





Abstract
During sexual reproduction in flowering plants, tip-growing pollen tubes travel from the stigma inside the maternal tissues of the pistil toward ovules. In maize (Zea mays L.), the stigma is highly elongated, forming thread-like strands known as silks. Only compatible pollen tubes successfully penetrate and grow through the transmitting tract of the silk to reach the ovules. Like pollen, fungal spores germinate at the surface of silks and generate tube-like structures (hyphae) penetrating silk tissue. To elucidate commonalities and differences between silk responses to these distinctive invading cells, we compared growth behavior of the various invaders as well as the silk transcriptome after self-pollination, cross-pollination, and infection using 2 different fungi. We report that self-pollination triggers mainly senescence genes, whereas incompatible pollen from Tripsacum dactyloides leads to downregulation of rehydration, microtubule, and cell wall–related genes, explaining the slower pollen tube growth and arrest. Invasion by the ascomycete Fusarium graminearum triggers numerous defense responses including the activation of monolignol biosynthesis and NAC as well as WRKY transcription factor genes, whereas responses to the basidiomycete Ustilago maydis are generally much weaker. We present evidence that incompatible pollination and fungal infection trigger transcriptional reprograming of maize silks cell wall. Pathogen invasion also activates the phytoalexin biosynthesis pathway.
Introduction
Maize (Zea mays L.) is a monoecious plant with unisexual female and male flowers (Dresselhaus et al. 2011). While the female inflorescence, also known as ear, is originating from axillary meristems, the male inflorescence or tassel is positioned at the apex of the plant and originates from the shoot apical meristem after transition to various flower meristems (Zhou et al. 2017; Bommert and Whipple 2018). Ears and tassels initially undergo a similar developmental program but eventually develop into distinct architectures. In developing ears, the lower floret is aborted, and only the upper floret continues to develop into a functional flower forming an ovary and stigma, the latter also known as silk (Abendroth et al. 2011). Maize silks are long thread-like strands exposed out of the ear shoot and function as the main pollen receptive organ. Numerous hairs (papilla hair cells or receptive trichomes) are distributed along the length of a silk and function as major surfaces for pollen capture and germination (Zhou et al. 2017). Alongside providing an extensive surface for capturing pollen, the main function of maize silks is to allow compatible pollen to hydrate, germinate, and successfully penetrate papilla hair cell structures, with the goal to carry their sperm cell cargo at long distances and high-speed growth inside the transmitting tract toward the female gametophyte or embryo sac inside the ovary (Lausser et al. 2010). However, maize silks also receive pollen from other plant species and are exposed to microorganisms including pathogens. While pollen from plant species outside the grass family are partly capable to germinate on papilla hair cells, they are usually not capable to penetrate. Pollen from other grass species like the maize relative Tripsacum dactyloides are capable to penetrate, but growth is not supported and arrested after about 2 to 3 cm inside the silk tissue (Lausser et al. 2010). Like pollen grains, spores of fungal pathogens germinate on silks generating tip-growing hyphae that use silks as a route to ultimately invade developing kernels (Thompson and Raizada 2018). Fungal hyphae growth inside silks is substantially slower compared with pollen tube growth and further inhibited if silks were simultaneously exposed to compatible pollen (Bircheneder and Dresselhaus 2016; Thompson and Raizada 2018).
Flowering plants have evolved sophisticated molecular responses to reject alien (incompatible) pollen and pathogens. While similarities of recognition mechanisms between alien pollen and pathogens have been proposed to be widespread in plants, few studies have compared these processes (Kessler et al. 2010; Mondragón-Palomino et al. 2017). Most of the common associated responses have been narrowed down to receptor-like kinases (RLKs) and phytohormones during defense mechanisms and pollen incompatibility (Mondragón-Palomino et al. 2017; Shi et al. 2017). Although our understanding of how plants (especially Arabidopsis) respond to pollination (Sella Kapu and Cosgrove 2010; Mondragón-Palomino et al. 2017; Shi et al. 2017; Mandrone et al. 2019; Rozier et al. 2020; Kodera et al. 2021) and pathogens (Mondragón-Palomino et al. 2017; Ning and Wang 2018; Zhou et al. 2018; Li et al. 2019; Yadav et al. 2020; Kumar et al. 2022) has improved significantly in recent years, it is unclear whether both processes trigger similar or different responses. Moreover, comparable studies of pollination and pathogen infection in maize have to our knowledge not been performed to determine whether models from Arabidopsis are generalizable to monocots and grasses.
We used maize as a model in this study as its elongated stigmas (silks) are easily accessible, can be harvested in large quantities, and allow the invasion of different biotic invaders to be followed over long distances and time. GFP marker lines were used to follow the growth of pollen tubes and fungal hyphae, respectively. A dataset of 11 different samples was generated including controls that included both aging silks and vegetative tissues. To identify common and specific responses toward the various invaders, we compared molecular signatures of maize silks after exposure to fungal infections using the hemibiotrophic ascomycete Fusarium graminearum and the biotrophic basidiomycete Ustilago maydis as well as compatible pollination (self-pollination) and incompatible pollination (cross-pollination) using the close maize relative T. dactyloides. While expression of relative few genes was changed after self-pollination, cross-pollination and especially infection by F. graminearum led to drastic gene expression changes. A number of processes especially related to genes involved in cell wall and lignin biosynthesis and modification, gene regulation as well as phytoalexin production have been studied in more detail.
Results and discussion
Maize silks are direct highways used by pollen tubes and fungal hyphae to enter ovaries
The elongated silks of maize represent an ideal cereal model system to explore and compare cellular and molecular strategies used by invading pollen tubes and pathogens during reproduction and defense responses, respectively. Even though some similarities between reproductive and defense responses have been reported in Arabidopsis (Mondragón-Palomino et al. 2017), it is unclear to which extent this can be translated to cereals and whether responses of the silk to incompatible pollen are similar to fungal pathogen invasion. To test this hypothesis, we used the fungal pathogens F. graminearum and U. maydis to study defense responses. To elucidate reproductive responses, we used pollen of the maize relative T. dactyloides and Z. mays (maize) as incompatible and compatible materials, respectively (Fig. 1A).
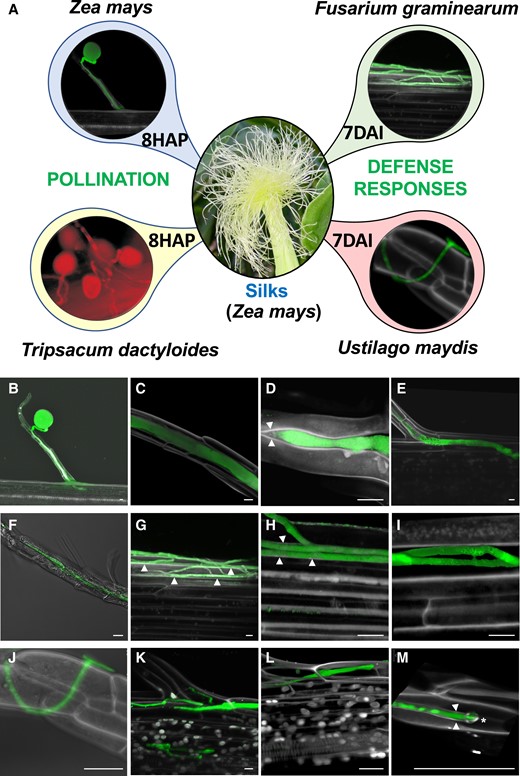
Invasion of papilla hair cells and silks of maize by own and alien pollen tubes as well as by 2 different fungi. A) Experimental setup: maize silks were pollinated with Z. mays B73 (self-pollination) and T. dactyloides (incompatible pollination), respectively, and samples collected at 8HAP. Additionally, silks of maize plants were infected with the facultative hemibiotrophic pathogen F. graminearum and the biotrophic pathogen U. maydis. Samples were collected at 7DAI. B) to E) A GFP-labeled maize pollen tube germinated at the apex of a papilla hair cell structure and grows through the papilla (B) in the intercellular space between hair cells (C) while pushing hair cells apart (D) and growing straight inside the silk intracellularly toward the transmitting tract (right) after reaching into the bottom of the hair cell structure (E). Arrowheads indicate cell walls. F) to I) GFP-expressing F. graminearum hyphae invade silks either after penetration of papilla hairs and intercellular growth like pollen tubes (F) or after initial growth at the silk surface (G). Growth inside the silk occurs first intercellular by pushing cell walls apart (H) before cells are penetrated and die (I) Arrowheads indicate cell walls. J) to M) GFP-labeled U. maydis growth through the silk “ignoring” cell walls. An appressorium is visible at the surface of a papilla hair (J) and further fungal growth occurs intracellularly (K and L) Cell wall material is always detectable surrounding intracellular growing fungal hyphae (M) The tip of hyphae is marked by an asterisk. The white color in all images results from propidium iodide counterstaining. Bars: 10 μm.
