |
| Cataplexy—Surprise | 60.00 | 50.00 | 0.526 |
| Cataplexy—Meeting someone unexpectedly | 40.00 | 30.00 | 0.502 |
| Frequency | % | % | P-Value |
| Cataplexy Frequency: <1/y | 0.00 | 3.30 | 0.597 |
| Cataplexy Frequency: 1/y-1/m | 13.30 | 13.30 | |
| Cataplexy Frequency: 1/m-1/w | 26.70 | 16.70 | |
| Cataplexy Frequency: 1/w-1/d | 40.00 | 26.70 | |
| Cataplexy Frequency: >1/d | 20.00 | 40.00 | |
| Duration | % | % | P-Value |
| Cataplexy Duration: <10 s | 13.30 | 53.30 | 0.008 |
| Cataplexy Duration: 10s–2min | 33.30 | 36.70 | |
| Cataplexy Duration: 2–10 min | 26.70 | 3.30 | |
| Cataplexy Duration: >10 min | 26.70 | 6.70 | |
| Distribution | % | % | P-Value |
| Cataplexy—Generalized | 73.30 | 60.00 | 0.378 |
| Cataplexy—Face/Jaw | 40.00 | 86.70 | 0.001 |
| Cataplexy—Head | 26.70 | 46.70 | 0.197 |
| Cataplexy—Upper Limbs | 66.70 | 76.70 | 0.475 |
| Cataplexy—Lower Limbs | 60.00 | 53.30 | 0.671 |
| Cataplexy—Unilateral | 6.60 | 0.00 | 0.153 |
Self-Reported Features of Cataplexy. In bold: P-Value < 0.05.
| Pseudocataplexy (n = 15) | NT1 (n = 30) | ||
|---|---|---|---|
| Relation with emotional triggers | % | % | P-Value |
| Cataplexy triggered by emotions—Never | 13.30 | 0.00 | 0.008 |
| Cataplexy triggered by emotions—Most of times | 60.00 | 30.00 | |
| Cataplexy triggered by emotions—Always | 26.70 | 70.00 | |
| Triggers | % | % | P-Value |
| Cataplexy—Laughter | 33.30 | 100 | <0.0001 |
| Cataplexy—Telling Jokes | 13.30 | 46.70 | 0.028 |
| Cataplexy—Anger | 66.70 | 50.00 | 0.289 |
| Cataplexy—Surprise | 60.00 | 50.00 | 0.526 |
| Cataplexy—Meeting someone unexpectedly | 40.00 | 30.00 | 0.502 |
| Frequency | % | % | P-Value |
| Cataplexy Frequency: <1/y | 0.00 | 3.30 | 0.597 |
| Cataplexy Frequency: 1/y-1/m | 13.30 | 13.30 | |
| Cataplexy Frequency: 1/m-1/w | 26.70 | 16.70 | |
| Cataplexy Frequency: 1/w-1/d | 40.00 | 26.70 | |
| Cataplexy Frequency: >1/d | 20.00 | 40.00 | |
| Duration | % | % | P-Value |
| Cataplexy Duration: <10 s | 13.30 | 53.30 | 0.008 |
| Cataplexy Duration: 10s–2min | 33.30 | 36.70 | |
| Cataplexy Duration: 2–10 min | 26.70 | 3.30 | |
| Cataplexy Duration: >10 min | 26.70 | 6.70 | |
| Distribution | % | % | P-Value |
| Cataplexy—Generalized | 73.30 | 60.00 | 0.378 |
| Cataplexy—Face/Jaw | 40.00 | 86.70 | 0.001 |
| Cataplexy—Head | 26.70 | 46.70 | 0.197 |
| Cataplexy—Upper Limbs | 66.70 | 76.70 | 0.475 |
| Cataplexy—Lower Limbs | 60.00 | 53.30 | 0.671 |
| Cataplexy—Unilateral | 6.60 | 0.00 | 0.153 |
| Pseudocataplexy (n = 15) | NT1 (n = 30) | ||
|---|---|---|---|
| Relation with emotional triggers | % | % | P-Value |
| Cataplexy triggered by emotions—Never | 13.30 | 0.00 | 0.008 |
| Cataplexy triggered by emotions—Most of times | 60.00 | 30.00 | |
| Cataplexy triggered by emotions—Always | 26.70 | 70.00 | |
| Triggers | % | % | P-Value |
| Cataplexy—Laughter | 33.30 | 100 | <0.0001 |
| Cataplexy—Telling Jokes | 13.30 | 46.70 | 0.028 |
| Cataplexy—Anger | 66.70 | 50.00 | 0.289 |
| Cataplexy—Surprise | 60.00 | 50.00 | 0.526 |
| Cataplexy—Meeting someone unexpectedly | 40.00 | 30.00 | 0.502 |
| Frequency | % | % | P-Value |
| Cataplexy Frequency: <1/y | 0.00 | 3.30 | 0.597 |
| Cataplexy Frequency: 1/y-1/m | 13.30 | 13.30 | |
| Cataplexy Frequency: 1/m-1/w | 26.70 | 16.70 | |
| Cataplexy Frequency: 1/w-1/d | 40.00 | 26.70 | |
| Cataplexy Frequency: >1/d | 20.00 | 40.00 | |
| Duration | % | % | P-Value |
| Cataplexy Duration: <10 s | 13.30 | 53.30 | 0.008 |
| Cataplexy Duration: 10s–2min | 33.30 | 36.70 | |
| Cataplexy Duration: 2–10 min | 26.70 | 3.30 | |
| Cataplexy Duration: >10 min | 26.70 | 6.70 | |
| Distribution | % | % | P-Value |
| Cataplexy—Generalized | 73.30 | 60.00 | 0.378 |
| Cataplexy—Face/Jaw | 40.00 | 86.70 | 0.001 |
| Cataplexy—Head | 26.70 | 46.70 | 0.197 |
| Cataplexy—Upper Limbs | 66.70 | 76.70 | 0.475 |
| Cataplexy—Lower Limbs | 60.00 | 53.30 | 0.671 |
| Cataplexy—Unilateral | 6.60 | 0.00 | 0.153 |
Patients with NT1 more frequently recognized a triggering role of laughter (100.0% vs. 33.0%, p < 0.0001), and telling jokes (46.7% vs. 13.3%, p = 0.028) than those with pseudocataplexy. Other emotional triggers (anger, surprise, unexpected meeting), as well as spells frequency, did not differ between the two groups. The episodes were more commonly of short duration in NT1 (lasting < 10 seconds in 53.3% and between 10 seconds and 2 minutes in 36.7% of NT1 patients; lasting between 2 and 10 minutes in 26.7% and above 10 minutes in 26.7% of pseudocataplexy participants; p = 0.008). However, episode duration showed meaningful overlaps with 13.3% and 33.3% of pseudocataplexy patients reporting very short (<10 seconds) or short-lasting (10 seconds–2 minutes) attacks’ duration, respectively.
