





Abstract
Clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR associated protein (Cas)-mediated gene disruption has revolutionized biomedical research as well as plant and animal breeding. However, most disease-causing mutations and agronomically important genetic variations are single base polymorphisms (single-nucleotide polymorphisms) that require precision genome editing tools for correction of the sequences. Although homology-directed repair of double-stranded breaks (DSBs) can introduce precise changes, such repairs are inefficient in differentiated animal and plant cells. Base editing and prime editing are two recently developed genome engineering approaches that can efficiently introduce precise edits into target sites without requirement of DSB formation or donor DNA templates. They have been applied in several plant species with promising results. Here, we review the extensive literature on improving the efficiency, target scope, and specificity of base editors and prime editors in plants. We also highlight recent progress on base editing in plant organellar genomes and discuss how these precision genome editing tools are advancing basic plant research and crop breeding.
Introduction
CRISPR/Cas9 derived from the adaptive immune system in prokaryotes has been repurposed for genome engineering in a wide range of living organisms (Knott and Doudna, 2018). The Cas9 nuclease recognizes target sequence under the guidance of a single-guide RNA (sgRNA) and induces a double-stranded break (DSB) three bases upstream of a protospacer adjacent motif (PAM) sequence. The DSB triggers either a cellular nonhomologous end joining (NHEJ) DNA repair pathway to generate small insertions and deletions (indels) that disrupt gene function, or a homology-directed repair (HDR) pathway to install precise edits when a donor repair template exists (Hsu et al., 2014; Komor et al., 2017a). The NHEJ-mediated gene knockout is highly efficient in mammalian cells and plants and has been harnessed to disrupt disease-related genes in animal models and patients and to improve crop agronomic traits (Hua et al., 2019c; Zeballos and Gaj, 2021). However, most disease-causative mutations and agronomically important variations are SNPs that require precision genome editing tools for correction or installation. The HDR-mediated repair of DSBs can introduce precise modifications at target sites, but the efficiency is quite low in differentiated animal and plant cells (Steinert et al., 2016; Yeh et al., 2019). Although much progress has been made in improving the efficiency of HDR-mediated editing, the results are far from satisfaction and the HDR process usually comes with high levels of indels and other undesired byproducts (Yeh et al., 2019; Čermák, 2021). Therefore, both gene therapy in humans and crop improvement call for alternative precision genome engineering tools.
Base editing (BE) is a breakthrough technology that can precisely and efficiently install point mutations at target sites without requirement of DSB generation and donor DNA templates (Rees and Liu, 2018). The first developed cytosine base editor (CBE) is composed of an SpCas9 (D10A) nickase fused to a cytidine deaminase and an uracil glycosylase inhibitor (UGI) that enables C•G to T•A transition (Figure 1A; Komor et al., 2016). The sgRNA guides the CBE to the target genomic DNA and induces R-loop formation. The cytidine deaminase then deaminates the cytosine (C) into uracil (U) in the exposed nontarget DNA strand, and subsequent DNA repair and replication results in C to T base conversion (Box 1). The adenine base editor (ABE) shows a similar architecture as CBE (Figure 1B) and employs a laboratory-evolved Escherichia coli transfer RNA adenosine deaminase (ecTadA) to deaminate adenine (A) into inosine (I) in the exposed nontarget strand, and subsequent DNA repair and replication cause A to G base conversion (Box 1; Gaudelli et al., 2017). Both CBEs and ABEs have been applied in different plant species to interrogate gene function and improve crop traits (Molla and Yang, 2019; Mishra et al., 2020). However, the CBEs and ABEs mainly induce base transitions, but not base transversions (Kim, 2018). The glycosylase base editors (GBEs) were recently developed to induce base transversions (C to A or C to G) by replacing UGI with a uracil-DNA glycosylase (UNG) in certain CBEs (Figure 1A; Kurt et al., 2020; Zhao et al., 2020). Previous work in rice (Oryza sativa) had already noted that removal of UGI from BE3 leads to an increase in C to A and C to G substitutions at some target sites (Lu and Zhu, 2017). In addition, the use of human activation-induced deaminase (hAID) instead of the rat apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 1 (rAPOBEC1) deaminase also somehow enhances the rates of C to non-T base conversions in rice (Ren et al., 2018). A recent study assessing three GBE platforms in rice, tomato (Solanum lycopersicum), and poplar (populus) revealed that each GBE has distinct sequence preferences and shows varied editing efficiencies in different plant species, indicating that no single GBE fits all needs (Sretenovic et al., 2021a).
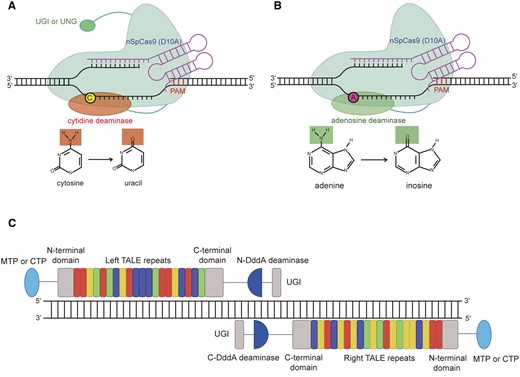
The architecture of base editors. A, Schematic view of cytosine base editor and glycosylase base editor. The cytosine base editor contains an UGI at the C-terminus, whereas the glycosylase base editor has an UNG at the C-terminus. The chemical structures of cytosine and uracil are shown and the deaminated amino group is highlighted in red. B, Schematic view of adenine base editor. The chemical structures of adenine and inosine are shown and the deaminated amino group is highlighted in green. C, Architecture of the DddAtox-derived cytosine base editor targeting to plant organelles. DddA deaminase, a dsDNA cytidine deaminase from Burkholderia cenocepacia; MTP, mitochondrion transit peptide; CTP, chloroplast transit peptide.
Prime editing (PE) is another DSB-independent precision genome editing tool, which can install all possible base conversions, small indels as well as their combinations at target sites (Anzalone et al., 2019). PE is achieved by a Cas9 nickase-reverse transcriptase fusion protein and a prime editing guide RNA (pegRNA), which has a 5′-spacer sequence that specifies the target site and a 3′-extension of primer binding site (PBS) sequence and reverse transcription (RT) template encoding the desired edit (Figure 2A). The pegRNA guides the fusion protein to the target site and then induces a nick at the nontarget strand, which anneals with PBS and primes the RT of RT template that ultimately copies the desired sequence into the target site after a complex DNA repair process (Anzalone et al., 2019). The prime editor 1 (PE1) has a wild-type moloney murine leukaemia virus reverse transcriptase (M-MLV-RT) fused at the C-terminus of an SpCas9 (H840A) nickase, whereas PE2 uses an engineered M-MLV-RT pentamutant with much higher RT performance and thus improves the PE efficiency three-fold in human cells. The PE3 requires a nick gRNA (ngRNA) to further boost the editing efficiency, whereas the PE3b nicks the edited DNA sequences to improve the purity of editing products. The PE system has been tested in several plant species with promising results (Butt et al., 2020; Hua et al., 2020a; Jiang et al., 2020; Li et al., 2020b; Lu et al., 2020; Tang et al., 2020; Wang et al., 2021a; Xu et al., 2020a, 2020c).