First, we analyzed invasive growth of pollen tubes. Silks were pollinated using maize pollen carrying a GFP marker (Fig. 1, B to E). After adhesion, pollen grains germinated within about 10 min and penetrated papilla hair cell structures between cell boundaries. Further growth occurred intercellularly in the center of hair cell structures (Fig. 1, B and C), apparently by pushing cells apart (Fig. 1D). After entering the main body of the silk, maize pollen tubes grew intercellularly toward the transmitting tract (Fig. 1E). While T. dactyloides pollen tubes entered papilla hair cell structures using the same strategy, they often did not enter the transmitting tract, and growth was arrested after 2 to 3 cm growth as reported previously (Lausser et al. 2010).
The overall pattern of silk infection was investigated for both pathogens also using GFP marker lines. Macroconidia or ascospores of F. graminearum germinated on exposed silks and entered silk hair cell structures using the intercellular pollen tube route and by pushing cells apart. After entering the body of the silk, further hypha growth occurred initially intercellularly, before cells were penetrated and killed. Later growth occurred intracellularly (Fig. 1, F to I). U. maydis entered maize silks after forming appressoria (Fig. 1J). Further growth occurred exclusively intracellularly without killing cells (Fig. 1, K and L). Fungal hyphae were always surrounded within cells by a plasma membrane and thin cell wall with cellulose content, as detected by propidium iodine stain (Fig. 1M).
Pathogen infection and foreign pollination elicit unique and only slightly overlapping pattern of molecular responses in maize silks
To characterize the reproductive and defense responses at the molecular level, we explored the transcriptional changes in all above-described scenarios. Silks of the maize inbred line B73 were self-pollinated and analyzed 8 h after pollination (8HAP) before they reach the ovules using own B73 pollen (referred to hereafter as “silks self-poll”). Incompatible pollination was studied after pollinating maize silks with T. dactyloides pollen (“silks Tripsacum”). As controls, we included mature unpollinated silks (“silks un-poll”) of the same age (8H) and pollen tubes collected after 45 min (45 m) in vitro germination (“pollen tubes”) in order to capture silk-specific responses. To elucidate fungal defense responses, maize silks were infected either with F. graminearum (“silks Fusarium”) or U. maydis (“silks Ustilago”), and material was collected 7 d after infection (7DAI). Two types of controls were included: mature silks 7 d (7D) after mock treatment (“silks mock”) and untreated mature silks 7D (“silks untreated”) as aging control. It was previously reported that silk aging is initiated at about 7D after silk emergence (Šimášková et al. 2022); thus, 7DAI was chosen for sampling showing maximum fungal infection with still minor aging-related responses. As additional controls for both groups, we included seeds, leaves, and roots of maize to elucidate silk-specific responses. Supplementary Table S1 summarizes the sample material, average of total reads per condition, and percentage of mapped reads. A detailed list containing the median expression values of treatment/tissue for the entire maize genome is provided in Supplementary Data Set 1.
A principal component analysis (PCA) of all samples showed clear separation of the different tissues and close association of the 3 biological replicates used per sample for RNA-seq analysis. This indicated that the samples were highly reproducible. PCA on the gene expression data showed that PCA1 largely separates silk samples from pollen tubes, seeds, and roots (Fig. 2A). To validate RNA-seq data, we randomly selected 33 genes that were expressed in more than one treatment (Supplementary Fig. S1). We found a 0.823 correlation coefficient between the RNA-seq results and the quantitative PCR (qPCR) data confirming the high quality and reproducibility of the data. A comparison of all genes expressed in silks under the different conditions studied showed similar percentages of total transcriptional expression (∼55% of all maize genes). A comparative percentage of expression was observed in seeds, leaves, and roots. Notably, pollen tubes expressed only ∼20% of all maize genes (Fig. 2B). This can be explained by the fact that they consist of only 2 cell types of highly specific functions compared with the other very complex tissues containing many different cell types, and it is consistent with findings of other studies investigating pollen transcriptomes (Rutley and Twell 2015; Warman et al. 2020). We further identified clusters of genes characteristic for all samples (Fig. 2C). While silks unpollinated and self-pollinated showed a similar pattern, the pattern after T. dactyloides pollination was quite distinctive. Infection with F. graminearum triggered the largest response in maize silks. Notably, silks harvested 7D after mock treatment and untreated showed a highly similar pattern, but different from young unpollinated silks, indicating aging effects.
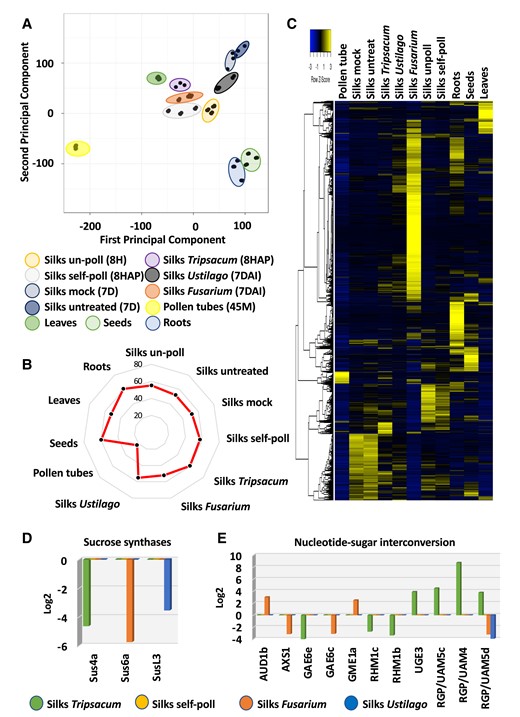
Transcriptome relationships between pollinated and infected maize silks. A) PCA of RNA-seq data from the 11 tissue samples used in this study (see Supplementary Table S1 for sample overview). Three biological replicates were used for each sample that include various pollinated, infected, and control samples as indicated. B) Radar (spider) chart displaying the percentage of transcripts from the maize genome expressed in each tissue. C) Heat map of the whole maize genome in response to self-pollination, incompatible pollination, and fungal infection. D) Genes involved in sucrose synthase and E) nucleotide sugar interconversion are strongly regulated after invasion of foreign pollen tubes and fungal hyphae, respectively.
We next studied whether there exist gene groups that are differentially regulated in response to foreign invaders, but which are not regulated by invasion of maize pollen tubes. Three sucrose synthase genes were identified, for example, with silk-preferred expression that were downregulated in response to incompatible pollination and fungal invasion (Fig. 2D). While sucrose synthase 4a (Sus4a) was downregulated in response to T. dactyloides, Sus6a was repressed after F. graminearum infection and SusL3 in response to U. maydis. Sucrose synthases are considered as markers for sink strength in crops (Xu et al. 2019); thus, their downregulation may indicate a reduction of starch, cellulose, or callose biosynthesis, but also reduced tolerance to stress conditions (Stein and Granot 2019) and biotic invaders.
One of the first steps during cell wall biosynthesis is substrate generation by nucleotide sugar interconversion pathways. Eleven gene families have been identified that encode enzymes of the nucleotide sugar interconversion pathways involved in the formation of the basic sugar building blocks of many plant cell walls (Penning et al. 2009; Yin et al. 2011). Although a number of different gene family members are regulated, especially in response to invading F. graminearum hyphae and T. dactyloides pollen tubes (Fig. 2E), there is almost no overlap in regulated genes, except for the UAM5d gene encoding a reversibly glycosylated protein which is downregulated in response to both F. graminearum and U. maydis. Altogether, these findings indicate already highly specific responses toward the various invaders.
Incompatible pollination triggers downregulation of rehydration and cell wall–associated genes
To explore the molecular mechanisms underlying compatible and incompatible pollen–silk interactions, respectively, we compared the transcriptional responses of maize silks after 8 h of pollination with B73 pollen (compatible) and T. dactyloides pollen (incompatible). To capture only maize silks’ responses, we used transcriptomic samples of in vitro grown maize pollen tubes to remove from consideration the majority of genes expressed in the male component. Notably, incompatible pollination resulted in a significant larger specific transcriptomic response in maize silks (32 vs. 2,232 genes; Fig. 3A). Notably, the expression pattern of 113 common differentially expressed genes (DEGs; Fig. 3A) after compatible and incompatible pollination was exactly the same (Fig. 3B, Supplementary Data Set 2). Of the 113 genes, only 1 gene (LHY1—late hypocotyl elongation protein ortholog1) was downregulated in response to both conditions. The other 112 genes were upregulated (Fig. 3B). These results suggest that the common genes are important for recognizing the presence of pollen grains and/or pollen tubes but are not necessarily involved in the differentiation between compatible and incompatible pollen.