Concerning self-reported topographical distribution of weakness, NT1 patients more frequently experienced involvement of the facial district than participants with pseudocataplexy (86.7% vs. 40.0%, p = 0.001), while all other focal (and generalized) localizations items did not differ between groups. Attacks were described as bilateral/symmetric by all NT1 and all but one pseudocataplexy patient (n.s.).
Psychiatric assessment
Patients with pseudocataplexy showed higher scores on BDI, STAI-Y1, STAI-Y2, and PHQ-15 compared to control group, although there were no statistically significant differences (Figure 1). Higher rate of moderate state anxiety (40.0% vs. 10.0%, p = 0.018), medium level of somatic symptoms (66.7% vs. 16.7%, p = 0.003), and a trend towards moderate to severe depressive symptoms (33.3% vs. 10%, p = 0.054) were found in patients with pseudocataplexy compared to NT1 group (Figure 2). Considering personality domains evaluated with PID-5, no differences emerged in pseudocataplectic patients compared with the control group.
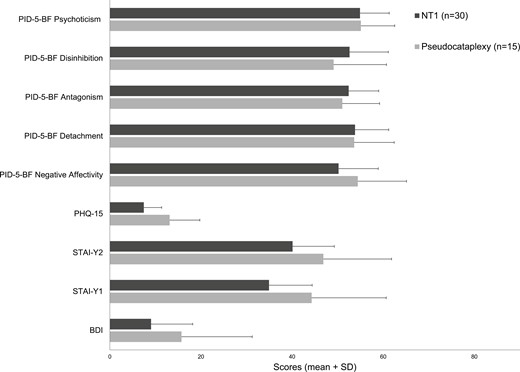
Mean scores in Beck Depression Inventory (BDI), State-Trait Anxiety Inventory for state anxiety (STAI-Y1) and trait anxiety (STAI-Y2), Patient Health Questionnaire-15 (PHQ-15) and Personality Inventory for DSM-5 Brief Form (PID-5-BF) (* P-value < 0.05).
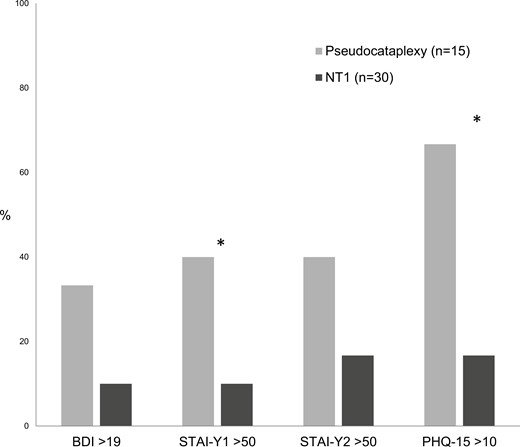
Percentage of participants with impaired scores (moderate-to-severe) in Beck Depression Inventory (BDI), State-Trait Anxiety Inventory for state anxiety (STAI-Y1) and trait anxiety (STAI-Y2) and Patient Health Questionnaire-15 (PHQ-15) (* P-value < 0.05).
Patients with pseudocataplexy presented significantly worse scores in all domains of SF-36, except mental health. Also, the PCS and MCS resulted in significantly lower in participants with pseudocataplexy (Figure 3). Full results of the psychiatric assessment are reported in Supplementary Material 1.
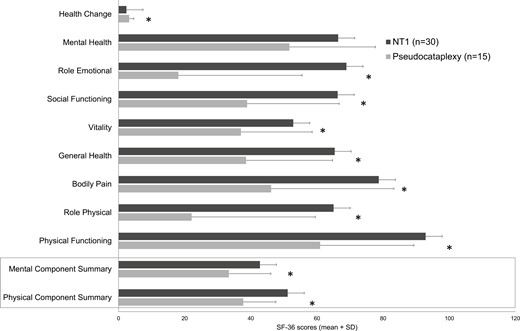
Mean scores in domains of Medical Outcome Study 36-item Short Form health survey (SF-36) and in Mental and Physical Component Summary (* P-value < 0.05).
Discussion
The identification of cataplexy and the differential diagnosis with its mimics are essentially based on the history of the episodes referred by the patients and their caregivers, as stated in the ICSD-3 criteria for narcolepsy [1], and further recommended by expert opinion focusing on the distinction between typical and “atypical” features of cataplexy [9].
Overall, distinguishing cataplexy from pseudocataplexy, among the other mimics, can be problematic in individual cases, given a partial clinical overlap, and point to the need for the documentation of the episode and of multidisciplinary assessment for proper differential diagnosis and subsequent management.
In this study, consistent with literature, some of the self-reported characteristics of the attacks differ between cataplexy and pseudocataplexy: the latter can occur unrelated to emotions, less frequently triggered by laughter and telling jokes, less frequently involve focal face or jaw weakness, and tend to have a longer duration. However, the clinical overlap between self-reported spells features prevents the definite diagnosis of cataplexy/pseudocataplexy based on interview only [2, 4]. In a previous study, we addressed the semiology of cataplectic and pseudocataplectic episodes documented with video recording and found a set of peculiarities that can guide the clinician to an objective positive diagnosis [2]. Differences in HLA-DQB1*06-02 status, CSF-hrct-1 level, and polysomnographic features mostly depend on inclusion and exclusion criteria and correspond to distinctive characteristics of NT1 as well as to the absence of NT1 cases presenting both cataplexy and pseudocataplexy in our series [27].
Focusing on triggers, laughter is associates with pseudocataplectic attacks in a minority of cases, whereas it is more often triggered by anger and surprise (without significant difference with NT1). Similarly, a recent investigation on triggered FMDs (not directly describing pseudocataplexy, but with paroxysmal weakness) found that in 37.5% of cases, the motor symptoms could be provoked by negative emotional stimuli [28]. The authors defined some of these FMDs as paroxysmal FMD, characterized by sudden episodes of FMD lasting for a brief but variable duration with periods during which the movement was noted by the patient to be absent. However, a non-negligible overlap of features appears between cataplexy and pseudocataplexy: in up to half of NT1 patients, cataplexy can be triggered by anger and emotional stimuli other than laughter, while prolonged attacks can occur in rare NT1 cases, though they are considered more suggestive for somatization. On the other hand, pseudocataplectic attacks can present a focal distribution. Likewise, Overeem et al. investigated features of cataplexy in patients with proven CSF-hrct-1 deficiency and highlighted how the “atypical” elements of cataplexy (e.g. prolonged attacks, spontaneous or triggered by non-humorous emotions) are frequently reported by patients together with also “typical” ones [14]. Table 3 provides an overview of the features that help the differential diagnosis between cataplexy, pseudocataplexy, and the other possible mimics in clinical practice.