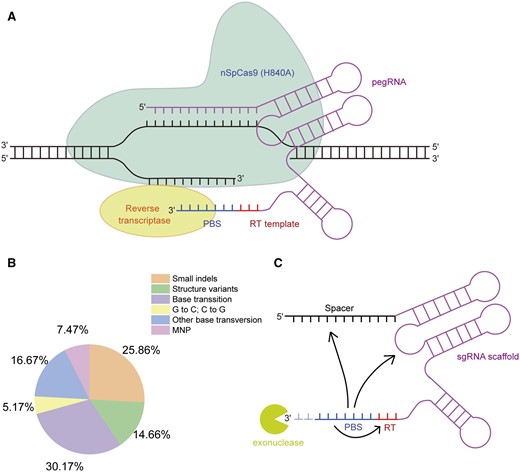
The power and limitation of prime editing. A, Schematic view of the prime editing system. B, The 384 known causative mutations regulating important agronomic traits in rice (data from Wei et al., 2021b), classified by mutation types. MNP, multiple nucleotide polymorphisms. C, The nucleotides in the 3′-extension of pegRNA can affect pegRNA folding, and the 3′-extension is susceptible to exonuclease degradation in cells. The arrows indicate possible base pairing within the 3′-extension or between the 3′-extension and sgRNA scaffold.
The BE and PE tools have been extensively reviewed in recent years (Abdullah et al., 2020; Anzalone et al., 2020; Kantor et al., 2020; Azameti and Dauda, 2021; Li et al., 2021f; Molla et al., 2021). In this update, we mainly focus on the strategies that help improve the efficiency, target scope, and specificity of base editors and prime editors in plants, and summarize the applications of BE and PE in basic plant research and crop improvement.
Expanding the target scope of base editors
The base editors containing SpCas9 nickase have a BE window of 3–5nucleotides and only the target bases within this window can be edited with high efficiency (Komor et al., 2016; GaudelLi et al., 2017; Kurt et al., 2020). The SpCas9 nickase in base editors requires an appropriate NGG (N=any nucleotide and G=guanine) PAM sequence to place the target bases within the BE window. Therefore, such PAM requirement severely restricts the genomic sites that can be accessed by the base editors. This constraint can be addressed by developing new base editors with Cas9 orthologs or variants that recognize alternative or relaxed PAM sequences.
The SpCas9 variants SpCas9-VQR (NGA PAM), SpCas9-VRER (NGCG PAM), SpCas9-NRRH (NRRH PAM), SpCas9-NRCH (NRCH PAM) and SpCas9-NRTH (NRTH PAM), iSpyMacCas9 (NAAR PAM), a Cas9 hybrid from SpCas9 and SmacCas9, and the Cas9 orthologs Nm1Cas9 (NNNNGATT PAM), Nm2Cas9 (NNNNCC PAM), SaCas9 (NNGRRT PAM), and its variant SaKKH (NNNRRT PAM) only slightly expanded the target range of base editors in rice due to their complex PAM sequence requirements (Hua et al., 2019b; Li et al., 2021a; Qin et al., 2019; Sretenovic et al., 2021b; Xu et al., 2021c; Zhang et al., 2020a). The Cas9 ortholog ScCas9 and its variant ScCas9++, and three Cas9 variants, xCas9, SpCas9-NG, and SpG, recognize the more relaxed NNG and NG PAM sequences, respectively, which substantially expanded the target range of base editors in plants (Hua et al., 2019a; Negishi et al., 2019; Ren et al., 2019; Zeng et al., 2019; Zhang et al., 2019; Zhong et al., 2019; Wang et al., 2020; Liu et al., 2021b; Ma et al., 2021). Nevertheless, their editing activities and PAM preferences varied in plants. The more recently engineered SpRY variant enables BE in almost PAM-less fashion in rice, although it recognizes NY PAM sites less efficiently than NR PAM sites (Li et al., 2021a; Ren et al., 2021b; Xu et al., 2021d; Zhang et al., 2021a). The relaxed-PAM Cas9 variants in some cases raise the concern of self-targeting and increased off-target editing; these risks can be mitigated by selecting highly specific sgRNAs and modifying the sgRNA scaffold sequence downstream of the spacer sequence (Qin et al., 2020a).
Improving the BE efficiency
The DNA deaminase activity is one of the key determinants of BE efficiency. Selecting DNA deaminase with high enzyme activity at various sequence contexts is a straightforward way to improve BE efficiency. The widely distributed cytidine deaminase orthologs in nature show diverse sequence preferences, different deaminase activities and distinct affinities to single-stranded DNA or RNA (ssDNA or ssRNA) substrates (Koblan et al., 2018; Cheng et al., 2019; Yu et al., 2020), which together have a large impact on BE outcomes. Many cytidine deaminases such as rAPOBEC1 (Li et al., 2017; Lu and Zhu, 2017; Zong et al., 2017), human APOBEC3A (A3A; Zong et al., 2018), human APOBEC3B (A3B; Jin et al., 2020), Petromyzon marinus cytidine deaminase 1 (PmCDA1; Shimatani et al., 2017), Lethenteron japonicum CDA1-like 4 (LjCDA1-L4), and hAID (Ren et al., 2018; Xu et al., 2021a) have been used for cytosine BE in plants. The rAPOBEC1 in BE3 shows a strong sequence preference for TC, with low or no editing at the GC (G=guanine and C=cytosine) context (Komor et al., 2016; Zong et al., 2017), whereas hAID, PmCDA1, and A3A show less sequence bias in plants (Shimatani et al., 2017; Ren et al., 2018; Zong et al., 2018). In general, CBEs containing the human A3A cytidine deaminase or its mutant form A3A-Y130F show high editing activities at all sequence contexts and wide BE windows in both monocot and dicot plants (Zong et al., 2018; Cheng et al., 2021; Randall et al., 2021; Ren et al., 2021a). Moreover, directed evolution has been used to improve the editing efficiency and target sequence compatibility of natural cytidine deaminases (Thuronyi et al., 2019). Three evolved cytidine deaminases, evorAPOBEC1, evoFERNY, and evoCDA1, show robust cytosine editing efficiencies in rice when combined with Cas9 orthologs or variants (Zeng et al., 2020). In contrast to the cytosine deamination enzymes, there is no natural enzyme capable of deaminating adenine in DNA. The sole adenosine deaminase used in ABE was derived from the laboratory-evolved ecTadA (Gaudelli et al., 2017). This situation restricts further improvement of ABEs. Fortunately, more efficient adenine deaminase variants, TadA8e and TadA8s, have been artificially evolved from TadA7.10 (Gaudelli et al., 2020; Richter et al., 2020). The ABE8e catalyzes adenine deamination 1,100-fold faster than ABE7.10 (Lapinaite et al., 2020) and thus boosts the adenine BE efficiency remarkably in rice, wheat (Triticum aestivum), and Nicotiana benthamiana (Han et al., 2021; Wang et al., 2021c; Wei et al., 2021a; Yan et al., 2021). The ABE8e and ABE8s also exhibit a high compatibility with different Cas9 orthologs and variants, and a side-by-side comparison in rice showed that ABE8e outperforms ABE8s (Yan et al., 2021). Interestingly, incorporating the two mutations V82S and Q154R from TadA8s into TadA8e generates a more efficient adenosine deaminase, TadA9, in rice (Yan et al., 2021).