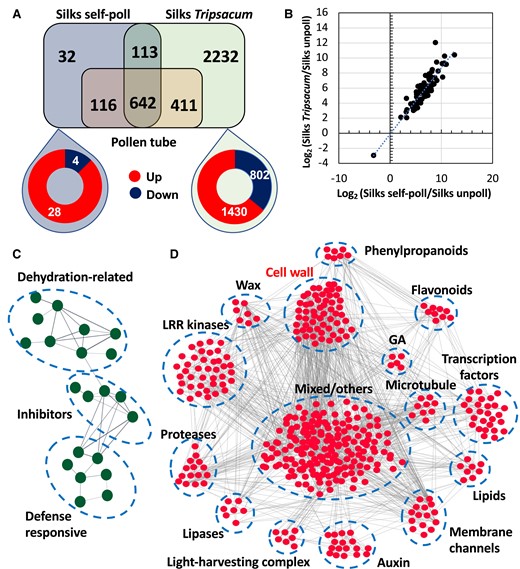
Comparison of transcriptional response in maize silks after compatible and incompatible pollination. A) Comparison of DEGs between self-pollinated (compatible) and T. dactyloides pollinated (incompatible) silks. The pollen tube transcriptome was included as a control to identify silk-responsive genes to pollination. B) Scatter plot representing log2 (fold change expression) for common DEGs in self-pollinated maize (x axis) vs. maize pollinated with T. dactyloides (y axis). Network analysis of downregulated DEGs after C) compatible and D) incompatible pollination of maize silks, respectively.
The strong transcriptional response in maize silks elicited by T. dactyloides pollen tubes (2,232 regulated genes) showed that ∼36% genes were downregulated, suggesting a strategy by maize silks to shut down key pathways for pollen tube growth support, ultimately leading to arrest of alien pollen tubes (Supplementary Data Set 3). With this possibility in mind, we then asked which biological processes were downregulated by T. dactyloides pollination that could lead to specific hypotheses for which processes are important for successful (i.e. compatible) pollen tube growth. We conducted a cluster correlation analysis using the “cor” function implemented in the R package WGCNA to identify groups of genes with shared expression profiles. Since the maize genome is not well annotated and functions of many genes are categorized as unknown, it was likely that our gene network correlation analysis would contain a large portion of uncharacterized genes. Therefore, we combined our gene cluster correlation analysis with the STRING app (Szklarczyk et al. 2023) of Cytoscape (Shannon et al. 2003) which provides coexpression interaction data to be able to extract pathways that are affected by incompatible pollination. In the compatible pollination analysis, we identified genes associated with dehydration-related, inhibitors and defense responsive as biological processes associated with compatible pollination (Fig. 3C, Supplementary Data Set 4).
For incompatible pollination, our differential expression analysis yielded 803 downregulated genes. After removal of genes without interaction (unrooted genes = 384), the remaining downregulated DEGs were queried in STRING, which allows to infer protein–protein associations from coexpression data. We identified 419 interacting genes associated with incompatible pollination. Using a high edge confidence of at least 0.7, we constructed a gene network that functionally grouped downregulated genes into 15 main hubs (Fig. 3D, Supplementary Data Set 4). The largest group is formed by genes with a variety of functions. The remaining 14 clusters could be assigned to functional categories using the NEAT package (Signorelli et al. 2016), including genes involved in phenylpropanoid, flavonoids, cell wall, and wax biosynthesis, but encode also microtubule components, transcription factors, lipids, membrane channels, light-harvesting complex components, lipases and lipid biosynthesis enzymes, proteases, and kinases including leucine-rich repeat (LLR) kinases. Gibberellin and auxin biosynthesis genes were also identified. Plant hormones appear to play roles during pollen–pistil interactions in maize, but their detailed functions are unclear. For instance, ethylene and auxin have been shown to rapidly increase in maize silks after compatible pollination (Mól et al. 2004). Increased auxin levels, followed by a spike of gibberellic acid (GA) biosynthesis, were also reported after fertilization (Dorcey et al. 2009). In our study, incompatible pollination with T. dactyloides induced a strong downregulation of auxin biosynthesis and response genes (Fig. 3D), but not of ethylene genes. A decrease in transcriptional expression of auxin and GA points toward a switch from development and growth to defense responses.
Water transport from stigmatic pistil cells to desiccated pollen grains is essential for pollen germination (Firon et al. 2012). Noteworthy, 4 clusters (lipid, wax biosynthesis, aquaporins, and LLR kinases) potentially involved in pollen rehydration and germination were downregulated by T. dactyloides pollination. These genes that encode, for example, enzymes for lipid and wax biosynthesis, specialized water channels or aquaporins, and LLR kinases have been shown previously to play a positive role in regulating pollen rehydration and initial stages of pollen tube growth in the female reproductive tract (Lee and Goring 2021; Windari et al. 2021; Abhinandan et al. 2022), indicating their role in recognition of alien pollen and activation of mechanisms to prevent invasion. We found genes of several members of the aquaporin gene family that are downregulated after T. dactyloides pollination. In Arabidopsis, it has been shown that SIP1;1 and PIP1;2, orthologs of the downregulated maize genes found in this study, are key regulators of water transport from papilla cells to pollen, thereby regulating pollen hydration (Windari et al. 2021). Similarly, in solanaceous and crucifer, but also lily plant species, pistil lipids and lipid transfer-like protein are required for pollen hydration and further penetration (Park et al. 2000; Sanchez et al. 2004; Thompson and Raizada 2018; Zheng et al. 2018). Furthermore, loss-of-function mutations in RKF1, an LLR kinase gene, reduced pollen hydration of wild-type pollen on mutant stigma (Lee and Goring 2021), indicating the importance of pollen recognition processes. In conclusion, although downregulation of LLR kinase, lipid, wax, and aquaporin genes after incompatible pollination is not sufficient to prevent initial germination and penetration of fast-growing T. dactyloides pollen tubes, they may be sufficient with other processes (see below) to prevent most incompatible pollen tubes to reach the ovaries.
The largest cluster of downregulated genes is associated with the cell wall (Fig. 3D, Supplementary Data Set 4). There were further sets of downregulated and related gene clusters formed by phenylpropanoid, flavonoids, and microtubule genes (Fig. 3D). Phenylpropanoids are a structurally diverse group of phenylalanine-derived metabolites involved in the synthesis of flavonoids and lignins used by plants to build cell walls. Flavonoids have been well studied in pollen grains as they are plant-specific compounds required for pollen germination and tube growth, and are constituents of the pollen cell wall and work as positive effectors for pollen tube elongation (Muhlemann et al. 2018; Dong et al. 2021; Wu et al. 2023; Xue et al. 2023). Thus, downregulation of phenylpropanoid and consequently flavonoid levels may arrest pollen tube growth as observed in incompatible pollinations of maize (Lausser et al. 2010). Similarly, cell wall and microtubule genes have been largely known for their importance during pollen tube/pistil interaction and pollen tube guidance (Dresselhaus and Franklin-Tong 2013; Riglet et al. 2020). During compatible pollination, these dynamic interactions facilitate pollen tube elongation. In plants of the Liliaceae family as well as in Arabidopsis, such interactions are mediated, for example, by microtubules, pectins, lipids, and a stylar cysteine-rich adhesin protein secreted in the secreted extracellular matrix of the pistils (Park et al. 2000; Onelli et al. 2015; Riglet et al. 2020). In summary, our data show downregulation of rehydration genes, microtubule, and cell wall–related genes explaining pollen tube growth arrest in incompatible pollinations.
Compatible pollination regulates expression of MYB transcription factor and senescence genes
Transcriptional responses in maize silks to compatible and incompatible pollination show only small overlap (Fig. 4A). Intriguingly, compatible pollination resulted only in 32 differentially expressed unique genes (Supplementary Table S2). We found that 10 of these genes encode transcription factors. Genes involved in transcriptional regulation are likely to be important components of pollen–pistil interaction, since they can precisely regulate large numbers of downstream genes. Especially MYB transcription factor genes (15% of the total differentially expressed unique genes) were regulated during compatible pollination. In Arabidopsis, MYB transcription factors have been reported as key regulators of pollen tube growth and sperm release (Leydon et al. 2013), pollen tube–synergid interaction (Liang et al. 2013), and senescence (Cao et al. 2023). In maize, MYB transcription factor genes were reported as targets of high temperature, leading to pollen sterility (Begcy et al. 2019). Functional studies are now necessary to evaluate the MYB transcription factor identified in this study and to elucidate their contribution to compatible pollen–pistil interaction(s) in maize.
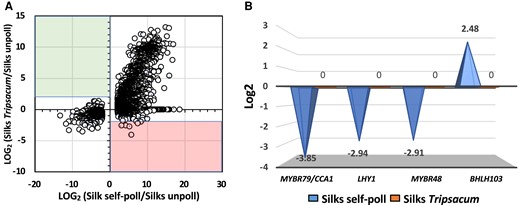
Self-pollination alters expression of senescence-associated transcription factor genes in maize silks. A) Self and incompatible pollination induce large transcriptional expression as showed in the scatter plot representing the log2 (fold change expression) for all DEGs in self-pollinated maize (x axis) vs. maize silks pollinated with T. dactyloides (y axis). B) Fold change expression changes of senescence-associated transcription factor genes.
Previous studies have reported that maize silks undergo an accelerated process of senescence in response to compatible pollination (Bassetti and Westgate 1993; Sella Kapu and Cosgrove 2010; Šimášková et al. 2022). Thus, we searched for senescence-associated genes within the DEGs in maize silks as a result of compatible pollination. We found that 34% of these genes, previously reported in maize and lettuce leaves, are indeed linked to senescence (He et al. 2020; Chase et al. 2024). Thus, our data indicate that silk and leaf senescence could be controlled by similar mechanisms.