Overview of Differential Diagnosis of Cataplexy, Pseudocataplexy, and Other Possible Mimicking Disorders
| Cataplexy (typical of NT1) | Pseudocataplexy | Syncope | Vertebrobasilar insufficiency | Epilepsy | Catatonia, hypokinetic subtype | Hyperekplexia | Psychomotor retardation | |
|---|---|---|---|---|---|---|---|---|
| Triggers | Emotions: mainly strong positive (e.g. laughing, telling jokes, and surprise). Spontaneous rarely Childhood phenotype: cataplectic facies; ataxic gate; falls unrelated to triggers. | Wide range of emotions (mostly negative). More attacks without clear triggers. | Neuro-mediated: (1) prolonged sitting or standing (2) emotions as fear, pain, hematophobia (3) Valsalva maneuver (4) Carotid sinus manipulation (5) Neck stretching Orthostatic hypotension: (6) drugs (7) volume depletion (8) rapid rising to upright posture. Cardiac syncope: (9) absent trigger (10) physical effort (11) drugs [29] | Typically absent. Favoured by reduced brain blood flow. | Often absent. Rarely triggered (reflex epilepsies): sensory stimuli, eating, complex tasks. Facilitating factors: stress, sleep deprivation, drug withdrawal, and fever. | Usually absent. Predisposing and precipitating factors are a wide range of psychiatric, neurological, metabolic, inflammatory conditions, or medications. | Unexpected visual, auditory, or tactile stimuli. | Usually absent. Startle reflex is possible. |
| Duration of episodes | Brief (seconds to 1–2 minutes). Rarely longer (status cataplecticus) | Variable, frequently lasting several minutes (>2 minutes) | Brief (seconds to 1–2 minutes) | Brief (seconds to minutes) | Ultra brief (msec) in negative myoclonus, very brief (<2 seconds) for isolated seizures. Prolonged in cases of status epilepticus. | From days to weeks. | Very brief (milliseconds to seconds) | Days to weeks |
| Recovery from episodes | Rapid and complete. Possible transition into sleep. | Variable (seconds to several minutes), may be followed by prolonged malaise. | Rapid (few seconds). Post-ictal signs: sweating, headache, nausea, and asthenia. | Rapid, although some patients may require several minutes to stand independently. | Variable, up to several minutes, with possible post-ictal confusion. | Gradual in days-to-weeks. Recovery often needs treatment and can be incomplete. | Immediate. | Gradual in days to weeks |
| Distribution/ laterality | Symmetric (mostly). Focal (especially face, limbs, and head-neck). Or generalized spells with typical rostro-caudal progression of hypotonia (the face involvement is a clue). | Often sudden generalized attacks (with preserved deep tendon reflexes). Focal spells rarely involving oro-facial district. | Generalized. | Usually generalized with possible fall (drop attack) | Usually axial distribution involving head nod or generalized with fall. Possible focal atonia (e.g. in Rasmussen syndrome). | Usually generalized, with immobility and waxy rigidity. | Variable: it can involve facial grimacing, abduction of the shoulders and flexion of the neck, trunk, elbows, and knees, resulting in uncontrolled falling. | Usually generalized |
| Associated symptoms | Preserved consciousness. Abolished deep tendon reflexes during generalized spells. | Multiple somatoform symptoms (headache, back pain, dizziness, fainting spells, and variable gastro-intestinal symptoms). | Loss of consciousness Prodromal vegetative signs and symptoms (nausea, palpitations, warm sensation, and lightheadedness), visual blurring. Possible limb irregular and brief shaking if syncope is prolonged. | Possible incoordination/ataxia, cranial nerve involvement. Consciousness usually preserved. | Consciousness can be altered or absent in atonic or absence seizures. Other types of seizures (spasms, myoclonic, tonic-clonic, absences, . . . ) or co-occurrence of different pattern in the same seizure (e.g. myoclonic-atonic, evolution to generalized tonic-clonic) Additional signs: tongue biting, urinary incontinence, cyanosis, and post-ictal confusion. | Negativism (including refusal to eat and drink) and mutism. Waxy flexibility, catalepsy and posturing in some cases. | Preserved consciousness. Generalized stiffness after birth; excessive startling to an unexpected stimulus. Cataplexy can overlap in Coffin Lowry syndrome [30]. | Slowed reaction time, speech abnormalities (in particular, speed), severe cognitive multi-domain deficits. |
| HLA status | Protective and associated HLA HLA-DQB1*0602 positive in 96% [31] HLA-DQB1*0602 negativity: red flag for secondary narcolepsy. | NA | NA | NA | NA | NA | NA | NA |
| Neurophysiological features | EEG: wake pattern during attacks. EMG: loss of tone and waxing-and waning EMG activity, rostro-caudal spreading of atonia. Sleep studies: (1) MSLT with reduced sleep latency and multiple SOREMPs, (2) night PSG with SOREMP, fragmented sleep, REM sleep without atonia. | EMG: persisting muscle tone during attacks. Sleep studies: although overlap with CNS hypersomnia is possible, MSLT and PSG exclude narcolepsy, | EEG: generalized slowing of the background activity during syncope. Autonomic function and cardiological tests: variably altered depending on the pathological mechanism; they can also be normal [32]. | Color-doppler studies may disclose flow abnormalities and stenosis. | EEG: focal or generalized epileptic activity during atonic phase. EMG: atonia synchronous with epileptic discharges. | EEG: normal. Possible abnormalities and seizures in neurological conditions | EEG: normal (except if in encephalopathy or encephalitis) EMG: bilaterally synchronous “shock-like set of movements” and generalized stiffness after a startle reflex lasting a few seconds. | EEG: normal, may reveal focal, or diffuse slowing. |
| Psychiatric morbidity | Depressive and anxiety symptoms are frequent. | Moderate-to-severe depressive and anxiety symptoms. | As in the general population. | As in the general population. | Possible cognitive and behavioral developmental delay or regression. | Depressive or manic episodes, | Cognitive developmental delay in some genetic syndromes (e.g. Coffin Lowry syndrome)[33] | Severe depressive episodes. |
| Demographic features | Typical onset in childhood-adolescence, but it can occur at any age. No gender effect. | Variable from childhood-adolescence to adulthood, more frequently female. | Any age, variably depending on the etiology. | Adult and elderly people [34, 59]. Associated with cardiovascular risk factors/comorbidity. | Typical onset during childhood. | Children and adolescents with psychiatric, neurodevelopmental and neurological disorders [35]. Adult (mainly elderly) with psychiatric and neurological disorder. | Onset during childhood-adolescence for genetic syndromes Variable onset in acquired forms (e.g. autoimmune/paraneoplastic, neurodegenerative, and post-hypoxic) [36, 37] | Possible childhood onset |