Since the DNA deaminases are efficient, the amount of functional base editor proteins targeted to the cell nucleus becomes a major bottleneck of BE efficiencies (Koblan et al., 2018; Zafra et al., 2018). The expression level of base editors and their nuclear targeting can be affected by many factors including codon-usage, regulatory elements driving their expression and the number, position and strength of nuclear localization signals (NLS; Koblan et al., 2018; Zafra et al., 2018). Through codon optimization of deaminase–Cas9 fusion and incorporating a strong bipartite NLS (bpNLS), the base editor Anc689BE4max showed much higher editing efficiencies compared to BE3 in rice (Wang et al., 2019). The simplified adenine base editor ABE-P1S with a monomer TadA7.10 fused with Cas9 improved adenine BE efficiency in rice, perhaps due to the higher expression level of ABE-P1S compared to the original ABE-P1 (Hua et al., 2020b). Coupling the production of base editor with selection marker expression through a P2A self-cleaving peptide is a simple way to ensure a high expression level of the base editor. This strategy substantially improves the adenine BE efficiency in rice (Li et al., 2020c).
During plant tissue culture, selecting the base edited cells from millions of transformed cells for regeneration is very challenging, especially for target sites with low editing efficiency. The use of a surrogate reporter is an effective approach that helps to enrich rare editing events. Xu et al. (2020b) developed a SurroGate system that enriched BE events at target sites on hygromycin selection medium when a defective hygromycin resistance gene in the T-DNA is corrected by BE. This strategy not only increased BE frequency but also enhanced the ratios of homozygous substitutions.
The base editors only deaminate target base(s) and create mismatch(es) (U:G or I:T) at target sites, so the editing results largely depend on the DNA repair pathway involved (Box 1). Modifying the DNA repair pathways can affect both BE outcomes and efficiencies (Chen et al., 2021a; Koblan et al., 2021; Komor et al., 2017b). Fusing UGI to rAPOBEC1-dCas9 can prevent UDG from removing U in U:G mismatches, which results in a three-fold higher cytosine BE efficiency (Komor et al., 2016). Moreover, incorporating DNA repair proteins into GBEs also affects C•G-to-G•C editing efficiencies and purities (Chen et al., 2021a; Koblan et al., 2021).
Improving the precision of BE
The BE process is usually accompanied with byproducts including bystander mutations, indels, and other undesired base conversions (Gaudelli et al., 2017; Komor et al., 2016; Kurt et al., 2020). The cytidine deaminase in CBEs and GBEs creates U:G mismatch(es) at target sites, which is predominately resolved by the error-prone BER pathway in cells, resulting in the random incorporation of nucleotides or occasional DSBs that lead to indel formation (Box 1). Fusing an UGI to rAPOBEC1-Cas9n decreases indel frequency and C to G/A substitution rates, but markedly increases C to T conversion rates (Komor et al., 2016). Appending more copies of UGI to CBE or recruiting more UGIs to the target site can further improve the product purity of CBEs (Qin et al., 2019, 2020b; Ren et al., 2021a). The ABEs do not induce any undesired products in stable transgenic plants because the inosine in DNA cannot be efficiently excised by cellular alkyl adenine DNA glycosylase in plants (Box 1).
Bystander mutations frequently arise when multiple target bases exist in the BE window (Komor et al., 2016; Gaudelli et al., 2017). The unintended bystander mutations can cause additional amino acid changes that may affect the gene function. This issue may be mitigated by adjusting the linker flexibility between the deaminase and the Cas9 nickase of the editor (Tan et al., 2019, 2020), engineering a deaminase with narrow BE window or strong sequence preference (Kim et al., 2017; Gehrke et al., 2018), and shifting BE window by using different circularly permutated Cas9 variants (Huang et al., 2019) or using different cytidine deaminases and/or configurations (Ren et al., 2021a). However, the efficacy of these strategies in plants remains to be tested in the future.