Many associated senescence genes have been identified, and transcription factors have been recognized as hub genes in senescence pathways in many plant species (Bengoa Luoni et al. 2019; Cao et al. 2023). We found 4 transcription factor genes in the list of DEGs in maize silks after self-pollination (Supplementary Table S2) previously associated with senescence (Fig. 4B). The homolog of ZmMYBR79, also known as CIRCADIAN CLOCK-ASSOCIATED 1 (CCA1), was previously shown to inhibit leaf senescence. mybr/cca mutants showed accelerated age-dependent senescence (Song et al. 2018). Notably, ZmMYBR79/CCA1 was downregulated 3.85-folds after compatible pollination. Since ZmMYBR79/CCA1 forms a core loop with LHY1 by repressing one another's expression (Song et al. 2018), we also studied LHY transcriptional expression after compatible pollination and found that ZmLHY was also downregulated (Fig. 4B). Similarly, ZmMYBR48, another gene that has been associated with the circadian clock (Huang et al. 2017) and is also likely contributes to the ZmMYBR79/CCA1-ZmLHY1 senescence regulatory network, was found downregulated after compatible pollination (Fig. 4B). Our data suggest that silk senescence in maize is likely accelerated due to downregulation of a ZmMYBR79/CCA1-ZmLHY1-ZmMYBR48 regulatory network. Mutant studies are now required to support this assumption.
Another transcription factor differentially expressed after compatible pollination is bHLH103 (fold change = 2.48, Fig. 4B). In petunia, for example, a basic helix–loop–helix (bHLH) transcription factor (FBH4) was shown to regulate flower senescence. While overexpression of FBH4 resulted in accelerated flower senescence, RNAi plants showed retarded senescence (Yin et al. 2015). Therefore, the coordinated downregulation of MYBs as senescence repressors and upregulation of bHLHs as likely senescence promoters indicate that precocious silk senescence is initiated after compatible pollination likely to prevent growth of other biotic invaders.
Downregulation of cellulose biosynthesis genes occurs during incompatible pollination and fungal invasion
Rigidification of the silks’ cell wall has been reported as one of the consequences in response to pollination in maize (Sella Kapu and Cosgrove 2010). Similarly, host cell wall damage has been observed during pathogen infection (Lorrai and Ferrari 2021). Therefore, we next investigated cell wall–related responses to incompatible pollination and fungal invasion (Fig. 5). We first explored the transcriptional expression of cellulose synthase (Ces) and cellulose synthase-like (Csl) genes (Fig. 5, A and B). In maize, the Ces family consists of 20 members. Incompatible pollination reduced the expression of cellulose synthase (CesA) genes, particularly that of CesA1, CesA2, CesA4, and Ces7b (Fig. 5A). Even though F. graminearum and U. maydis invasion downregulated members of the cellulose synthase gene family, these groups of genes were different than the ones downregulated by T. dactyloides. Both fungal pathogens reduced the transcriptional expression of CesA11a, CesA11b, and CesAL4, respectively. Infection by both fungal pathogens repressed CesA11b by 9.2-fold and 4.4-fold, respectively. No changes in expression were found during incompatible pollination (Fig. 5A). Since CesA11b, also known as brittle stalk-5, is critical for cell stiffness (Li et al. 2022), infection by F. graminearum and U. maydis might be associated with silk brittleness as a strategy to prevent fungal progression.
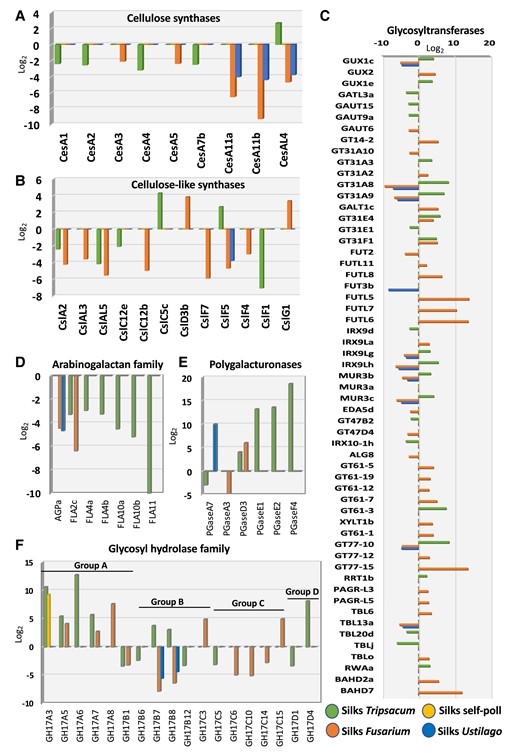
In contrast to self-pollination, incompatible pollination and fungal invasion alter cell wall–related genes families in maize silks. Fold change expression analysis of gene families related to cell wall formation is shown. Genes encoding cellulose synthases (A) and most genes for cellulose-like synthases (B) are downregulated. Genes encoding glycosyltransferases (C) are differentially expressed, while genes involved in arabinogalactans (D) are downregulated and polygalacturonases (E) are mostly upregulated. Genes for glycosyl hydrolases (F) show a differential expression pattern.
In addition to CesA genes, maize also has cellulose synthase-like (Csl) genes that synthesize other essential noncellulosic polysaccharides that are constituents of the cell wall (Li et al. 2019). Genes belonging to the Csl gene family were also downregulated after invasion of alien pollen tubes and fungal hyphae (Fig. 5B). In summary, these results suggest that cellulose biosynthesis is downregulated when noncompatible pollen or fungal pathogen interact and grow within maize silks, likely contributing to fast growth inhibition.
Glycosyltransferase and glycosyl hydrolase genes are strongly regulated in response to alien invaders
Another important gene superfamily primarily involved in cell wall formation encodes glycosyltransferases. The glycosyltransferases are divided into processive and nonprocessive enzymes. Within the nonprocessive glycosyltransferase gene families, 12 families showed differential responses to incompatible pollination and fungal invasion but remained unaltered after self-pollination (Fig. 5C). Glycosyltransferases are involved in pectin synthesis (Qin et al. 2013; Zabotina et al. 2021). Glucuronic acid substitution of xylan (GUX1c) was the only gene differentially expressed by incompatible pollination and fungal invasion (Fig. 5C). Notably, GUX1c was downregulated after infection by both F. graminearum and U. maydis and upregulated by T. dactyloides. GUX1c did not change in response to compatible pollination. In Arabidopsis, gux1/2/3 triple mutants cause a reduction in plant growth and in the mechanical strength of the stem (Lee et al. 2012).
Arabinogalactan proteins constitute a large highly glycosylated family of hydroxyproline-rich proteins involved in plant reproduction. Notably, both incompatible pollination and fungal invasion downregulate arabinogalactan genes (Fig. 5D). Since this gene family was predominately affected by incompatible pollination, they could be a good target for future incompatibility studies during pollination in maize.
Similarly, as observed with arabinogalactans, glycosyl hydrolases including polygalacturonases (pectinases) were differentially expressed in response to T. dactyloides (Fig. 5, E and F). Pectinases are enzymes that cleave pectin, a major cell wall component especially accumulated in the middle lamellae between plant cells, and which have been shown to participate in cell wall elongation and pollen tube growth (Minic 2008; Babu and Bayer 2014; Lu et al. 2021). We found that incompatible pollination enhanced expression of polygalacturonase genes (Fig. 5E). This suggests that T. dactyloides pollination may lead to softening of cell walls, opposite of what it is observed after compatible pollination, where cell wall undergoes structural modifications leading to its rigidification (Sella Kapu and Cosgrove 2010). Among other glycosyl hydrolases, U. maydis infection only downregulated GH14B7 and GH14B8 genes (Fig. 5F). F. graminearum and T. dactyloides induced mixed responses. While glycosyl hydrolases from Group A were upregulated, those from Group C were downregulated. Groups B and D showed both patterns of expression (Fig. 5F).
In summary, many genes encoding glycosyltransferases and hydrolases are regulated in response to incompatible pollination and fungal invasion, potentially contributing to cell wall modifications that suppress fast growth and cell elongation of alien invaders in silk tissue. Functional studies are now needed to support this assumption.
F. graminearum triggers the monolignol biosynthesis pathway in maize silks
Biosynthesis of lignin, one of the core components of cell walls that form a complex network of polymers of hydroxylated and methoxylated phenylpropane units, depends on phenylpropanoid and monolignol pathways. Both pathways are interconnected, since monolignol biosynthesis utilizes substrates generated by the phenylpropanoid pathway (Yadav et al. 2020). We found that F. graminearum induced high expression of key genes involved in the monolignol biosynthesis pathway (Fig. 6A) in maize silks. The conversion activity of phenylalanine ammonia lyases (PALs) into cinnamic acid is the first step of the general phenylpropanoid pathway. Seven maize PALs showed significant upregulation compared with both mock and untreated samples, particularly PAL8, PAL9, and PAL10, which showed 8-time fold increase (Fig. 6B). Similarly, the 3 genes showed high expression in response to U. maydis. In contrast, T. dactyloides pollination decreased PAL1, PAL2, and PAL3 expression by more than 2-fold. PAL genes were described as mediators for cell wall's first layer of immunity and involved in broad-spectrum disease resistance (Ning and Wang 2018; Zhou et al. 2018). Thus, upregulation of PAL genes after fungal invasion appears to be a mechanism of defense also used by maize silks to prevent the spread of pathogens. In contrast, incompatible pollination does not activate this immune response.