| Diagnoses | NT1, secondary narcolepsy (Niemann-Pick type C, demyelinating diseases, and paraneoplastic encephalitis) [30]. | Functional neurological disorder, malingering. | Vasovagal syncope, cardiac syncope, primary and secondary orthostatic hypotension, postural orthostatic tachycardia syndrome, stretch syncope. | Cerebrovascular disorders. | Epilepsy with myoclonic-atonic seizures, Lennox-Gastaut syndrome [35, 38]. | Schizophrenia, other psychotic disorders, neurodevelopmental disorders, encephalitis (e.g. anti-NMDA-receptor) [36, 39]. | Genetic syndromes involving alteration of glycine-receptor pathways. Brainstem lesions. Autoimmune diseases [37, 40]. | Neurodevelopmental disorders, |
| Cataplexy (typical of NT1) | Pseudocataplexy | Syncope | Vertebrobasilar insufficiency | Epilepsy | Catatonia, hypokinetic subtype | Hyperekplexia | Psychomotor retardation | |
|---|---|---|---|---|---|---|---|---|
| Triggers | Emotions: mainly strong positive (e.g. laughing, telling jokes, and surprise). Spontaneous rarely Childhood phenotype: cataplectic facies; ataxic gate; falls unrelated to triggers. | Wide range of emotions (mostly negative). More attacks without clear triggers. | Neuro-mediated: (1) prolonged sitting or standing (2) emotions as fear, pain, hematophobia (3) Valsalva maneuver (4) Carotid sinus manipulation (5) Neck stretching Orthostatic hypotension: (6) drugs (7) volume depletion (8) rapid rising to upright posture. Cardiac syncope: (9) absent trigger (10) physical effort (11) drugs [29] | Typically absent. Favoured by reduced brain blood flow. | Often absent. Rarely triggered (reflex epilepsies): sensory stimuli, eating, complex tasks. Facilitating factors: stress, sleep deprivation, drug withdrawal, and fever. | Usually absent. Predisposing and precipitating factors are a wide range of psychiatric, neurological, metabolic, inflammatory conditions, or medications. | Unexpected visual, auditory, or tactile stimuli. | Usually absent. Startle reflex is possible. |
| Duration of episodes | Brief (seconds to 1–2 minutes). Rarely longer (status cataplecticus) | Variable, frequently lasting several minutes (>2 minutes) | Brief (seconds to 1–2 minutes) | Brief (seconds to minutes) | Ultra brief (msec) in negative myoclonus, very brief (<2 seconds) for isolated seizures. Prolonged in cases of status epilepticus. | From days to weeks. | Very brief (milliseconds to seconds) | Days to weeks |
| Recovery from episodes | Rapid and complete. Possible transition into sleep. | Variable (seconds to several minutes), may be followed by prolonged malaise. | Rapid (few seconds). Post-ictal signs: sweating, headache, nausea, and asthenia. | Rapid, although some patients may require several minutes to stand independently. | Variable, up to several minutes, with possible post-ictal confusion. | Gradual in days-to-weeks. Recovery often needs treatment and can be incomplete. | Immediate. | Gradual in days to weeks |
| Distribution/ laterality | Symmetric (mostly). Focal (especially face, limbs, and head-neck). Or generalized spells with typical rostro-caudal progression of hypotonia (the face involvement is a clue). | Often sudden generalized attacks (with preserved deep tendon reflexes). Focal spells rarely involving oro-facial district. | Generalized. | Usually generalized with possible fall (drop attack) | Usually axial distribution involving head nod or generalized with fall. Possible focal atonia (e.g. in Rasmussen syndrome). | Usually generalized, with immobility and waxy rigidity. | Variable: it can involve facial grimacing, abduction of the shoulders and flexion of the neck, trunk, elbows, and knees, resulting in uncontrolled falling. | Usually generalized |
| Associated symptoms | Preserved consciousness. Abolished deep tendon reflexes during generalized spells. | Multiple somatoform symptoms (headache, back pain, dizziness, fainting spells, and variable gastro-intestinal symptoms). | Loss of consciousness Prodromal vegetative signs and symptoms (nausea, palpitations, warm sensation, and lightheadedness), visual blurring. Possible limb irregular and brief shaking if syncope is prolonged. | Possible incoordination/ataxia, cranial nerve involvement. Consciousness usually preserved. | Consciousness can be altered or absent in atonic or absence seizures. Other types of seizures (spasms, myoclonic, tonic-clonic, absences, . . . ) or co-occurrence of different pattern in the same seizure (e.g. myoclonic-atonic, evolution to generalized tonic-clonic) Additional signs: tongue biting, urinary incontinence, cyanosis, and post-ictal confusion. | Negativism (including refusal to eat and drink) and mutism. Waxy flexibility, catalepsy and posturing in some cases. | Preserved consciousness. Generalized stiffness after birth; excessive startling to an unexpected stimulus. Cataplexy can overlap in Coffin Lowry syndrome [30]. | Slowed reaction time, speech abnormalities (in particular, speed), severe cognitive multi-domain deficits. |
| HLA status | Protective and associated HLA HLA-DQB1*0602 positive in 96% [31] HLA-DQB1*0602 negativity: red flag for secondary narcolepsy. | NA | NA | NA | NA | NA | NA | NA |
| Neurophysiological features | EEG: wake pattern during attacks. EMG: loss of tone and waxing-and waning EMG activity, rostro-caudal spreading of atonia. Sleep studies: (1) MSLT with reduced sleep latency and multiple SOREMPs, (2) night PSG with SOREMP, fragmented sleep, REM sleep without atonia. | EMG: persisting muscle tone during attacks. Sleep studies: although overlap with CNS hypersomnia is possible, MSLT and PSG exclude narcolepsy, | EEG: generalized slowing of the background activity during syncope. Autonomic function and cardiological tests: variably altered depending on the pathological mechanism; they can also be normal [32]. | Color-doppler studies may disclose flow abnormalities and stenosis. | EEG: focal or generalized epileptic activity during atonic phase. EMG: atonia synchronous with epileptic discharges. | EEG: normal. Possible abnormalities and seizures in neurological conditions | EEG: normal (except if in encephalopathy or encephalitis) EMG: bilaterally synchronous “shock-like set of movements” and generalized stiffness after a startle reflex lasting a few seconds. | EEG: normal, may reveal focal, or diffuse slowing. |