Similar to other CRISPR–Cas-based genome editing tools, base editors can bind and edit certain genomic sites that show sequence homology to the sgRNA. The Cas-dependent off-target editing can be reduced by selecting sgRNAs with high specificity or by using high-fidelity Cas variants (Yeh et al., 2018; Jang et al., 2021). In addition, the deaminase moieties in base editors can cause Cas-independent genome-wide and/or transcriptome-wide off-target editing in mammalian cells and plants, because most deaminases have inherently nonspecific affinities to ssDNA or ssRNA substrates (Grünewald et al., 2019a; Jin et al., 2019; Rees et al., 2019; Zhou et al., 2019; Zuo et al., 2019). This raises concerns about the wide application of base editors in gene therapy and functional genomics studies. Three major strategies have been developed to mitigate Cas-independent deamination activity of base editors. One strategy is to limit the exposure time of base editors to the off-target sites. Delivering the base editors as a ribonucleoprotein (RNP) complex is an effective way to decrease both the Cas-dependent and Cas-independent off-target editing because the RNP is degraded rapidly in cells (Jang et al., 2021). Recently, a transformer BE (tBE) system was developed that maintains highly efficient on-target editing with negligible levels of genome-wide and transcriptome-wide off-target mutations (Wang et al., 2021b). The tBE was inactive at off-target sites because of a cleavable deoxycytidine deaminase inhibitor domain. When the tBE binds at on-target sites, it is activated by cleaving off a deoxycytidine deaminase inhibitor domain. Another strategy is to select deaminases with low off-target editing activity. A screen of hundreds of natural cytidine deaminases with diverse sequences led to several candidates that display promising on/off-target editing profiles (Yu et al., 2020). Structure-guided mutagenesis of the deaminase domains can change their affinity to ssDNA and ssRNA substrates and is widely used to reduce off-target effects of base editors on genomic DNA and/or cellular RNA (Grünewald et al., 2019b; Jin et al., 2020; Lee et al., 2020; Li et al., 2021b; Rees et al., 2019; Yu et al., 2020; Zhou et al., 2019; Zuo et al., 2020). A third strategy is to change the protein fusion configuration of base editors. Several studies demonstrated that embedding the deaminase domains in Cas9 nickase dramatically reduced DNA and RNA off-target editing, perhaps due to the steric hindrance imposed by such fusion configuration (Li et al., 2020d; Liu et al., 2020b; Nguyen Tran et al., 2020). It should be noted that ABEs are highly specific in plants and the number of unwanted single-nucleotide variations (SNVs) induced by CBEs in rice and tomato is comparable or even lower than that caused by somatic mutations during tissue culture (Jin et al., 2019; Randall et al., 2021; Ren et al., 2021a). Such unwanted SNVs may have a limited influence on crop breeding and can be further eliminated by using appropriate cytidine deaminases (Jin et al., 2020; Ren et al., 2021a).
Extending BE to plant organellar genomes
In addition to the nuclear genome, plants also have two organellar genomes in mitochondria and chloroplasts, which encode hundreds of essential genes for respiration and photosynthesis, respectively. Engineering organellar genomes thus holds great promise for improving plant metabolic and photosynthetic performance, which may increase crop yields (Li et al., 2021d). The plant mitochondrial genes can be disrupted by mitochondrion-targeted transcription activator-like effector nucleases (mitoTALENs), but repairing mitoTALEN-induced DSBs mainly generates large imprecise deletions, which may lead to many side effects (Kazama et al., 2019; Arimura et al., 2020). Chloroplast genomes of angiosperms can be engineered through homologous recombination by direct bombardment of donor DNA into chloroplasts (Svab et al., 1990). This method has been used for decades with series of improvements, but its application is largely limited to several species of dicots that do not include major cereal crops (Bock, 2015). The CRISPR-based precise genome editing tools have advanced rapidly, but these tools could not access organelle genomes due to the difficulty of delivering both the guide RNA and Cas proteins into organelles. Therefore, developing precise genome editing tools targeting organellar genomes of both monocots and dicots are highly desired.
Recently, Mok and coworkers made a breakthrough in precise organellar genome engineering (Mok et al., 2020). They found that the deaminase domain of the bacterial toxin DddA (DddAtox) shows structural homology with APOBEC enzymes, but deaminates the cytosines within double-stranded DNA (dsDNA). This feature raises the possibility of directly deaminating dsDNA in organellar genomes by fusing DddAtox with organelle targeted programmable dsDNA-binding proteins, such as transcription activator-like effector (TALE) repeat arrays. To avoid cellular toxicity of DddAtox, the researchers designed a split DddAtox strategy by fusing each half of DddAtox with a mitochondrion-targeting TALE array and a UGI (Figure 1C; Mok et al., 2020). The deaminase activity can be reconstituted when a pair of TALE arrays binds adjacently on target DNA in mitochondria. These DddAtox-derived cytosine base editors (DdCBEs) can edit mitochondrial genes with various efficiencies in human cells (Mok et al., 2020). Later, DdCBEs were used to model disease-causing mtDNA mutations in mice and zebrafish (Guo et al., 2021; Lee et al., 2021). Transient expression of DdCBEs in lettuce (Lactuca sativa) and rapeseed (Brassica napus) protoplasts also successfully induced C-to-T substitutions in several mitochondrial genes with up to 23% efficiencies (Kang et al., 2021). Interestingly, chloroplast transit peptides can guide the DdCBEs to plastids, which makes plastome base-editing feasible. A collection of studies have shown that chloroplast-targeting DdCBEs can edit chloroplast genes with varied efficiencies in both protoplasts and stable transgenic lines (Kang et al., 2021; Li et al., 2021c; Nakazato et al., 2021). The editing efficiencies of DdCBEs at different target sites were affected by the types of split and orientations of DddAtox, the cytosine positions and sequence contexts. It is quite surprising that a substantial portion of stable transgenic lines show homoplasmic or near homoplasmic mutations at target sites (Li et al., 2021c; Nakazato et al., 2021), considering that the plastome is highly allopolyploid in each plant cell. Moreover, the transgene-null, homoplasmic mutation lines can be obtained by segregating out nuclear T-DNA in subsequent generations (Nakazato et al., 2021).
Despite the successful application of DdCBEs in different plant species, several issues such as sequence preference of DddAtox deaminase, frequent bystander mutations and potential off-target editing, remain to be addressed to make organellar genome engineering precise and efficient in plants.
PE is at an early stage
The prime editors can install all possible base substitutions and small indels that enable over 90% disease-causing mutations in humans to be corrected or modeled (Anzalone et al., 2019). Analyzing a comprehensive catalog of 348 causative variants regulating important agronomic traits in rice suggests that over 85% of them can be precisely introduced into the rice genome by prime editors (Wei et al., 2021b; Figure 2B). Moreover, prime editors induce much lower frequencies of off-target editing than base editors at potential off-target sites because BE requires only target DNA–sgRNA spacer complementarity to install mutations (Komor et al., 2016; Gaudelli et al., 2017), whereas PE requires additional target DNA-pegRNA PBS region complementarity and PAM downstream sequence-RT product complementarity to achieve editing (Anzalone et al., 2019). In contrast to base editors, prime editors did not induce undesired bystander mutations and genome-wide and transcriptome-wide pegRNA-independent off-target editing (Anzalone et al., 2019; Kim et al., 2020; Jin et al., 2021). These features make PE potentially the most promising and powerful genome editing tool since the CRISPR technology was first applied in human cells.