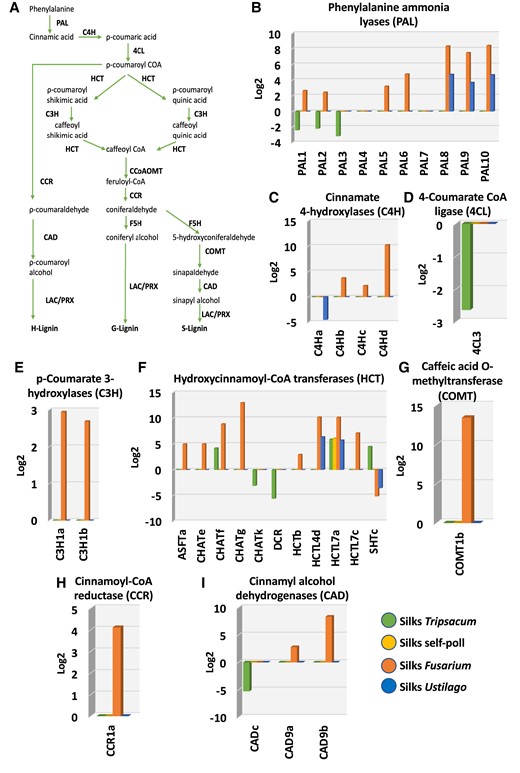
Genes involved in monolignol biosynthesis are highly upregulated after F. graminearum infection in maize silks. A) Overview of the H-/G-/S-monolignol biosynthesis pathway including products, educts, and respective enzymes. PALs, cinnamate 4-hydroxylases (C4H), 4-coumarate CoA ligases (4CL), hydroxycinnamoyl-CoA transferases (HCT), p-coumarate 3-hydroxylases (C3H), Caffeoyl-CoA 3-O-methyltransferase (CCoAOMT), cinnamoyl-CoA reductases (CCR), ferulate 5-hydroxylase (F5H), caffeic acid O-methyltransferases (COMT), CADs, LAC, peroxidase (PRX), syringyl (S), guaiacyl (G), and p-hydroxyphenyl (H) lignin. Expression pattern of monolignol biosynthesis genes in maize silks including genes encoding B) PAL, C) C4H, D) 4CL, E) C3H, F) HCT, G) COMT, H) CCR, and I) CAD.
Cinnamate 4-hydroxylase (C4H), involved in the reduction of cinnamic acid into ρ-coumaric acid at the beginning of the phenylpropanoid and monolignol biosynthesis pathway (Fig. 6A), showed also high upregulation in response to F. graminearum (Fig. 6C). Compatible and incompatible pollination did not alter any of the C4H genes. U. maydis infection led to downregulation of C4Ha but did not alter the expression of any other member of this gene family (Fig. 6C). Notably, neither pathogen infection nor compatible pollination changed 4-coumarate CoA ligase (4CL) expression. Only T. dactyloides pollination triggered decreased 4CL expression (Fig. 6D).
An important step in the general phenylpropanoid pathway involves the conversion of ρ-coumaroyl-CoA to caffeoyl-CoA by the p-coumarate3-hydroxylase (C3H) and hydroxycinnamoyl-CoA:shikimate/quinate hydroxycinnamoyltransferase (HCT). Gene expression changes of C3H by compatible and incompatible pollination as well as U. maydis infection were not observed. Only F. graminearum infection induced the expression of both sets of genes in maize silks. While C3H1a and C3H1b were induced compared with the mock treatment (Fig. 6E), respectively, 9 HCTs were differentially expressed (Fig. 6F). Eight of them were upregulated and 1 was downregulated (sHTc), respectively. Similarly, U. maydis invasion upregulated HCTL4d and HCTL7a in maize silks and downregulated SHTc. Incompatible pollination by T. dactyloides downregulated CHATk and DRC and upregulated CHATf, HCTL7a, and SHTc. Compatible pollination upregulated HCTL7a similarly as incompatible and fungal invasion (Fig. 6F).
Notably, Arabidopsis leaves challenged with several pathogens (Zhang et al. 2018; Vogel et al. 2021; Zhu et al. 2023; Entila et al. 2024) induced the expression of the caffeic acid (5-hydroxyconiferaldehyde) O-methyltransferase gene (COMT1), similarly as we observed in maize silks infected with F. graminearum (Fig. 6G). Regulation of COMT was suggested to be one of the first mechanisms of plant's pathogen defense and obviously occurs also in maize silks.
F. graminearum induced genes involved not only in the general phenylpropanoid pathway but also in the monolignol pathway. Sinapyl alcohol and coniferyl monolignol are the major building blocks of lignin, and they generate syringyl (S) and guaiacyl (G) units of lignin polymers, respectively. The minor monolignol unit, which is deposited more in monocots, is the p-hydroxyphenyl (H) unit derived from p-hydroxyphenyl (Yadav et al. 2020). Cinnamoyl-CoA reductase (CCR) converts 5 types of hydroxycinnamoyl-CoA into cinnamaldehyde, and their overexpression was shown to play roles in pathogen defense responses of plants (Lauvergeat et al. 2001; Cheng et al. 2017). Only F. graminearum infection induced CCR1a in maize silks (Fig. 6H). Additionally, F. graminearum infection upregulated CAD9a and CAD9b in maize silks (Fig. 6I). Cinnamyl alcohol dehydrogenases (CADs), which deoxidizes 5 types of cinnamaldehyde to the corresponding monolignols (Fig. 6A), were also associated with lignin synthesis and stress resistance (Guo et al. 2010).
Peroxidases (PRX) and laccases (LAC) are further classes of enzymes present in the apoplast of plant cells. Among their diverse functions, they are also important for lignin biosynthesis (Laitinen et al. 2017). In our study, 40 genes encoding PRX enzymes were misregulated in all conditions: 18 after T. dactyloides pollination, 27 after F. graminearum, and only 3 after U. maydis infection (Fig. 7A). Only one gene, PRDG7, was upregulated after compatible pollination. Noteworthy, the same gene was upregulated after T. dactyloides pollination, which suggests that it responds to pollination in general. Remarkably, the pattern of expression of PRXs is opposite when incompatible pollination and fungal invasion by F. graminearum are compared. While T. dactyloides pollination represses PRXs, F. graminearum infection enhances PRXs expression. Another gene family involved in the production of lignin is LAC. After F. graminearum infection, Lac5b, Lac5c, Lac7a, Lac7c, Lac7k, Lac15a, and Lac15b showed increased expression, whereas Lac7 h was downregulated (Fig. 7B). Only Lac7a increased in response to U. maydis and Lac15a decreased after T. dactyloides pollination. None of the LAC responded after compatible pollination. These results show that incompatible pollination negatively impacts the entire phenylpropanoid and monolignol pathways, whereas fungal infection promotes the expression of genes involved in the lignin biosynthesis pathway.
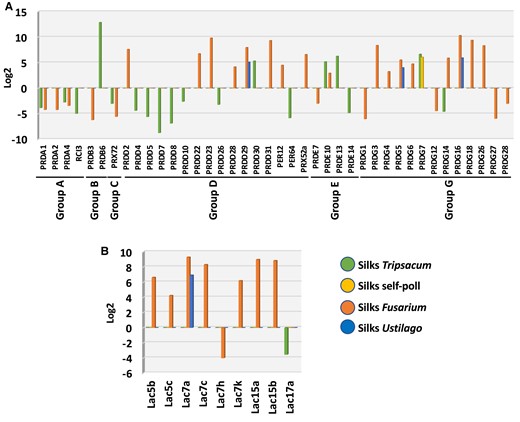
Genes encoding enzymes that facilitate reactive oxygen species reduction and lignin polymerization are only regulated in maize silks by foreign invaders. A) Peroxidase (PRX) and B) LAC genes are largely upregulated after F. graminearum infection, partly upregulated after U. maydis infection and tendentially downregulated by pollen tubes of T. dactyloides.
NAC and WRKY transcriptional regulators are highly upregulated in response to fungal infection, but not to pollination
Activation of monolignol biosynthesis pathways and polymerization of the monolignol units is genetically controlled by an evolutionarily conserved regulatory network of NAC and MYB transcription factors (Ohtani and Demura 2019). To elucidate whether the high expression of the monolignol pathway after F. graminearum infection is associated with the NAC-MYB gene regulatory network in maize silks, we next studied the expression of the 2 gene families. Four secondary wall–associated NACs (ZmSWN1, ZmSWN3, ZmSWN6, and ZmSWN7) have been described as members of the NAC-MYB gene regulatory network (Zhong et al. 2011). None of these genes is expressed in maize silks in treated or in control samples (Supplementary Fig. S2). Since the characterized secondary wall–associated NAC genes were not expressed in maize silks, we examined the entire NAC transcription gene family to identify putative NAC regulators in maize silks. In comparison with vegetative tissues such as roots and leaves, expression of NAC genes in unpollinated mature silks was low (Fig. 8A). Expression of NAC genes increased during silk aging but was especially high after fungal infection (Fig. 8, A and B). For instance, while ZmNAC109, ZmNAC75, and ZmNAC5 were particularly induced by F. graminearum, ZmNAC23, ZmNAC30, and ZmNAC4 were upregulated by both F. graminearum and U. maydis infection (Fig. 8B). These differences could explain why many more genes are detected as differentially expressed in maize silks after F. graminearum compared with U. maydis infection.
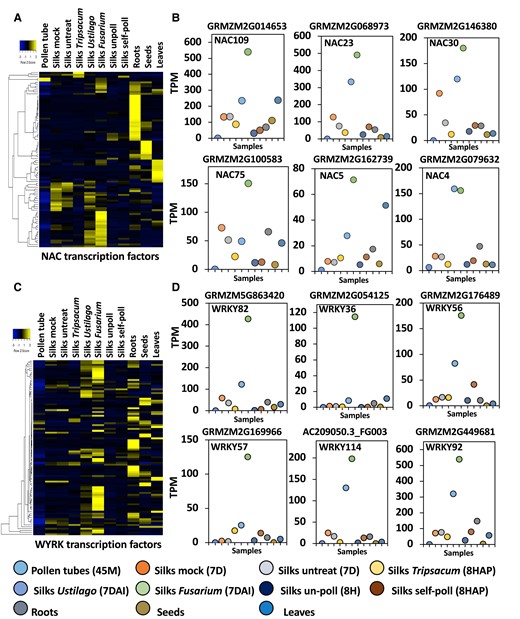
NAC and especially WRKY transcription factor genes are induced after fungal infection in maize silks. A) Heat map displaying induction of NAC transcription factor genes in maize silks at indicated conditions. B) Transcript per million reads (TPMs) of selected NAC transcription factor genes. C) Heat map displaying induction of WYRK transcription factor genes in maize silks at indicated conditions. D) TPMs of selected WYRK transcription factor genes indicate high induction especially after F. graminearum infection.
Some MYB transcription factors function downstream of NACs as a second layer of transcriptional regulation (Ohtani and Demura 2019). Targets of these MYB proteins include enzyme-encoding genes involved in xylan and lignin biosynthesis. Thus, we next investigated expression of the entire maize MYB transcription factor family and their responses to pollination and fungal invasion (Supplementary Fig. S3A). We found that, for example, ZmMYB20 and ZmMYB100 were highly upregulated after T. dactyloides pollination (Supplementary Fig. S3, B and C), but a strong correlation to specific invaders like above-described NAC transcription factor genes was not observed.
Another class of important gene regulators acting in plant defense mechanisms are WRKY transcription factors. So far, WRKYs have been comprehensively studied using mainly Arabidopsis as plant model organism (Seo et al. 2015). In maize silks, members of the WRKY gene family were highly induced in response to pathogen infection, particularly by F. graminearum (Fig. 8C). Three genes, ZmWRKY36, ZmWRKY57, and ZmWRKY82, are specifically upregulated after F. graminearum infection, whereas ZmWRKY56, ZmWRKY92, and ZmWRKY114 are upregulated by both pathogens, but significantly higher by F. graminearum (Fig. 8D). WRKY transcription factors have been shown to positively regulate jasmonate (JA)-mediated plant defense to necrotrophic fungal pathogens (Chen et al. 2021). Two JA-associated genes, octadecanoid-responsive Arabidopsis (ORA59) and plant defensin 1.2a (PDF1.2), were upregulated in AtWRKY75 (a homolog of ZmMWKY36) overexpressing Arabidopsis transgenic plants (Chen et al. 2021). The maize ortholog of Arabidopsis ORA59 has been annotated as Ereb58 (AP2-EREBP-transcription factor 58). F. graminearum induced its expression 6.5-fold in maize silks. Expression of the maize ortholog of PDF1.2 (defensin-like protein2, Def2), however, changed in response neither to pathogens nor to pollination (Supplementary Data Set 1). Further investigations are now necessary to identify targets of above-described NAC and WRKY transcription factors to better understand differences between the gene regulatory networks activated after F. graminearum and U. maydis infection.
F. graminearum and U. maydis provoke a diterpenoid response in maize silks
To further investigate the differential response of maize silks to F. graminearum and U. maydis infection, we next compared the global transcriptional response. Even though a large overlap of DEGs was detected (Fig. 9A), F. graminearum induced a significantly larger differentially expressed response compared with U. maydis (Fig. 9B). Gene ontology analysis showed enrichment of responses to stimuli, macromolecule, and protein modification as the primary functional processes affected by F. graminearum. Divergently, U. maydis mostly impacted metabolic processes associated with photosynthesis (Supplementary Fig. S4). Considering that these findings are very general, we searched for pathways that are associated with the processes and selected the terpenoid pathway, a major component of maize defenses to herbivores, pathogens, and other environmental challenges, as an example (Mafu et al. 2018). In maize, the antifungals, phytoalexins, kauralexins, and dolabralexins, but also the hormone, GA, were derived from a common precursor, ent-copalyl diphosphate (ent-CPP). Geranyl geranyl diphosphate (GGPP) is the precursor of ent-CPP (Fig. 9C). Two catalytically redundant diterpene synthase (diTPS) enzymes, ANTHER EAR 1 (ZmAn1) and ANTHER EAR 2 (ZmAn2), control the production of ent-CPP (Schmelz et al. 2011; Mafu et al. 2018; Murphy et al. 2021). Infection by F. graminearum resulted in a more than 14-fold increase in ZmAN2 expression (Fig. 9D). ZmAN1 and ZmAN2 did not change in response to U. maydis. It has been shown that ZmAN2 is critical for kauralexin and dolabralexin biosynthesis, whereas ZmAN1 controls GA biosynthesis. This allows a pathway partition by separating precursor flux toward primary and secondary diterpenoid pathways, respectively (Mafu et al. 2018; Ding et al. 2019; Murphy et al. 2021). ZmKSL2 (kaurene synthase-like2) and ZmKSL4 drive the production of kauralexin and dolabralexin biosynthesis, respectively. Genes for both enzymes are upregulated in response to F. graminearum infection (Fig. 9, E and F), while only ZmKSL2 is induced after U. maydis infection (Fig. 9E). Cytochrome P450 monooxygenase 16/18 (ZmCYP71Z16/18), another enzyme required to generate kauralexin and dolabralexin, was induced 12-fold and 8-fold in response to F. graminearum and U. maydis, respectively (Fig. 9G). Notably, the maize antifungal toxin zealexin is produced in an initially independent pathway using ZmdiTPS6/11 (diTPSs). ZmdiTPS6/11 was only upregulated in response to F. graminearum (Fig. 9H). In contrast to genes required for phytoalexin biosynthesis, ZmKSL5 and KO1 representing key genes for GA biosynthesis were downregulated after F. graminearum infection (Fig. 9, I and J). In summary, these results show that F. graminearum infection triggered the expression of phytoalexin biosynthesis genes, whereas genes involved in GA biosynthesis are downregulated. In contrast, only minor or no changes in terpenoid pathway genes was triggered after U. maydis infection. These findings are in agreement with a previous study showing that F. graminearum infection activates the generation of phytoalexins in root tissues of maize (Mafu et al. 2018).
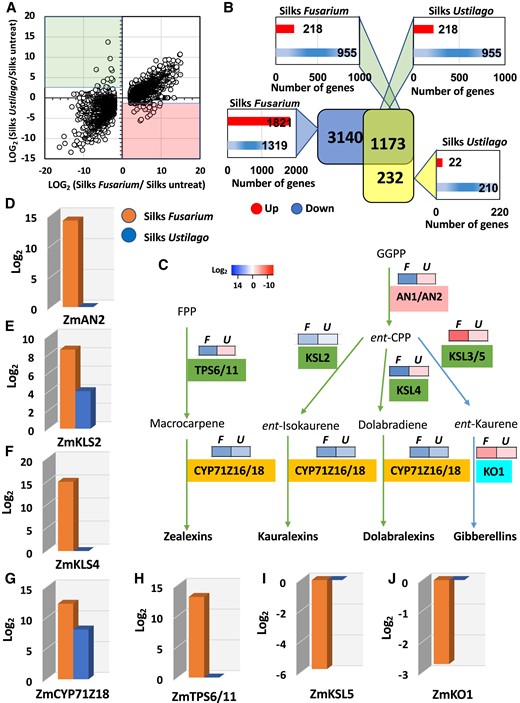
Activation of the defense system in maize silks to inhibit fungal growth. A) Scatter plot representing the log2 for all DEGs in the maize silks infected with F. graminearum (x axis) vs. infected with U. maydis (y axis) and B) comparison of corresponding DEGs. C) Overview of the biosynthetic pathway of diterpenoids (zealexins, kauralexins, and dolabralexins), major components of the maize defense system, and the growth hormone GA. Class I diTPSs are shown in pink, Class II diTPSs in green, and P450 enzymes in orange and cyan, respectively. Heat map represent pattern of expression being red high expression and blue low expression. Geranylgeranyl diphosphate (GGPP), ent-CPP, F. graminearum (F), and U. maydis (U). Fold change expression analysis of the various diterpenoid biosynthesis enzyme genes, namely D)ent-CPP synthase (AN2), E)ent-kaurene synthase2 (KSL2), F)ent-kaurene synthase4 (KSL4), G) cytochrome P450 monooxygenase (CYP71Z18), H) diTPSs (TPS6/11), I)ent-kaurene synthase5 (KSL5), and J)ent-kaurene oxidase (KO1).