| Psychiatric morbidity | Depressive and anxiety symptoms are frequent. | Moderate-to-severe depressive and anxiety symptoms. | As in the general population. | As in the general population. | Possible cognitive and behavioral developmental delay or regression. | Depressive or manic episodes, | Cognitive developmental delay in some genetic syndromes (e.g. Coffin Lowry syndrome)[33] | Severe depressive episodes. |
| Demographic features | Typical onset in childhood-adolescence, but it can occur at any age. No gender effect. | Variable from childhood-adolescence to adulthood, more frequently female. | Any age, variably depending on the etiology. | Adult and elderly people [34, 59]. Associated with cardiovascular risk factors/comorbidity. | Typical onset during childhood. | Children and adolescents with psychiatric, neurodevelopmental and neurological disorders [35]. Adult (mainly elderly) with psychiatric and neurological disorder. | Onset during childhood-adolescence for genetic syndromes Variable onset in acquired forms (e.g. autoimmune/paraneoplastic, neurodegenerative, and post-hypoxic) [36, 37] | Possible childhood onset |
| Diagnoses | NT1, secondary narcolepsy (Niemann-Pick type C, demyelinating diseases, and paraneoplastic encephalitis) [30]. | Functional neurological disorder, malingering. | Vasovagal syncope, cardiac syncope, primary and secondary orthostatic hypotension, postural orthostatic tachycardia syndrome, stretch syncope. | Cerebrovascular disorders. | Epilepsy with myoclonic-atonic seizures, Lennox-Gastaut syndrome [35, 38]. | Schizophrenia, other psychotic disorders, neurodevelopmental disorders, encephalitis (e.g. anti-NMDA-receptor) [36, 39]. | Genetic syndromes involving alteration of glycine-receptor pathways. Brainstem lesions. Autoimmune diseases [37, 40]. | Neurodevelopmental disorders, |
EEG, electroencephalography; EMG, electromyography; PSG, polysomnography; MSLT, multiple sleep latency test; SOREMP, sleep onset rapid eye movement sleep period.
Overview of Differential Diagnosis of Cataplexy, Pseudocataplexy, and Other Possible Mimicking Disorders
| Cataplexy (typical of NT1) | Pseudocataplexy | Syncope | Vertebrobasilar insufficiency | Epilepsy | Catatonia, hypokinetic subtype | Hyperekplexia | Psychomotor retardation | |
|---|---|---|---|---|---|---|---|---|
| Triggers | Emotions: mainly strong positive (e.g. laughing, telling jokes, and surprise). Spontaneous rarely Childhood phenotype: cataplectic facies; ataxic gate; falls unrelated to triggers. | Wide range of emotions (mostly negative). More attacks without clear triggers. | Neuro-mediated: (1) prolonged sitting or standing (2) emotions as fear, pain, hematophobia (3) Valsalva maneuver (4) Carotid sinus manipulation (5) Neck stretching Orthostatic hypotension: (6) drugs (7) volume depletion (8) rapid rising to upright posture. Cardiac syncope: (9) absent trigger (10) physical effort (11) drugs [29] | Typically absent. Favoured by reduced brain blood flow. | Often absent. Rarely triggered (reflex epilepsies): sensory stimuli, eating, complex tasks. Facilitating factors: stress, sleep deprivation, drug withdrawal, and fever. | Usually absent. Predisposing and precipitating factors are a wide range of psychiatric, neurological, metabolic, inflammatory conditions, or medications. | Unexpected visual, auditory, or tactile stimuli. | Usually absent. Startle reflex is possible. |
| Duration of episodes | Brief (seconds to 1–2 minutes). Rarely longer (status cataplecticus) | Variable, frequently lasting several minutes (>2 minutes) | Brief (seconds to 1–2 minutes) | Brief (seconds to minutes) | Ultra brief (msec) in negative myoclonus, very brief (<2 seconds) for isolated seizures. Prolonged in cases of status epilepticus. | From days to weeks. | Very brief (milliseconds to seconds) | Days to weeks |
| Recovery from episodes | Rapid and complete. Possible transition into sleep. | Variable (seconds to several minutes), may be followed by prolonged malaise. | Rapid (few seconds). Post-ictal signs: sweating, headache, nausea, and asthenia. | Rapid, although some patients may require several minutes to stand independently. | Variable, up to several minutes, with possible post-ictal confusion. | Gradual in days-to-weeks. Recovery often needs treatment and can be incomplete. | Immediate. | Gradual in days to weeks |
| Distribution/ laterality | Symmetric (mostly). Focal (especially face, limbs, and head-neck). Or generalized spells with typical rostro-caudal progression of hypotonia (the face involvement is a clue). | Often sudden generalized attacks (with preserved deep tendon reflexes). Focal spells rarely involving oro-facial district. | Generalized. | Usually generalized with possible fall (drop attack) | Usually axial distribution involving head nod or generalized with fall. Possible focal atonia (e.g. in Rasmussen syndrome). | Usually generalized, with immobility and waxy rigidity. | Variable: it can involve facial grimacing, abduction of the shoulders and flexion of the neck, trunk, elbows, and knees, resulting in uncontrolled falling. | Usually generalized |
| Associated symptoms | Preserved consciousness. Abolished deep tendon reflexes during generalized spells. | Multiple somatoform symptoms (headache, back pain, dizziness, fainting spells, and variable gastro-intestinal symptoms). | Loss of consciousness Prodromal vegetative signs and symptoms (nausea, palpitations, warm sensation, and lightheadedness), visual blurring. Possible limb irregular and brief shaking if syncope is prolonged. | Possible incoordination/ataxia, cranial nerve involvement. Consciousness usually preserved. | Consciousness can be altered or absent in atonic or absence seizures. Other types of seizures (spasms, myoclonic, tonic-clonic, absences, . . . ) or co-occurrence of different pattern in the same seizure (e.g. myoclonic-atonic, evolution to generalized tonic-clonic) Additional signs: tongue biting, urinary incontinence, cyanosis, and post-ictal confusion. | Negativism (including refusal to eat and drink) and mutism. Waxy flexibility, catalepsy and posturing in some cases. | Preserved consciousness. Generalized stiffness after birth; excessive startling to an unexpected stimulus. Cataplexy can overlap in Coffin Lowry syndrome [30]. | Slowed reaction time, speech abnormalities (in particular, speed), severe cognitive multi-domain deficits. |
| HLA status | Protective and associated HLA HLA-DQB1*0602 positive in 96% [31] HLA-DQB1*0602 negativity: red flag for secondary narcolepsy. | NA | NA | NA | NA | NA | NA | NA |