The PE system has been tested in several plant species (Butt et al., 2020; Hua et al., 2020a; Jiang et al., 2020; Li et al., 2020b; Lin et al., 2020; Lu et al., 2020; Tang et al., 2020; Wang et al., 2021a; Xu et al., 2020a, 2020c). The average editing frequencies in plants was much lower than that reported in mammalian cells and many sites even failed to be edited, especially in dicot species (Lu et al., 2020; Wang et al., 2021a). Although PE events can be detected in stable transgenic lines of rice and tomato, the ratio of homozygous and biallelic edits is extremely low (Hua et al., 2020a; Li et al., 2020b; Lin et al., 2020; Lu et al., 2020; Xu et al., 2020a, 2020c), which also indicates the inefficiency of PE in plants. Another interesting general finding in plants is that in most cases the editing efficiency of PE3 is comparable or even lower than that of PE2 (Hua et al., 2020a; Lin et al., 2020; Tang et al., 2020; Xu et al., 2020a), which is not consistent with the results in mammalian cells (Anzalone et al., 2019,).
While the BE efficiency was substantially improved through codon-optimization of base editors, changing NLS configuration and selecting strong promoters for fusion protein expression (see above), the PE efficiency was only modestly improved by these strategies in plants and mammalian cells (Lin et al., 2020; Liu et al., 2021a; Lu et al., 2020; Tang et al., 2020). Screening diverse reverse transcriptase orthologs with varied catalytic activities also could not boost the PE efficiency in rice (Lin et al., 2020). In addition, leveraging a surrogate reporter system to enrich prime-edited cells showed only a limited success in improving PE efficiency in rice (Xu et al., 2020a).
The pegRNA is a key determinant of PE efficiency, and optimal PBSs and RT templates are crucial for efficient editing (Anzalone et al., 2019). The difficulty for pegRNA design is that the PBS sequence and part of the RT sequence at the 3′-end of pegRNA (10–17 nt) is complementary to the spacer region at the 5′-end of pegRNA, such that their potential annealing is expected to cause pegRNA self-circularization (Figure 2A), which may hamper the PE process. Indeed, pegRNAs showed a compromised ability to induce Cas9-mediated DNA indels in mammalian cells and tomato (Liu et al., 2021c; Van Vu et al., 2021), perhaps due to their self-circularization. The moloney-murine leukaemia virus (M-MLV) reverse transcriptase in PE2 and PE3 tolerates high temperature for RT because it incorporates multiple mutations that improve protein thermostablility (Anzalone et al., 2019). Moreover, high temperature may destabilize the in vivo PBS-spacer intramolecular or intermolecular annealing, which can help pegRNA to fold correctly. However, increasing the temperature of tissue culture had no effect on PE efficiencies in rice and tomato (Lin et al., 2020; Lu et al., 2020; Tang et al., 2020; Van Vu et al., 2021). Surprisingly, it was reported that appending a 20-nt Csy4 recognition site with a hairpin structure to the 3′-end of pegRNA can potentially inhibit the pegRNA self-circularization and thus markedly boosts the PE efficiency in mammalian cells (Liu et al., 2021c).
The self-circularization issue of pegRNA underscores the importance of selecting an optimal length and GC content for PBSs, which determine the melting temperature (Tm) between PBS and spacer sequence. A recent study in rice suggests that designing PBSs with a Tm of 30°C leads to the highest editing frequencies (Lin et al., 2021). Perhaps, the 30°C Tm ensures an efficient annealing between PBS and nicked nontarget strand while maximally avoids pegRNA misfolding in rice. Interestingly, introducing mismatches between the PBS and nontarget strand can somehow increase the PE efficiency in mammalian cells (Kim et al., 2020). The mismatches may also disrupt the intramolecular PBS-spacer annealing of pegRNA. However, whether these strategies can be expanded to other plant species is unclear. Preliminary results from tomato showed that modifying the PBS length or introducing mismatches between PBS and spacer has no effect on the PE efficiency (Van Vu et al., 2021). Another strategy to mitigate the potential pegRNA misfolding is to boost the expression level of pegRNA. Although the percentage of correctly folded pegRNA remains unchanged, their quantities were substantially increased due to the extremely high levels of expression. This approach was recently used to improve PE efficiency in maize (Zea mays) (Jiang et al., 2020). Interestingly, expressing a pair of pegRNAs that encode the same edits to target both sense and antisense DNA strands can enhance PE efficiency in rice and human cells (Lin et al., 2021; Zhuang et al., 2021).
In addition to the potential pegRNA self-circularization issue, the sequence in the 3′-extension of pegRNA may form secondary structures that interfere with the folding of the sgRNA scaffold (Figure 2C). A well-known example is that a cytosine at the first position in the RT template nearly abolished the editing activity of PEs because this cytosine can pair with G81 in the sgRNA scaffold, which affects Cas9 binding to target site (Anzalone et al., 2019). Moreover, the 3′-extension of the pegRNA is susceptible to exonuclease degradation in cells since it cannot be protected by Cas9 binding (Figure 2C). The partially degraded pegRNAs can bind to target site but lose the ability to introduce the desired edit. Appending the structured RNA motifs to the 3′-terminus of pegRNAs can prevent the degradation of the 3′-extension and thus boost PE efficiency in human cell lines (Nelson et al., 2021).
The pegRNA design is much more complicated than the sgRNA design for other CRISPR-based editing tools because the pegRNA design needs to follow many basic rules and the plausible combinations of different PBS and RT templates increase the difficulty for selecting optimal 3′-extensions of pegRNA (Anzalone et al., 2019). Therefore, the manual design of pegRNA is error-prone and limited in throughput. Recently, several programs and web tools were developed that can automatically design the pegRNAs with high-throughput by following general design rules, and some web tools even provide sequences of oligonucleotides for pegRNA cloning (Table 1).
Web-based tools and command lines for pegRNA design
| Software | Supported enzyme | Off target analysis | Input | Prime design for pegRNA cloning | Support pegRNA design for plant | Special features | References |
|---|---|---|---|---|---|---|---|
| Multicrispr | SpCas9 | Yes | – | No | Yes | – | Bhagwat et al. (2020) |
| Prime editing design tool | SpCas9 SpCas9-NG | Yes | – | No | No | Designing pegRNAs for correcting pathogenic variants in ClinVar database | Morris et al. (2020) |
| PnB Designer | SpCas9 | No | DNA sequence or genomic coordinates | No | No | – | Siegner et al. (2021) |
| PINE-CONE | SpCas9 | No | DNA sequence, edit types, PBS and RT length | No | No | – | Standage-Beier et al. (2021) |
| pegFinder | SpCas9 SpCas9-NG SpRY | Yes (not for plants) | DNA sequence | Yes | Yes | _ | Chow et al. (2020) |
| PE-Designer | SpCas9 SpCas9-VQR SpCas9-VRERSpCas9-NG | Yes | DNA sequence | No | Yes | – | Hwang et al. (2021) |
| pegIT | SpCas9 SpCas9-NG CjCas9 SaCas9 SaCas9-KKH | Yes | DNA sequence or genomic coordinates | Yes | Yes | – | Anderson et al. (2021) |
| Easy-Prime | SpCas9 | No | DNA sequence | No | Yes | Considering pegRNA secondary structure and predicting the prime editing efficiency | Li et al. (2021e) |
| PrimeDesign | SpCas9 | Yes (not for plants) | DNA sequence | No | Yes | Calculating pegRNA secondary structure and designing pegRNAs for saturation mutagenesis | Hsu et al. (2021) |
| DeepPE | SpCas9 | No | DNA sequence | No | Yes | Predicting the prime editing efficiency | Kim et al. (2021) |
| PlantPeg Designer | SpCas9 SpCas9-NG Cas9 orthlogs | No | DNA sequence | Yes | Yes | Designing dual pegRNAs | Lin et al. (2021) |
| Software | Supported enzyme | Off target analysis | Input | Prime design for pegRNA cloning | Support pegRNA design for plant | Special features | References |
|---|---|---|---|---|---|---|---|
| Multicrispr | SpCas9 | Yes | – | No | Yes | – | Bhagwat et al. (2020) |
| Prime editing design tool | SpCas9 SpCas9-NG | Yes | – | No | No | Designing pegRNAs for correcting pathogenic variants in ClinVar database | Morris et al. (2020) |
| PnB Designer | SpCas9 | No | DNA sequence or genomic coordinates | No | No | – | Siegner et al. (2021) |
| PINE-CONE | SpCas9 | No | DNA sequence, edit types, PBS and RT length | No | No | – | Standage-Beier et al. (2021) |
| pegFinder | SpCas9 SpCas9-NG SpRY | Yes (not for plants) | DNA sequence | Yes | Yes | _ | Chow et al. (2020) |
| PE-Designer | SpCas9 SpCas9-VQR SpCas9-VRERSpCas9-NG | Yes | DNA sequence | No | Yes | – | Hwang et al. (2021) |
| pegIT | SpCas9 SpCas9-NG CjCas9 SaCas9 SaCas9-KKH | Yes | DNA sequence or genomic coordinates | Yes | Yes | – | Anderson et al. (2021) |
| Easy-Prime | SpCas9 | No | DNA sequence | No | Yes | Considering pegRNA secondary structure and predicting the prime editing efficiency | Li et al. (2021e) |
| PrimeDesign | SpCas9 | Yes (not for plants) | DNA sequence | No | Yes | Calculating pegRNA secondary structure and designing pegRNAs for saturation mutagenesis | Hsu et al. (2021) |
| DeepPE | SpCas9 | No | DNA sequence | No | Yes | Predicting the prime editing efficiency | Kim et al. (2021) |
| PlantPeg Designer | SpCas9 SpCas9-NG Cas9 orthlogs | No | DNA sequence | Yes | Yes | Designing dual pegRNAs | Lin et al. (2021) |
Note: The dash in the table indicates that the data are not available.
Web-based tools and command lines for pegRNA design
| Software | Supported enzyme | Off target analysis | Input | Prime design for pegRNA cloning | Support pegRNA design for plant | Special features | References |
|---|---|---|---|---|---|---|---|
| Multicrispr | SpCas9 | Yes | – | No | Yes | – | Bhagwat et al. (2020) |
| Prime editing design tool | SpCas9 SpCas9-NG | Yes | – | No | No | Designing pegRNAs for correcting pathogenic variants in ClinVar database | Morris et al. (2020) |
| PnB Designer | SpCas9 | No | DNA sequence or genomic coordinates | No | No | – | Siegner et al. (2021) |
| PINE-CONE | SpCas9 | No | DNA sequence, edit types, PBS and RT length | No | No | – | Standage-Beier et al. (2021) |
| pegFinder | SpCas9 SpCas9-NG SpRY | Yes (not for plants) | DNA sequence | Yes | Yes | _ | Chow et al. (2020) |
| PE-Designer | SpCas9 SpCas9-VQR SpCas9-VRERSpCas9-NG | Yes | DNA sequence | No | Yes | – | Hwang et al. (2021) |
| pegIT | SpCas9 SpCas9-NG CjCas9 SaCas9 SaCas9-KKH | Yes | DNA sequence or genomic coordinates | Yes | Yes | – | Anderson et al. (2021) |
| Easy-Prime | SpCas9 | No | DNA sequence | No | Yes | Considering pegRNA secondary structure and predicting the prime editing efficiency | Li et al. (2021e) |
| PrimeDesign | SpCas9 | Yes (not for plants) | DNA sequence | No | Yes | Calculating pegRNA secondary structure and designing pegRNAs for saturation mutagenesis | Hsu et al. (2021) |
| DeepPE | SpCas9 | No | DNA sequence | No | Yes | Predicting the prime editing efficiency | Kim et al. (2021) |
| PlantPeg Designer | SpCas9 SpCas9-NG Cas9 orthlogs | No | DNA sequence | Yes | Yes | Designing dual pegRNAs | Lin et al. (2021) |
| Software | Supported enzyme | Off target analysis | Input | Prime design for pegRNA cloning | Support pegRNA design for plant | Special features | References |
|---|---|---|---|---|---|---|---|
| Multicrispr | SpCas9 | Yes | – | No | Yes | – | Bhagwat et al. (2020) |
| Prime editing design tool | SpCas9 SpCas9-NG | Yes | – | No | No | Designing pegRNAs for correcting pathogenic variants in ClinVar database | Morris et al. (2020) |
| PnB Designer | SpCas9 | No | DNA sequence or genomic coordinates | No | No | – | Siegner et al. (2021) |
| PINE-CONE | SpCas9 | No | DNA sequence, edit types, PBS and RT length | No | No | – | Standage-Beier et al. (2021) |
| pegFinder | SpCas9 SpCas9-NG SpRY | Yes (not for plants) | DNA sequence | Yes | Yes | _ | Chow et al. (2020) |
| PE-Designer | SpCas9 SpCas9-VQR SpCas9-VRERSpCas9-NG | Yes | DNA sequence | No | Yes | – | Hwang et al. (2021) |
| pegIT | SpCas9 SpCas9-NG CjCas9 SaCas9 SaCas9-KKH | Yes | DNA sequence or genomic coordinates | Yes | Yes | – | Anderson et al. (2021) |
| Easy-Prime | SpCas9 | No | DNA sequence | No | Yes | Considering pegRNA secondary structure and predicting the prime editing efficiency | Li et al. (2021e) |
| PrimeDesign | SpCas9 | Yes (not for plants) | DNA sequence | No | Yes | Calculating pegRNA secondary structure and designing pegRNAs for saturation mutagenesis | Hsu et al. (2021) |
| DeepPE | SpCas9 | No | DNA sequence | No | Yes | Predicting the prime editing efficiency | Kim et al. (2021) |
| PlantPeg Designer | SpCas9 SpCas9-NG Cas9 orthlogs | No | DNA sequence | Yes | Yes | Designing dual pegRNAs | Lin et al. (2021) |
Note: The dash in the table indicates that the data are not available.