Conclusion
The first comparative studies on fungal and pollen tube invasion have shown that partly the same members of RLK families (catharanthus roseus receptor-like kinases, leucine-rich repeats and RLKs), their ligands (rapid alkalinization factors and other small cysteine-rich proteins), as well as membrane channels and reactive oxygen species production/removal are involved in recognition and reception/rejection of the various biotic invaders (Kessler et al. 2010; Bircheneder and Dresselhaus 2016; Mondragón-Palomino et al. 2017; Zhou and Dresselhaus 2019; Gao et al. 2022). Much less is known about rejection and defense of unwanted pollen tubes and fungi after successful penetration. We have expanded previous studies using maize as a grass model species and characterized the molecular signatures of maize stigmas (silks) to 4 different invaders including own pollen tubes. The major findings are summarized in Fig. 10. Although invasion occurs mainly via stigmatic papillae hair cell structures, further growth of each invader significantly varies and triggers a different molecular response. (i) Growth of own pollen tubes occurs through the transmitting tract of the maize silk triggering regulation of relatively few genes including those involved in senescence processes. (ii) T. dactyloides pollen tubes penetrate the same route but often do not target the transmitting tract and arrest after growing 2 to 3 cm triggering many responses including modifications of the cell wall of silk cells and defense reactions. Similar to pollen tubes, (iii) hyphae of F. graminearium grow initially intercellularly, until they penetrate and kill the cells. The invaded silk by F. graminearium elicits the strongest response, which appears to be mainly directed to inhibit fungal growth and to defend itself from the invader. In contrast, (iv) U. maydis grow exclusively intracellularly without killing host cells. Compared with F. graminearium, the silk response is much weaker and can be considered in our opinion as a “half-hearted” defense reaction.
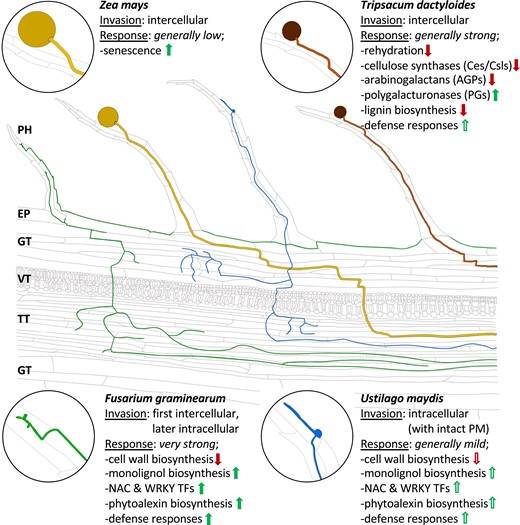
Summary of invasion pattern and major transcriptional responses of maize silks to own and alien pollen tubes as well as to 2 different types of fungi. Ocher, own maize pollen tubes; brown, alien T. dactyloides pollen tubes; green, F. graminearum; and blue, U. maydis hyphae. Enlargements show germinated pollen (left) and fungal spores/conidia (right). PH, papilla hair cell; EP, epidermis; GT, ground tissue; VT, vascular tissue; TT, transmitting track. Green arrows represent upregulation. Red arrows depict downregulation. Solid arrows indicate strong response. Unfilled arrows show mild response.
The molecular signatures associated with each of the different invaders now enable approaches to target and enhance the specific molecular responses to defend invading fungal pathogens but also to lower rejection processes during incompatible pollen tube invasion. This could help reach the goal to overcome hybridization barriers and to increase the gene pool for plant breeding and seed production. The analysis and identification of the transcription factor regulatory network controlling cell wall development and defense responses in this study serves already as a very promising starting point for many future studies in grass crops to achieve plant pathogen resilience. Moreover, the comprehensive data set provided opens the windows also for other groups to identify, for example, more and novel genes involved in response to biotic invaders, to obtain stress-induced promoters and to identify further pathways involved in the described biotic interaction processes in maize and other grasses. Finally, we further propose that the highly elongated maize silk, which consists of only few cell types and is a relative transparent tissue, is also very useful as a grass model for pathogen studies at both the genomic and cell biological levels.
Materials and methods
Plant material and growth conditions
Seeds of maize (Z. mays L.) inbred line B73 as well as a pUbi::CerTN-L15 marker line (see below) were germinated in germination trays containing soils and then transferred to pots (10 cm diameter, 10 seedlings per pot) containing a standard substrate and soil mixture (1:1, v/v) in the greenhouse (Begcy et al. 2019). After 3 wk in the greenhouse, all genotypes were transferred to 10 L pots and grown under controlled conditions of 16 h of light at 26 ± 2 °C and 8 h of darkness at 22 ± 2 °C and a constant air humidity of 60%. Supplemented light of ∼20,000 lux was provided to adjust day length duration. An automated temperature–water-based irrigation system was used to supply water according to plant consumption in a time-based preprogrammed schedule. Plants were fertilized twice a week with 2% (w/v) Hakaphos and monitored throughout their vegetative and reproductive development. Diploid T. dactyloides lines were grown in the greenhouse using the same growth conditions as maize. Plants were fertilized every second week.
Maize developmental stages were determined as previously described (Abendroth et al. 2011). Plant tissues were collected as follows: third leaves and whole roots were collected from maize seedlings at Stage V3. Fresh pollen was collected at Stage R1 (anthesis; Begcy and Dresselhaus 2017) and either directly frozen or germinated for 2 h on Petri dishes containing pollen germination medium (Begcy et al. 2019; Li et al. 2024). Silks at Stage R1 were collected 5 d after emergence from ear husks. Silks of the same stage were harvested 8HAP using pollen of maize inbred line B73 and diploid T. dactyloides, respectively. Whole developing seeds were excised 11 d after pollination. After collection, all samples were immediately frozen in lipid N2 until further usage.
Fungal material and growth conditions
F. graminearum wild-type strain 8/1 expressing GFP (Jansen et al. 2005) was used to visualize fungal growth in maize silks. Induction of conidia was carried out in wheat medium (7.5 g of 100% Bio-Weizengras LEBEPUR, Lebepur GmbH in 1 L of distilled water) on a shaker (150 rpm) in the dark at 28 °C for 5 to 6 d and then filtered through a 70-μm sieve (Easytrainer, Greiner Bio-One). The filtrate was centrifuged at 4,000 rpm for 10 min at 4 °C, and the supernatant was discarded. Inoculation medium for infection of maize cobs consisted of conidia resuspended in cold sterile distilled water to a final concentration of 1 to 2 × 105 conidia per milliliter.
The GFP-expressing U. maydis strain used in this study was SG200-GFP, deriving from the solo-pathogenic wild-type strain SG200 (Kämper et al. 2006). U. maydis was grown on a shaker (200 rpm) at 28 °C to an OD600 of 1 in YEPS-light medium (1% yeast extract, 0.4% peptone, and 0.4% sucrose), washed 3 times with sterile distilled water and centrifuged at 3,500 rpm for 5 min each time (Woriedh et al. 2015). Spores were finally resuspended in sterile distilled water to an OD600 of 3, and the suspension was used for infecting plant material as described below.
In vitro pollination of maize silks
Maize ears of ∼8 to 10 cm in length from inbred line B73 were harvested and first cut longitudinally and then transversally (without wounding of silks) to obtain segments of ∼5 cm in length. Segments were placed with the cut site on half MS agar medium in small Petri dishes (5.5 cm diameter) that were then placed in bigger Petri dishes (13.5 cm diameter) lacking medium. All silks were orientated toward one direction, cut to the same length, and kept at room temperature before further processing. Silks were pollinated with freshly collected pollen from B73 as well as a homozygous transgenic line containing a pUbi::CerTN-L15 marker construct (see below). Pollen tube growth was examined up to 24 h after pollination by confocal laser scanning microscopy.
Fungal infection of maize silks
Maize silks were infected with the F. graminearum strain 8/1 and U. maydis strain SG200, both expressing GFP, using the silk channel inoculation method (Maier et al. 2006). In brief, fungal infection of maize silks was performed by applying 4 mL suspension with a syringe containing either F. graminearum conidia or U. maydis SG200-GFP spores, respectively. We monitored that silks were completely covered with the infection suspension solutions. After inoculation, samples were incubated at 28 °C in the dark, and cobs were covered with plastic bags for 3 d to favor fungal development. Infected silks were harvested 7DAI. A parallel set of untreated maize silks were harvested 7D after silk emergence (DASE) as an aging control. Senescence of unpollinated silk has been associated with tissue disintegration. Microscopically, the disintegration of unpollinated aging silks appears shrunken and resembles the tissue alterations associated with silk abscission following fertilization. Aging and disintegration of silks are initiated about 7 DASE (Šimášková et al. 2022). Similarly, maize silks inoculated with sterile water were used as mock infection control.
Generation of a fluorescent maize pollen tube marker line
The calcium sensor line pDONR-CerTN-L15 (Denninger et al. 2014) containing cerulean and citrine fluorescent proteins was recombined into the p7i-Ubi-GW destination vector via attL x attR recombination (LR) reaction to generate pUbi::CerTN-L15. This expression vector was transformed into maize HiIIA/B hybrid via Agrobacterium-mediated maize transformation as previously described (Frame et al. 2002). Transformants were selected by selection markers and fluorescent properties.
Microscopy analyses
Confocal images were taken using a SP8 confocal laser scanning microscope (CLSM; Leica). Cell walls were visualized using propidium iodide staining (Sigma-Aldrich). Infected maize silks were incubated with 50 mm propidium iodide staining solution for 15 min under dark conditions and mounted directly on objective slides before microscopy. Maize silks were incubated with propidium iodide, and the various GFP fluorescent reporter lines were imaged using an inverted Leica TCS SP8 CLSM equipped with a monochrome high-speed Leica DFC365 FX camera and a hybrid detector. The following Leica objectives were used: HC PL FLUOTAR 10x/0.30, PL APO 20x/0.7 CS, and HCX PL APO 40x/1.3 oil. GFP fluorescence was detected at 500 to 550 nm using a 488-nm argon laser and propidium iodide at 570 to 650 nm after 561 nm excitation with a DPSSLaser (Antosz et al. 2020) using mounted living silks samples. Images were taken and processed using Leica Application Suite X (LAS X) software.
RNA isolation and reverse transcription qPCR
Total RNA was extracted using the Pure Link RNA Mini Kit (Ambion, Life Technologies, Carlsbad, CA, USA) and treated with RNase-Free DNase (Qiagen) according to the manufacturer's instructions. Complementary DNA (cDNA) synthesis was performed using the Invitrogen SuperScript II reverse transcription system and oligo(dT) primers.
RNA quantification and quality control were assessed using a 2100 Bioanalyzer (Agilent Technologies). Reverse transcription qPCRs were carried out using KAPA SYBR Fast qPCR master mix (Peqlab) using an Eppendorf Mastercycler Realplex Real-Time detection system. Amplification efficiencies were calculated based on standard curves as previously described (Kim et al. 2024). Primer sequences with their corresponding melting temperatures and amplification efficiencies are provided in Supplementary Table S3. Cycling parameters consisted of 5 min at 95 °C followed by 40 cycles of 95 °C for 15 s, 60 °C for 30 s, and 70 °C for 30 s. Resulting Cq values were imported into qbase + v3.1 (Biogazelle). Quality control settings required that Cq values from technical replicates differ by <0.5 cycles. Reactions were performed in triplicate for each RNA sample using at least 3 biological replicates. Normalization was based on 3 most stably expressed internal reference genes GRMZM2G000531, GRMZMG003734, and GRMZM2G003631 detected with the GeNorM program implemented by qBASE + v3.1. The full list of primers used in this study is included in Supplementary Table S3. The resulting calibrated normalized relative quantities were then exported into Microsoft Excel for further analysis. Log2-transformed and calculation of fold change values were performed using the 2-ΔΔCt method.
Library preparation and RNA-seq
Library preparation and RNA-seq were performed at the Regensburg University genomics service facility KFB (Competence Center for Fluorescent Bioanalytics) as described in the Illumina TruSeq Stranded mRNA Sample Preparation Guide, the Illumina HiSeq 1000 System User Guide (Illumina), and the KAPA Library Quantification Kit-Illumina/ABI Prism User Guide (Kapa Biosystems). Library construction was generated from cDNA fragments adenylated at their 3′ ends and ligated with indexing adapters, and specific cDNA libraries were subsequently created after PCR enrichment (Begcy et al. 2019). Libraries were quantified using the KAPA SYBR FAST ABI Prism Library Quantification Kit (Kapa Biosystems). Equimolar amounts of each library were used for cluster generation on the cBot using the Illumina TruSeq Paired-End Cluster Kit v3. Sequencing was performed on a HiSeq 1000 Illumina instrument using the indexed, 50-cycle paired-end read (PR) protocol and the TruSeq SBS v3 Reagents according to the Illumina HiSeq 1000 System User Guide. Image analysis and base calling resulted in bcl-files, which were converted into fastq-files using the bcl2fastq v2.18 software (https://www.illumina.com.cn/content/dam/illumina-support/documents/downloads/software/bcl2fastq/bcl2fastq2-v2-18-software-guide-15051736-01.pdf).
Data processing, mapping, and differential expression
PRs were trimmed and filtered using Trimmomatic v.0.35 (Bolger et al. 2014). Data quality was assessed using FastQC. Processed PRs were aligned to the high-quality maize reference genome sequence AGPv3 assembly using annotation release-5b+ (Gramene AGPv3.27, https://www.maizegdb.org/assembly/) and a mapping program, STAR v. 2.5.2a (Dobin et al. 2013). Reads overlapping with more than one gene region were excluded from the analysis. To test for the presence/absence of batch effect and outliers, variance stabilizing transformation of raw counts and analyzed nonmerged technical replicates was performed using R package DESeq2 (Love et al. 2014). For pairwise comparisons, DEGs were identified using DEseq2 by analyzing the number of reads aligned to the genes. The thresholds for differential expression were set at fold change >2 and P < 0.05 (after the false discovery rate adjustment for multiple testing) for the null hypothesis.
Gene network analysis
To build gene expression networks, log2-transformed normalized expression values from DEGs were analyzed using the cor function in the R package WGCNA (Langfelder and Horvath 2008). Genes without shared expression profile (unrooted genes) were removed from the network analyses. Then, the blockwiseModules function was implemented to generate a network under a thresholding power of 12, minimum module size of 10, and merged cut height of 0.005. To evaluate the predicted gene network analysis, Pearson correlation coefficients were estimated for each node, and a P-value of 0.05 was used as cutoff. Genes with shared expression profiles were considered as seed candidates and used to obtain direct and indirect interactions using the STRING app (Szklarczyk et al. 2023) of Cytoscape (Shannon et al. 2003) that allows to infer protein–protein associations from coexpression data. We implemented a deep learning model method called functional associations using variational autoencoders to infer the STRING's coexpression networks. A high edge confidence of at least 0.7 was used as a threshold. Functional categories for each hub were assigned using the R package NEAT (Signorelli et al. 2016). Only interactions with high levels of confidence were considered as valid.
Accession numbers
A detailed list of genes and IDs mentioned in this study is provided in Supplementary Data Set 5.
Acknowledgments
The authors are grateful to Cathrin Kröger and Wilhelm Schäfer for providing the F. graminearum 8/1-GFP strain and to Vera Göhre and Michael Feldbrügge for the U. maydis SG200-GFP strain and for their valuable support to optimize fungal growth conditions. The authors thank Mayada Woriedh for sample collection and Armin Hildebrand for plant care.
Author contributions
T.D. designed the project. K.B. and T.D. conceived the experiments and wrote the article with input from all authors. K.B., M.M.-P., and T.D. analyzed the data. K.B. and M.M.-P. performed the bioinformatics analyses. L.Z.-Z., P.L.-S., and M.L.-M. performed the experimental assays. All authors read and approved the article.
Supplementary data
The following materials are available in the online version of this article.
Supplementary Table S1. Summary of samples generated for this study.
Supplementary Table S2. List of DEGs in maize silks after self-pollination.
Supplementary Table S3. List of primers used in this study.
Supplementary Figure S1. Validation of RNA-seq data using qPCR.
Supplementary Figure S2. Key secondary wall–associated NAC transcription factor genes known as ZmSWNs are not upregulated in maize silks in response to F. graminearum and U. maydis.
Supplementary Figure S3. MYB transcription factors gene expression in response to pollination and fungal invasion.
Supplementary Figure S4. Biological processes associated with fungal invasion in maize silks.
Supplementary Data Set 1. Median expression values of treatment/tissue of genes mapped to the maize genome (AGPv3, version 82.6).
Supplementary Data Set 2. Common DEGs between compatible and incompatible pollination.
Supplementary Data Set 3. Genes differentially expressed in response to incompatible pollination with T. dactyloides.
Supplementary Data Set 4. Downregulated rooted genes after compatible and incompatible pollination used for gene network analyses.
Supplementary Data Set 5. Detailed gene names and IDs described in this study.
Funding
The German Research Foundation (DFG) is acknowledged for financial support via SFB924 (to T.D.). This work was partially supported by the USDA National Institute of Food and Agriculture, Hatch project FLA-ENH-005853 to K.B.
Data availability
Data will be available in the NCBI Sequence Read Archive (SRA) under the bioproject accession number PRJNA1098880. All other materials are available from the corresponding authors upon request.
Dive Curated Terms
The following phenotypic, genotypic, and functional terms are of significance to the work described in this paper:
References
Author notes
The authors responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (https://dbpia.nl.go.kr/plphys/pages/General-Instructions) are: Thomas Dresselhaus ([email protected]) and Kevin Begcy ([email protected]).
Conflict of interest statement. The authors declare no conflict of interests.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}