| Neurophysiological features | EEG: wake pattern during attacks. EMG: loss of tone and waxing-and waning EMG activity, rostro-caudal spreading of atonia. Sleep studies: (1) MSLT with reduced sleep latency and multiple SOREMPs, (2) night PSG with SOREMP, fragmented sleep, REM sleep without atonia. | EMG: persisting muscle tone during attacks. Sleep studies: although overlap with CNS hypersomnia is possible, MSLT and PSG exclude narcolepsy, | EEG: generalized slowing of the background activity during syncope. Autonomic function and cardiological tests: variably altered depending on the pathological mechanism; they can also be normal [32]. | Color-doppler studies may disclose flow abnormalities and stenosis. | EEG: focal or generalized epileptic activity during atonic phase. EMG: atonia synchronous with epileptic discharges. | EEG: normal. Possible abnormalities and seizures in neurological conditions | EEG: normal (except if in encephalopathy or encephalitis) EMG: bilaterally synchronous “shock-like set of movements” and generalized stiffness after a startle reflex lasting a few seconds. | EEG: normal, may reveal focal, or diffuse slowing. |
| Psychiatric morbidity | Depressive and anxiety symptoms are frequent. | Moderate-to-severe depressive and anxiety symptoms. | As in the general population. | As in the general population. | Possible cognitive and behavioral developmental delay or regression. | Depressive or manic episodes, | Cognitive developmental delay in some genetic syndromes (e.g. Coffin Lowry syndrome)[33] | Severe depressive episodes. |
| Demographic features | Typical onset in childhood-adolescence, but it can occur at any age. No gender effect. | Variable from childhood-adolescence to adulthood, more frequently female. | Any age, variably depending on the etiology. | Adult and elderly people [34, 59]. Associated with cardiovascular risk factors/comorbidity. | Typical onset during childhood. | Children and adolescents with psychiatric, neurodevelopmental and neurological disorders [35]. Adult (mainly elderly) with psychiatric and neurological disorder. | Onset during childhood-adolescence for genetic syndromes Variable onset in acquired forms (e.g. autoimmune/paraneoplastic, neurodegenerative, and post-hypoxic) [36, 37] | Possible childhood onset |
| Diagnoses | NT1, secondary narcolepsy (Niemann-Pick type C, demyelinating diseases, and paraneoplastic encephalitis) [30]. | Functional neurological disorder, malingering. | Vasovagal syncope, cardiac syncope, primary and secondary orthostatic hypotension, postural orthostatic tachycardia syndrome, stretch syncope. | Cerebrovascular disorders. | Epilepsy with myoclonic-atonic seizures, Lennox-Gastaut syndrome [35, 38]. | Schizophrenia, other psychotic disorders, neurodevelopmental disorders, encephalitis (e.g. anti-NMDA-receptor) [36, 39]. | Genetic syndromes involving alteration of glycine-receptor pathways. Brainstem lesions. Autoimmune diseases [37, 40]. | Neurodevelopmental disorders, |
| Cataplexy (typical of NT1) | Pseudocataplexy | Syncope | Vertebrobasilar insufficiency | Epilepsy | Catatonia, hypokinetic subtype | Hyperekplexia | Psychomotor retardation | |
|---|---|---|---|---|---|---|---|---|
| Triggers | Emotions: mainly strong positive (e.g. laughing, telling jokes, and surprise). Spontaneous rarely Childhood phenotype: cataplectic facies; ataxic gate; falls unrelated to triggers. | Wide range of emotions (mostly negative). More attacks without clear triggers. | Neuro-mediated: (1) prolonged sitting or standing (2) emotions as fear, pain, hematophobia (3) Valsalva maneuver (4) Carotid sinus manipulation (5) Neck stretching Orthostatic hypotension: (6) drugs (7) volume depletion (8) rapid rising to upright posture. Cardiac syncope: (9) absent trigger (10) physical effort (11) drugs [29] | Typically absent. Favoured by reduced brain blood flow. | Often absent. Rarely triggered (reflex epilepsies): sensory stimuli, eating, complex tasks. Facilitating factors: stress, sleep deprivation, drug withdrawal, and fever. | Usually absent. Predisposing and precipitating factors are a wide range of psychiatric, neurological, metabolic, inflammatory conditions, or medications. | Unexpected visual, auditory, or tactile stimuli. | Usually absent. Startle reflex is possible. |
| Duration of episodes | Brief (seconds to 1–2 minutes). Rarely longer (status cataplecticus) | Variable, frequently lasting several minutes (>2 minutes) | Brief (seconds to 1–2 minutes) | Brief (seconds to minutes) | Ultra brief (msec) in negative myoclonus, very brief (<2 seconds) for isolated seizures. Prolonged in cases of status epilepticus. | From days to weeks. | Very brief (milliseconds to seconds) | Days to weeks |
| Recovery from episodes | Rapid and complete. Possible transition into sleep. | Variable (seconds to several minutes), may be followed by prolonged malaise. | Rapid (few seconds). Post-ictal signs: sweating, headache, nausea, and asthenia. | Rapid, although some patients may require several minutes to stand independently. | Variable, up to several minutes, with possible post-ictal confusion. | Gradual in days-to-weeks. Recovery often needs treatment and can be incomplete. | Immediate. | Gradual in days to weeks |
| Distribution/ laterality | Symmetric (mostly). Focal (especially face, limbs, and head-neck). Or generalized spells with typical rostro-caudal progression of hypotonia (the face involvement is a clue). | Often sudden generalized attacks (with preserved deep tendon reflexes). Focal spells rarely involving oro-facial district. | Generalized. | Usually generalized with possible fall (drop attack) | Usually axial distribution involving head nod or generalized with fall. Possible focal atonia (e.g. in Rasmussen syndrome). | Usually generalized, with immobility and waxy rigidity. | Variable: it can involve facial grimacing, abduction of the shoulders and flexion of the neck, trunk, elbows, and knees, resulting in uncontrolled falling. | Usually generalized |
| Associated symptoms | Preserved consciousness. Abolished deep tendon reflexes during generalized spells. | Multiple somatoform symptoms (headache, back pain, dizziness, fainting spells, and variable gastro-intestinal symptoms). | Loss of consciousness Prodromal vegetative signs and symptoms (nausea, palpitations, warm sensation, and lightheadedness), visual blurring. Possible limb irregular and brief shaking if syncope is prolonged. | Possible incoordination/ataxia, cranial nerve involvement. Consciousness usually preserved. | Consciousness can be altered or absent in atonic or absence seizures. Other types of seizures (spasms, myoclonic, tonic-clonic, absences, . . . ) or co-occurrence of different pattern in the same seizure (e.g. myoclonic-atonic, evolution to generalized tonic-clonic) Additional signs: tongue biting, urinary incontinence, cyanosis, and post-ictal confusion. | Negativism (including refusal to eat and drink) and mutism. Waxy flexibility, catalepsy and posturing in some cases. | Preserved consciousness. Generalized stiffness after birth; excessive startling to an unexpected stimulus. Cataplexy can overlap in Coffin Lowry syndrome [30]. | Slowed reaction time, speech abnormalities (in particular, speed), severe cognitive multi-domain deficits. |