Although the in vitro biochemical experiments provide support for the early RT process of PE (Anzalone et al., 2019), the downstream cellular DNA repair pathways that determine the PE outcomes remain poorly understood. Recent studies in human cells indicated that the DNA mismatch repair (MMR) pathway impedes PE and transient inhibition of specific MMR genes can remarkably boost PE efficiencies in several mammalian cell types (Chen et al., 2021b; Ferreira da Silva et al., 2021).
Applications of BE and PE in plants
As DSB-independent genome editing tools, the editing outcomes of BE and PE are more predictive than that of the DSB-dependent editing tools (Komor et al., 2016; Gaudelli et al., 2017; Anzalone et al., 2019), which provides many advantages in gene function analysis and precision crop breeding. The CRISPR–Cas9-induced DSBs in the coding sequence can potentially induce chromosomal rearrangements and large deletions, and the predominant frameshift indel events can lead to the transcription of abnormal transcripts that support the translation of aberrant proteins (Tuladhar et al., 2019; Zuccaro et al., 2020). Such aberrant proteins may confound downstream analysis of phenotypes. In contrast, BE and PE can disrupt genes by precisely introducing premature stop codons or inactivating highly conserved splicing sites in coding regions, which largely avoid undesired mutations and the formation of aberrant proteins (Billon et al., 2017; Kluesner et al., 2021; Li et al., 2019; Ren et al., 2021a). In addition, potential gene regulatory elements such as miRNA binding sites, upstream open reading frames, transcription factor binding sites, and posttranslational modification sites can be precisely edited by the BEs and PEs to infer the functions of the elements (Xing et al., 2020; Ren et al., 2021a).
The causative mutations for most agronomically important traits are single nucleotide polymorphisms and small indels (Wei et al., 2021b), which are suitable for correction by BEs and PEs for precision crop breeding. At the present time, over 35% and 85% causative variants corresponding to 225 quantitative traits in rice can be precisely introduced by BEs and PEs, respectively (Figure 2B), and many examples of successful editing have been reported by different groups (Azameti and Dauda, 2021; Li et al., 2021f; Molla et al., 2021). Moreover, the BEs and PEs can create novel and valuable germplasms in a relative short time. Editing the uORF of a bZIP gene in strawberry (Fragaria vesca) by CBE creates many novel genotypes with a continuum of sugar content variation (Xing et al., 2020). The herbicide tolerance trait can be rapidly introduced into different elite crop varieties by BEs and PEs, which have been summarized in recent reviews (Azameti and Dauda, 2021; Li et al., 2021f; Molla et al., 2021). The highly efficient ABE can even simultaneously install point mutations in four endogenous herbicide target genes in a commercial rice cultivar, which holds great promise for weed control in the field (Yan et al., 2021). Multiplex BE in rice generates the P171F/G628E/G629S triple mutations in the acetolactate synthase (ALS) gene, thus expanding the tolerance spectrum to ALS inhibitor herbicides (Zhang et al., 2020b).
For crop genes for which functional SNPs are not known, BEs and PEs can perform targeted saturation mutagenesis that produces valuable genetic variants with improved agronomic performance (Molla et al., 2021). Such in vivo directed evolution of plant genes can be achieved by combining BEs and PEs with an sgRNA library tiling the user-defined coding region. Employing CBE and ABE with an sgRNA library covering the full-length coding region of OsALS1 generates multiple base substitutions that confer tolerance to ALS inhibitors (Kuang et al., 2020). Combining BEs with an sgRNA tiling library targeting the CCT domain of OsACC gene also results in multiple herbicide resistance mutations (Li et al., 2020a; Liu et al., 2020a). Interestingly, combining CBE with a low coverage sgRNA design covering the key amino acids responsible for enzymatic activity of the rice Waxy gene generates a range of mutations that can fine-tune the amylose content in rice grains (Xu et al., 2020d). The saturation mutagenesis will benefit from high editing efficiencies at different sequence contexts, increased sgRNA coverage, wide BE window, and more flexible base conversion types. BEs and PEs with the SpCas9-NG or SpRY variants can significantly increase the sgRNA coverage (Li et al., 2020a). The dual deaminase base editor (DuBE), which fuses both the cytidine and adenosine deaminases with Cas9 nickase and UGI, can simultaneously introduce C to T and A to G changes at target sites and thus increases the mutation potential and the number of possible amino acid changes (Grünewald et al., 2020; Li et al., 2020a; Sakata et al., 2020; Xie et al., 2020; Zhang et al., 2020d). Incorporating the more efficient deaminases such as TadA8e into the DuBE can further increase the editing frequencies and concurrent C to T and A to G conversion rates (Xu et al., 2021a). While the BEs have restrictions in base conversion scopes and induce limited types of amino acid changes, PEs can install all types of small genetic changes that can be harnessed for producing all possible substitutions for key amino acids when combined with a well-designed pegRNA library (Xu et al., 2021b). Recently, the T7 RNA-polymerase-guided base editors was developed to facilitate in vivo continuous gene evolution in bacteria, yeast, and human cells (Box 2; Moore et al., 2018; Álvarez et al., 2020; Chen et al., 2020; Cravens et al., 2021; Park and Kim, 2021), which has the potential for evolving plant genes in the future.
Conclusion and perspectives
Since the first report of BE in 2016, there has been remarkable progress in precision genome engineering tools in plants. The efficiency and accuracy of base editors and prime editors have improved considerably, but some challenges remain to be addressed (see Outstanding Questions box). Successful applications of BE and PE in plants are limited to several plant species only and many important crops remain inaccessible to these tools largely due to difficulties in plant transformation (Li et al., 2021f; Molla et al., 2021). Additionally, the construct design for these engineering tools requires extensive optimization for each crop species (Hassan et al., 2021). It should be noted that a slight change in expression modules in the binary vector could lead to a large improvement of editing efficiency, as exemplified by the improvement of ABE efficiencies in dicot species (Kang et al., 2018; Niu et al., 2021). Many useful strategies established in mammalian cells to improve BE and PE efficiencies deserve to be tested in plants (Park et al., 2021; Zhang et al., 2020c). Moreover, the editing efficiencies of BEs and PEs can vary widely at different target sites in plants. Considering that the plant transformation process is labor-extensive for most plant species, designing highly efficient sgRNAs or pegRNAs for successful editing is very important. In mammalian cells, the machine learning models driven by large data can predict the editing outcomes and frequencies for BEs and PEs, which can guide the sgRNA or pegRNA design (Arbab et al., 2020; Kim et al., 2021; Song et al., 2020; Koblan et al., 2021; Marquart et al., 2021; Yuan et al., 2021). It is unclear whether these models can be used in plants, since the differences in cell types and cell cycles can severely affect BE and PE results. Answers to these issues will enable broad applications of BEs and PEs in plants that can facilitate basic research and precision crop breeding.
CBEs and ABEs can induce four base transitions in different plant species.
Available CBEs and ABEs target plant genomes with little PAM constraints and work efficiently with negligible levels of off-target editing.
DdCBEs can induce C to T conversions in plant organellar genomes.
Prime editors can install all possible point mutations and small indels in different plant species with varied efficiencies.
Base editors and prime editors facilitate basic plant research and precision crop breeding.
How can BE efficiencies be improved at pyrimidine-rich PAM sites?
What is the best GBE to install C•G-to-G•C base transversions in plants?
How can PE efficiencies be improved in plants, especially in dicot species?
How can we design highly efficient pegRNAs for different plants?
Can BE and PE outcomes and efficiencies be predicted in plants?
Base editors require Cas9 binding to the target site, which induces the formation of an RNA–DNA ternary R-loop. The cytosine in the exposed nontarget DNA strand of the R-loop is deaminated by the tethered cytidine deaminase in CBE and GBE, thus creating a U:G mismatch at target site (Figure for Box 1; Komor et al., 2016; Kurt et al., 2020). The U:G mismatch is primarily resolved by the base excision repair (BER) pathway, which removes the uracil through cellular UNG activity and generates an abasic site. The CBE inhibits this process by fusing a UGI at the C terminus of Cas9 nickase (Komor et al., 2016). In addition, the Cas9 protein in CBE nicks the target strand, stimulating the cellular DNA mismatch repair pathway to use a uracil-containing strand as template to replace the G in the U:G mismatch with an A. This elegant design results in an efficient conversion of the U:G mismatch to the desired U:A product, which becomes T:A after DNA replication. In contrast, the GBE promotes BER by fusing a UNG at the C-terminus of Cas9 nickase and preferentially creates an abasic lesion at target site (Kurt et al., 2020). However, the mechanistic details governing error-prone incorporation of A or G at the abasic site remain unclear. Moreover, as Cas9 nicks the target strand, a DSB is formed when the abasic site on the nontarget strand is converted into a nick by an apurinic or apyrimidinic site lyase (AP lyase). The DSB results in indel formation at the target site. The adenosine deaminase in ABE creates an I:T mismatch at the target site (Figure for Box 1; Gaudelli et al., 2017). In contrast to CBE, the cellular alkyl adenine DNA glycosylase removes inosine in DNA quite inefficiently, which obviates the requirement of inhibiting base excision repair activity in ABE. Similar to CBE, Cas9 in ABE nicks the target strand that leads to much more efficient conversion of the I:T intermediate to the desired G:C base-pairing. Therefore, the adenine BE yields products of high purity and with extremely low levels of indel formation.
Figure for Box 1: Schematic illustration of cellular DNA repair mechanisms that determine the editing outcomes of base editors. The deaminases in base editors can create U:G or I:T mismatch. Resolving U:G mismatch by different DNA repair pathways lead to diverse editing outcomes. The I:T mismatch is converted into G:C base paring by DNA mismatch repair pathway. nCas9, Cas9 nickase; AP lyase, apurinic/apyrimidinic site lyase.
T7 RNA polymerase-guided BE utilizes the DNA deaminase-T7 RNA polymerase fusion to continuously introduce mutations downstream of a T7 promoter during transcription (Figure for Box 2; Moore et al., 2018). The bacteriophage-derived monomeric T7 RNA polymerase and T7 promoter can form an orthogonal RNA transcription system in bacteria and eukaryotes, which independently controls gene expression without interfering with the host gene transcription (Zhang et al., 2021b). When the T7 promoter is specifically recognized by T7 RNA polymerase, the downstream multiple kilobases of coding DNA can be transcribed with high efficiency. The single-stranded DNA exposed during the T7 RNA polymerase transcription can serve as the substrate for DNA deaminase, which randomly introduces mutations within the transcribed region of any relevant length. Thus, the T7 RNA polymerase, through fusion with different deaminases (cytidine and adenosine deaminase), substantially widens the mutational spectrum (Cravens et al., 2021). Similar to CBEs, inhibiting the cellular UNG activity improves the mutation rates of cytidine deaminase-T7 RNA polymerase fusion (Chen et al., 2020). The T7 RNA polymerase-guided BE can be exploited as a platform for directed gene evolution in vivo with minimal off-target mutations. Compared to ABE- and CBE-based directed evolution of genes, this platform has several advantages: (1) It obviates the need to design an sgRNA library for each target gene. (2) It edits target base(s) without PAM constraint. (3) It mutates the target gene continuously leading increased sequence diversity. (4) The mutation process can be controlled by fine-tuning the expression of DNA deaminase-T7 RNA polymerase fusion.
Figure for Box 2: Schematic illustration of T7 RNA polymerase-guided BE. T7 RNA polymerase in the DNA deaminase-T7 RNA polymerase fusion transcribes the target gene downstream of the T7 promoter and the DNA deaminase can convert cytosine to thymine (or adenine to guanine) on the exposed coding strand. The introduced mutations are marked in red color.
K.H., P.H., and J.-K.Z. wrote the manuscript. P.H. prepared the figures.
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (https://dbpia.nl.go.kr/plphys/pages/General-Instructions) is: Jian-Kang Zhu ([email protected]).
Acknowledgments
We apologize to the authors whose work was not cited here due to space limitation. This work was supported by the National Natural Science Foundation of China (No.U19A2022 and No.32001058), Science and Technology Commission of Shanghai Municipality (17391900200), and the Chinese Academy of Sciences. K.H. was supported by China Postdoctoral Science Foundation (No.226528) and Shanghai Super Postdoctoral Incentive Program.
Funding
This work was supported by the National Natural Science Foundation of China (Nos. U19A2022 and 32001058), Science and Technology Commission of Shanghai Municipality (17391900200), and the Chinese Academy of Sciences. K.H. was supported by China Postdoctoral Science Foundation (No. 226528) and Shanghai Super Postdoctoral Incentive Program.
Conflict of interest statement. The authors declare no conflict of interests.
References
Author notes
Senior author.
These authors contributed equally (K.H., P.H.).

{kind=link}
{kind=link}