| HLA status | Protective and associated HLA HLA-DQB1*0602 positive in 96% [31] HLA-DQB1*0602 negativity: red flag for secondary narcolepsy. | NA | NA | NA | NA | NA | NA | NA |
| Neurophysiological features | EEG: wake pattern during attacks. EMG: loss of tone and waxing-and waning EMG activity, rostro-caudal spreading of atonia. Sleep studies: (1) MSLT with reduced sleep latency and multiple SOREMPs, (2) night PSG with SOREMP, fragmented sleep, REM sleep without atonia. | EMG: persisting muscle tone during attacks. Sleep studies: although overlap with CNS hypersomnia is possible, MSLT and PSG exclude narcolepsy, | EEG: generalized slowing of the background activity during syncope. Autonomic function and cardiological tests: variably altered depending on the pathological mechanism; they can also be normal [32]. | Color-doppler studies may disclose flow abnormalities and stenosis. | EEG: focal or generalized epileptic activity during atonic phase. EMG: atonia synchronous with epileptic discharges. | EEG: normal. Possible abnormalities and seizures in neurological conditions | EEG: normal (except if in encephalopathy or encephalitis) EMG: bilaterally synchronous “shock-like set of movements” and generalized stiffness after a startle reflex lasting a few seconds. | EEG: normal, may reveal focal, or diffuse slowing. |
| Psychiatric morbidity | Depressive and anxiety symptoms are frequent. | Moderate-to-severe depressive and anxiety symptoms. | As in the general population. | As in the general population. | Possible cognitive and behavioral developmental delay or regression. | Depressive or manic episodes, | Cognitive developmental delay in some genetic syndromes (e.g. Coffin Lowry syndrome)[33] | Severe depressive episodes. |
| Demographic features | Typical onset in childhood-adolescence, but it can occur at any age. No gender effect. | Variable from childhood-adolescence to adulthood, more frequently female. | Any age, variably depending on the etiology. | Adult and elderly people [34, 59]. Associated with cardiovascular risk factors/comorbidity. | Typical onset during childhood. | Children and adolescents with psychiatric, neurodevelopmental and neurological disorders [35]. Adult (mainly elderly) with psychiatric and neurological disorder. | Onset during childhood-adolescence for genetic syndromes Variable onset in acquired forms (e.g. autoimmune/paraneoplastic, neurodegenerative, and post-hypoxic) [36, 37] | Possible childhood onset |
| Diagnoses | NT1, secondary narcolepsy (Niemann-Pick type C, demyelinating diseases, and paraneoplastic encephalitis) [30]. | Functional neurological disorder, malingering. | Vasovagal syncope, cardiac syncope, primary and secondary orthostatic hypotension, postural orthostatic tachycardia syndrome, stretch syncope. | Cerebrovascular disorders. | Epilepsy with myoclonic-atonic seizures, Lennox-Gastaut syndrome [35, 38]. | Schizophrenia, other psychotic disorders, neurodevelopmental disorders, encephalitis (e.g. anti-NMDA-receptor) [36, 39]. | Genetic syndromes involving alteration of glycine-receptor pathways. Brainstem lesions. Autoimmune diseases [37, 40]. | Neurodevelopmental disorders, |
EEG, electroencephalography; EMG, electromyography; PSG, polysomnography; MSLT, multiple sleep latency test; SOREMP, sleep onset rapid eye movement sleep period.
To the best of our knowledge, this study is the first investigation of psychiatric correlates of pseudocataplexy in comparison with cataplexy in patients with NT1. Participants with pseudocataplexy presented a higher level of psychopathology including both somatoform and affective symptoms. These differences in the psychopathological profile appeared despite NT1 by itself having been extensively associated with significant psychiatric comorbidity, especially mood and anxiety disorder [40]. Depressive and anxiety symptoms are widely reported as typical comorbidity of FND (both FMD and PNES) [41, 42] and are considered to play a role in the pathophysiology of FND as predisposing and precipitating risk factors [43].
Somatoform symptoms have never been described in NT1, making them a useful hint to suspect the functional nature of cataplectic attacks. Indeed, somatization is an important feature of other FNDs and correlates with the long-term outcome [44, 45]. PHQ-15 score has been linked to impaired functional connectivity between centromedial amygdala and insula, suggesting a possible neural substrate for somatic symptoms in FND [46]. Conversely, several MRI and functional MRI studies pointed to an altered structure, connectivity, and function of the amygdala in NT1 patients, especially evident in protocols encompassing emotional processing, possibly linking some aspects of cataplexy to pseudocataplexy [47]. Also, the female prevalence in patients with pseudocataplectic spells is consistent with solid evidence of gender asymmetry in FND [48].
No specific personality traits emerged in the comparison with narcoleptic patients. Up to date, little evidence for personality profiles of patients with narcolepsy is available, and it suggests an increased prevalence of abnormal profile: Baker et al. employed the Minnesota Multiphasic Personality Inventory in patients with NT1 compared to those with idiopathic central hypersomnia and found a higher prevalence of abnormal scores in hypochondriasis, hypomania, depression, schizophrenia, psychasthenia, and psychopathic deviate scores [49]. Given the lack of data, patients with functional and “true” cataplexy could potentially share common alterations, but the reported scores in the various domains do not differ from the average of the general reference population, suggesting the absence in both conditions of a personality profile predisposing to a disorder. Based on previous reports of pseudocataplexy [16], we hypothesized to find high scores in the domains of negative affectivity and disinhibition. It is possible that the instrument used in its brief form was not able to fully capture the personality profile of very complex patients, while the complete version of PID-5 with 220 items might be more accurate. Given the recent introduction of PID-5, we have no comparison data in other studies with patients with functional or somatic symptoms.
However, it must be also considered that the relation between FND and personality traits is not well established. Psychiatric profiles and personality traits seem to differ between PNES and FMD [50]. Some studies described patients with FMD as having personality traits not different from healthy controls [50], while various personality clusters have been identified within groups of patients with PNES [51].
Patients with pseudocataplexy also reported markedly worse quality of life than those with NT1, broadly affecting all areas investigated by SF-36, including both physical and mental indexes. Again, low health-related quality of life in FND has been widely reported [52], with studies showing an impairment comparable to organic neurological conditions such as Parkinson’s disease [53] and emphasizing the impact of anxiety-depressive and somatic symptoms on functioning [54]. It is conceivable that a substantial part of this poor quality of life is related to the lack of a specific pathway to care and suggests the importance of recognizing and treating pseudocataplexy and its psychiatric comorbidity, with a potential improvement of health impacts, similar to other more common and widely studied FND [55]. Surprisingly, the quality-of-life impairment of pseudocataplexy patients was even worse than those of NT1, a clinical population that has repeatedly shown a poor profile [56].
Overall, pseudocataplexy is associated with psychological features distinct from NT1 and mostly in line with other FMDs. Pseudocataplexy could be therefore included among the sub-group of paroxysmal FMD. Despite more evidence is needed, these findings bring some potential clinical implications. In cases of “atypical” cataplexy presentation, it is useful to approach the patient with multistep neuropsychological assessment suggested for other suspected FNDs in a recent review and expert opinion from American Neuropsychiatric Association Committee for Research [57], yet recognizing the central diagnostic role of sleep neurophysiological exams and CSF-hcrt-1 analysis.
Based on the result of our audit, BDI, STAI, and PHQ-15 could be administered to patients with “atypical” cataplexy presentations pending the results of sleep investigations. Indeed, PHQ-15 could be a useful screening tool, due to the higher impact of somatoform symptoms in pseudocataplexy patients, to be further tested in larger consecutive series. Emphasizing functional symptoms and rapidly promoting multi-professional assessment can make the patient feel less stigmatized than referral to a psychiatrist for a residual clinical problem after the diagnostic work-up for suspected narcolepsy. Psychiatric assessment could be therefore inserted in the diagnostic work-up after a screening with self-reported questionnaires through a visit, either in person or by means of telemedicine approaches, to patients with evidence of psychopathological traits. Since negative emotions are key triggers, psychological interventions focused on emotion regulation and stress tolerance could also improve patients’ symptoms and reduce pseudocataplexy episodes. Additionally, the impact of antidepressant drugs, often used for somatoform disorders, or mood stabilizers should be evaluated in further longitudinal studies on the evolution of this fascinating disorder. Therefore, a multidisciplinary follow-up could improve patients’ management, both considering cases with pseudocataplexy and cases with NT1 with psychological/psychiatric comorbidity.
We acknowledge some limitations in the present study that pave the way for future research on this topic. First, we did not investigate past personal stressors (e.g. adverse life events, childhood maltreatment, sexual abuse) that are recognized in literature as potential predisposing and precipitating factors of FND [43], and did not investigate whether there were sources of information about narcolepsy in the pseudocataplexy cohort (although none of these patients had relatives with a diagnosis of narcolepsy). Future studies assessing this dimension are needed to improve the understanding of psychopathological mechanisms of pseudocataplexy in comparison with other FND manifesting with different semiology (e.g. movement disorders). We did not include data regarding follow-up treatment for NT1 and pseudocataplexy, given that the relatively low dimension of our cohort of pseudocataplexy patients would not allow a stratification by treatment or outcome. Indeed, we followed up in clinical practice patients with pseudocataplexy and evidence of hypersomnia, while pure pseudocataplexy patients were mostly followed up in the psychiatric department. Further longitudinal studies including a larger number of NT1 and FND patients are warranted to disentangle the effect of different pharmacological agents on psychological features in parallel to the evolution of the occurrence and of the semiology of the spells. Indeed, personality traits and mood disorders have a bidirectional relation with symptom control, especially in FND. For instance, participants with persisting PNES were found to present lower scores in life satisfaction and extraversion, higher scores in inhibitedness, and higher levels of anxiety, depressive, dissociative, obsessive-compulsive, and somatic symptoms compared to those with resolved PNES [58]. Lastly, based on inclusion criteria, none of our pseudocataplexy cases occurred in patients with an established diagnosis of NT1, as was described by Meinen et al. 2018 [18] and by Plazzi et al. 2010 [3], whereas it is not possible to exclude that part of the attacks reported by patients with NT1 are of functional origin. This overlap condition is potentially more than anecdotal (similar to PNES in patients with epilepsy) and deserves to be addressed in further studies on greater numbers.
The occurrence of subjective EDS and, in a non-negligible minority of cases, of objective EDS in this cohort with pseudocataplexy is not clear. EDS could represent another symptom of somatization in predisposed individuals, possibly influenced by the suggestion of NT1. Alternatively, it could depend on a selection bias: while patients with transient paralysis and EDS complaint are more easily referred to a center specialized in narcolepsy in the suspect of NT1, it is possible that patients with pseudocataplexy but without EDS complaint are preferentially evaluated by other neurology units (e.g. specialized in epilepsy or movement disorders).
In conclusion, pseudocataplexy in FND has clinical similarities and differences with cataplexy in NT1, making the diagnosis difficult when based on history taking alone and thus limiting the management options. The assessment of somatoform and affective dimensions could help to better describe these patients with pseudocataplexy and claims for patient-centered multidisciplinary care. Patients with pseudocataplexy share several features with other FND, a pattern that differs from NT1. Further studies are needed to better characterize pseudocataplexy features (also in possible comorbidity with NT1), its response to pharmacological and non-pharmacological interventions, and its relations with other FND manifesting mainly as movement disorders.
Supplementary material
Supplementary material is available at SLEEP online.
Acknowledgments
We are indebted to all the participants of the study, most notably the Italian association of narcoleptic patients (Associazione Italiana Narcolettici ed Ipersonni, ONLUS). Without their contributions, this study would not have been possible.
Disclosures Statement
Financial disclosure: none. Nonfinancial disclosure: none.
Data Availability
Anonymized data not published within this article will be made available by request from any qualified investigator.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments