





Abstract
Disease resistance genes encoding nucleotide-binding and leucine-rich repeat (NLR) intracellular immune receptor proteins detect pathogens by the presence of pathogen effectors. Plant genomes typically contain hundreds of NLR-encoding genes. The availability of the hexaploid wheat (Triticum aestivum) cultivar Chinese Spring reference genome allows a detailed study of its NLR complement. However, low NLR expression and high intrafamily sequence homology hinder their accurate annotation. Here, we developed NLR-Annotator, a software tool for in silico NLR identification independent of transcript support. Although developed for wheat, we demonstrate the universal applicability of NLR-Annotator across diverse plant taxa. We applied our tool to wheat and combined it with a transcript-validated subset of genes from the reference gene annotation to characterize the structure, phylogeny, and expression profile of the NLR gene family. We detected 3,400 full-length NLR loci, of which 1,560 were confirmed as expressed genes with intact open reading frames. NLRs with integrated domains mostly group in specific subclades. Members of another subclade predominantly locate in close physical proximity to NLRs carrying integrated domains, suggesting a paired helper function. Most NLRs (88%) display low basal expression (in the lower 10 percentile of transcripts). In young leaves subjected to biotic stress, we found up-regulation of 266 of the NLRs. To illustrate the utility of our tool for the positional cloning of resistance genes, we estimated the number of NLR genes within the intervals of mapped rust resistance genes. Our study will support the identification of functional resistance genes in wheat to accelerate the breeding and engineering of disease-resistant varieties.
The status of wheat (Triticum spp.) as the world’s most widely grown and important food crop (FAOSTAT, 2014) is threatened by the emergence and spread of new and old diseases. For example, wheat stem rust, long considered a vanquished foe of the past, has in the last 20 years caused devastating epidemics in Africa (Pretorius et al., 2000; Olivera et al., 2015) and eastern Russia (Shamanin et al., 2016), while in 2013 and 2016, large outbreaks occurred for the first time in more than 50 years in western Europe (Bhattacharya, 2017; Firpo et al., 2017). The spread of disease into new regions can be attributed to the very success of wheat as a globally traded commodity. In that context, wheat blast, a relatively new disease of wheat, which had until recently been confined to Brazil and other countries in South America, appeared in 2016 in Bangladesh, possibly as a result of importing contaminated grain (Islam et al., 2016; Abu Sadat and Choi, 2017). The warm, wet climates in the wheat belts of India and China, which supply ∼30% of the world’s wheat (FAOSTAT, 2014), favor the further proliferation of this devastating disease. In temperate regions, Septoria tritici blotch (caused by Zymoseptoria tritici) has become a major foliar disease of wheat, which with the emergence of fungicide-resistant strains (Fraaije et al., 2012; Hayes et al., 2016) and the forthcoming restrictions and bans on the remaining effective chemicals (Jess et al., 2014), threatens to cause an almost complete collapse of wheat production in some regions (McCormack, 2018). The breeding of new wheat cultivars with improved genetic resistance to a diverse array of pathogens is therefore of great importance to ensure future wheat production.
In plants, heritable genetic variation for disease resistance is often controlled by dominant Resistance (R) genes encoding intracellular immune receptors with nucleotide-binding and leucine-rich repeat (NLR) domains (Kourelis and van der Hoorn, 2018). NLRs detect the presence of pathogen effector molecules that are delivered into the plant cell by the pathogen to promote virulence (Jones and Dangl, 2006). NLRs either detect effectors directly or, more frequently, indirectly through the modification of a host target by the pathogen effector. In the latter case, the NLR is often said to be guarding the pathogenicity target (Swiderski and Innes, 2001). Some NLRs contain an integrated domain, which has been proposed to act as a decoy to the intended effector pathogenicity target (Cesari et al., 2014; Sarris et al., 2016; Baggs et al., 2017).
NLRs belong to one of the largest multigene families in plants. A plant genome may contain several hundred NLRs (Sarris et al., 2016). Many NLRs are under extreme diversifying selection, to the extent that two accessions from the same species can display significant NLR copy number and sequence variation, the result of the selection of new variants based on duplication, deletion, unequal crossing over, and mutation (Noël et al., 1999; Kuang et al., 2004; Chavan et al., 2015; Seo et al., 2016). In wheat, 19 cloned R genes against wheat rust and powdery mildew encode NLRs (Supplemental Table S1).
A major impediment to studying NLRs in wheat and the cloning of functional R genes has been the lack of a contiguous reference genome sequence. Although the genome of bread wheat (Triticum aestivum) is one of the most challenging crop genomes to study due to its large size (15.4–15.8 Gb; International Wheat Genome Sequencing Consortium, 2018), repetitiveness (∼85%), and hexaploid nature, recent technological advances (Bevan et al., 2017) as well as a strong interest by governmental and industrial funding sources have allowed completion of the genome sequence. Starting in 2012 with a low-coverage survey sequence (Brenchley et al., 2012), improved assemblies underpinned by different strategies have been published (International Wheat Genome Sequencing Consortium, 2014; Chapman et al., 2015; Clavijo et al., 2017; Zimin et al., 2017). In 2017, the first version of a wheat genome sequence with chromosome-sized scaffolds (IWGSC RefSeq v1.0) was made publicly available. The major results of this project are summarized in the study by the International Wheat Genome Sequencing Consortium (2018) and have resulted in a large number of additional studies including the analysis of the transcriptional landscape of wheat (Ramírez-González et al., 2018). In addition, high-quality reference genome sequences have been recently published for the wild diploid progenitor of the wheat D genome, Aegilops tauschii (Luo et al., 2017; Zhao et al., 2017), the AB tetraploid progenitor, wild emmer (Triticum diccocoides; Avni et al., 2017), and its domesticated brethren, pasta wheat (Triticum durum; Maccaferri et al., 2019).
The new high-quality wheat and wild wheat reference genomes will undoubtedly facilitate the study of wheat NLR structure, function, and evolution. However, the study of NLRs, including those in wheat, is complicated by (1) the absence of a tool for accurate and high-throughput NLR annotation and (2) the extremely low basal level of expression for many NLR genes. Previous NLR annotation pipelines (Meyers et al., 2003; Li et al., 2010; Steuernagel et al., 2015; Pal et al., 2016; Yang and Wang, 2016) have mostly relied on a preceding gene annotation. We expected such an annotation method to be potentially not sensitive enough to detect NLR genes. In addition, orthologous NLR loci that are not functional genes in the reference accession would be left out. To overcome these challenges, we first performed NLR exome capture and sequencing on cDNA (Andolfo et al., 2014) isolated from different tissues and developmental stages. These libraries were supplied to the annotation team of the wheat reference and included in the general gene annotation. Second, we developed NLR-Annotator, a tool for de novo genome annotation of loci associated with NLRs. By applying NLR-Annotator and our expression data to the genome of the wheat reference cv Chinese Spring, we found 3,400 loci that may be functional NLR genes or pseudogenes. We show here that wheat NLRs predominantly occur toward the telomeres and in close proximity to each other, display pronounced copy number and sequence variation between the A, B, and D subgenomes (yet maintain conservation of intron-exon structure), contain clade-specific integration of exogenous domains, and that NLR expression is modulated by biotic stress.
RESULTS
Motif-Based Annotation of NLR Loci in Whole-Genome Assemblies
We set out to develop a method for the annotation of NLRs independent from gene calling. The recently published pipeline NLR-Parser (Steuernagel et al., 2015) uses combinations of short motifs of 15 to 50 amino acids to classify a sequence as NLR related. These motifs had been defined based on manual curation of a training set of known NLR sequences by Jupe et al. (2012) and mainly resemble the substructures of NLR protein domains also described in other studies (Supplemental Fig. S1). Since these motifs may occur randomly in a genome, NLR-Parser searches for combinations of doublets or triplets of motifs that often occur in the same order. The drawback of NLR-Parser, however, is that it can only classify a sequence, not distinguish the border between two NLRs within the same sequence. In the extreme case of a whole chromosome with multiple NLRs, this would be classified as a single complete NLR. NLR-Parser thus needs predefined gene models or another method to delimit the borders it searches within.
Here, we present a new tool, NLR-Annotator, as an extension of NLR-Parser to annotate NLR loci in genomic sequence data. In this study, we define the term NLR locus as a section of genomic sequence associated with a single NLR (i.e. one NB-ARC [nucleotide-binding adaptor shared by APAF-1, R proteins, and CED-4] domain potentially followed by one or more leucine-rich repeats [LRRs]). An NLR locus may be a gene with an intact open reading frame but may also be the trace of a sequence pseudogenized in the observed genome. Our pipeline dissects genomic sequences into overlapping fragments and then uses NLR-Parser to preselect those fragments potentially harboring NLR loci. In this step, the nucleotide sequence of each fragment is translated in all six frames to search for motifs. Subsequently, the positions of each motif are transferred to nucleotide positions. NLR-Annotator then integrates data from all fragments, evaluates motif positions and combinations, and suggests a list of loci likely to be associated with NLRs. For an overview, see Figure 1. The general concept is to search for a series of consecutive motifs that are associated with the NB-ARC domain (Supplemental Fig. S1). This is used as a seed, which is then elongated into the pre-NB region and the LRRs by searching for additional motifs associated with those regions.
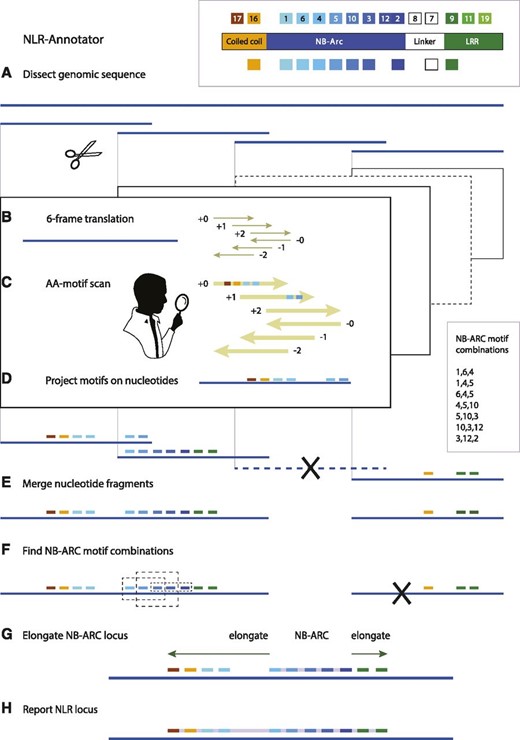
NLR-Annotator step-by-step. A, A genomic sequence is dissected into 20-kb fragments overlapping by 5 kb. B, Each fragment is translated in all six reading frames. C, Amino acid sequences are screened for NLR-associated motifs (see inset). D, The positions of motifs are projected back onto their originating 20-kb genomic fragments. E, Motif-containing genomic fragments are merged, and redundant motifs resulting from overlaps are removed. F, Motif combinations associated with an NB-ARC domain (inset, right side) are identified in the genomic sequence (stippled boxes). Overlapping combinations are merged. G, The NB-ARC locus is used as a seed to search the DNA sequence upstream and downstream for additional NLR-associated motifs (e.g. those indicative of a coiled coil or LRR). H, The final NLR locus is reported.
We tested NLR-Annotator on the Arabidopsis (Arabidopsis thaliana) genome sequence using Arabidopsis gene models (TAIR10; http://www.arabidopsis.org) as a gold standard. Because NLR-Annotator identifies NLR loci, which can be active genes or pseudogenes, the Arabidopsis gold standard annotation is not perfect for benchmarking, but it is nonetheless a good option. Independently of NLR-Annotator, we classified the 27,416 protein sequences from TAIR10 using hidden Markov models from Pfam domains. We found 166 protein sequences with an NB-ARC domain. Using NLR-Annotator, we found 171 loci in the TAIR10 genome assembly. Of the 166 NLR annotated genes, only eight were not overlapping with one of our loci. We manually investigated those eight protein sequences and found that four were Activated Disease Resistance1 (ADR1) or ADR1-like. We had already reported that NLR-Parser does not detect the ADR1 family genes (Steuernagel et al., 2015). Similar to ADR1, the remaining four proteins were not detected because for at least one motif, the similarity to the consensus motif was below the default threshold. Of the 171 loci called by NLR-Annotator, 162 overlapped with our gold standard gene models. In four of these cases, the protein contained two NB-ARC domains, resulting in two separate loci annotated by NLR-Annotator. We aligned the remaining nine NLR loci, which had no overlap to an NB-ARC-containing protein, to National Center for Biotechnology Information nonredundant proteins using BlastX (https://blast.ncbi.nlm.nih.gov). In all cases, we found homology to NLR proteins from other species, which suggests that although those loci were not called as genes in the Arabidopsis reference, NLR-Annotator accurately identified sequences associated with NLRs. Finding 158 out of 166 NLR genes in TAIR10, we conclude a sensitivity (ratio of identified NLR genes to all NLR genes) of 95% for NLR-Annotator. Every one of the loci found with NLR-Annotator was validated to be associated with NLRs, supporting a specificity (ratio of correctly identified NLRs to all identified NLRs) of 100%.
To further test the functionality of NLR-Annotator, we annotated the published genomes of eight additional plant species for NLR loci (Supplemental Table S2). We found 932 loci in robusta coffee (Coffea canephora [Denoeud et al., 2014]; http://coffee-genome.org), 154 loci in maize (Zea mays [Jiao et al., 2017]; http://ensembl.gramene.org), 50 in papaya (Carica papaya [Ming et al., 2008]; https://phytozome.jgi.doe.gov), 71 in cucumber (Cucumis sativus; https://phytozome.jgi.doe.gov), 527 in soybean (Glycine max [Schmutz et al., 2010]; https://phytozome.jgi.doe.gov), 694 in potato (Solanum tuberosum [Xu et al., 2011]; https://solgenomics.net, PGSC_DM_v3), 284 in tomato (Solanum lycopersicum [Sato et al., 2012]; https://solgenomics.net, SL3.00), and 342 in purple false brome (Brachypodium distachyon [Vogel et al., 2010]; https://phytozome.jgi.doe.gov). In a previously published study of tomato, 355 NLRs were identified (Andolfo et al., 2014). The lower number of NLRs identified by NLR-Annotator (284) is mostly due to the higher stringency of NLR-Annotator for minimal number and organization of motifs and a requirement for the presence of an NB-ARC domain.
Using NLR-Annotator, we screened various assemblies of hexaploid bread wheat cv Chinese Spring. With the annotated loci, we distinguished between partial and complete loci, where a complete locus contains the P-loop/motif1 that marks the start of an NB-ARC domain as well as at least one LRR-associated motif (Supplemental Fig. S1). In the assemblies from 2014 (International Wheat Genome Sequencing Consortium, 2014) and 2015 (Chapman et al., 2015), we found 12,441 and 15,315 loci, respectively, but only 1,050 and 1,249 were complete. The assemblies made available in 2017 by Clavijo et al. (2017), Zimin et al. (2017), and the International Wheat Genome Sequencing Consortium (2018) have considerably smaller numbers of total NLR loci (3,251, 3,568, and 3,400, respectively) but concomitantly larger numbers of complete NLR loci (2,465, 2,731, and 2,580, respectively). Possible reasons for the differences in the number of detected NLR loci include differences in the genome completeness and continuity as well as assembly mistakes such as collapsed regions or falsely duplicated regions. For the analyses described in this study, we used the IWGSC RefSeq v1.0 to take advantage of the pseudochromosomes and the gene annotation (International Wheat Genome Sequencing Consortium, 2018).
Physical Positions of NLR Loci
With the availability of pseudochromosomes, which represent 94% of the predicted wheat genome, most NLR loci can now be placed in the physical context of their location on the chromosome. The foremost practical implication of this is to take advantage of available mapping data and other resources to accelerate the identification of functional disease resistance genes. Annotated NLR loci were found to preferentially locate at the telomeres of each chromosome (Fig. 2) and cluster together. More than 400 of the 3,400 observed NLR loci are in a proximity of less than 5 kb to another NLR locus. Half of all loci are in a distance of less than 50 kb to another locus (Fig. 2). The NLR loci are distributed over all chromosomes, and the number of NLR loci per chromosome ranges between 29 (chromosome 4D) and 280 (chromosome 4A). No clear preference of NLR numbers toward one homeologue within each group could be observed (Supplemental Table S3).
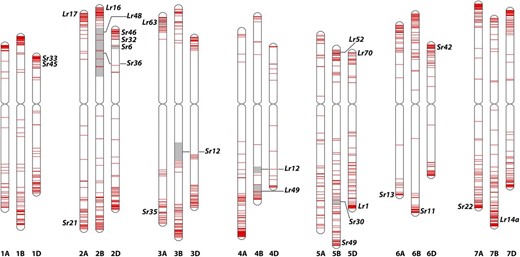
Physical positions of NLR-associated loci for stem rust (Sr) and leaf rust (Lr) within the wheat genome. Red bars on chromosomes mark the presence of NLR-associated loci. Gray regions depict intervals between flanking markers of fine-mapped rust R genes.
To demonstrate the potential practical advantage of our resource (i.e. annotated NLR loci with a physical position on pseudochromosomes), we searched the literature for leaf rust and stem rust resistance genes that have been genetically mapped in wheat but not yet cloned. Most genes have been mapped in accessions other than the reference accession cv Chinese Spring, and most resistance genes may not have a functional allele in cv Chinese Spring. Nevertheless, we hypothesized that in many cases, a nonfunctional allele or close homolog may be present, the sequence of which could then be used to speed up the cloning of the functional allele from the resistant accession.
To explore the suitability of this approach, we searched the cv Chinese Spring genome for homologs of exemplarily cloned R genes, namely the stem rust resistance genes Sr22, Sr33, Sr35, Sr45, and Sr50, the powdery mildew resistance genes Pm2, Pm3, and Pm8, the leaf rust resistance genes Lr1, Lr10, and Lr22a, and the yellow rust resistance genes Yr5, Yr7, and YrSP (Supplemental Table S1). In eight cases (Sr22, Sr33, Sr45, Pm2, Pm3, Lr1, Yr5, and Yr7), the best alignment of the protein sequence to cv Chinese Spring was on the same chromosome where the gene was cloned from. For Sr35 and YrSp, the second best alignment was on the right chromosome and the best alignment was on a homeologous chromosome. For Pm8, which was introgressed from rye (Secale cereale), and Lr22a, the second best alignment was on the right chromosome, whereas the best alignment was on a scaffold yet to be assigned to a chromosome. Sr50 was cloned from rye as well (Mago et al., 2015) but characterized to be an ortholog of Mla, on group 1 chromosomes. As expected, the best three alignments for Sr50 were therefore with chromosomes 1A, 1D, and 1B. For Lr10, we did not find an alignment on 1A but on the homeologous chromosomes. However, this was expected, since cv Chinese Spring was previously characterized to be a deletion haplotype of Lr10 (Isidore et al., 2005). Next, we aligned sequences of flanking markers from 39 mapped, but not yet cloned, Sr and Lr genes to cv Chinese Spring, defined the physical interval, and counted the number of NLR-associated loci within the interval. The loci can be used for candidate gene approaches or further delimiting of map intervals in positional cloning projects. In 32 cases, we could determine a physical interval, either by using the exact flanking markers or by using chromosome ends or the centromere as surrogate, to determine a number of candidate NLRs. We found between zero and 61 NLRs as candidates (average of 10; Supplemental Table S4). Interesting examples include Sr6 (physical interval 15.8 Mb, one candidate), Lr13 (physical interval 60 Mb, three candidates), and Lr49 (physical interval 33 Mb, three candidates). Notably, these are large intervals with only a few candidate NLR loci, providing the opportunity to explore candidates before embarking on the more laborious tasks to further delimit or sequence the interval in the donor accession.
Comparison of NLR Loci Identified by NLR-Annotator with Automated Gene Annotation in IWGSC RefSeq v1.1
We expected a substantial number of our independently predicted NLR loci to be overlapping with genes from the automated gene annotation v1.1 of the cv Chinese Spring reference sequence (IWGSC RefSeq v1.0). Of the 3,400 loci we predicted by NLR-Annotator, 2,955 overlap with genes annotated in RefSeq v1.1. In total, 578 NLR loci defined by NLR-Annotator correspond to more than one gene in RefSeq v1.1. We looked at these 578 loci in more detail. In 70 cases, we found gaps (stretches of unassigned nucleotides) in the assembly potentially interfering with transcript mapping and thus hampering gene calling and giving rise to two falsely called genes. In 116 cases, we found one of the gene models resembling a complete NLR gene (i.e. the P-loop, at least three consecutive NB-ARC motifs, and at least one LRR motif), indicating a potential overextension of the NLR locus, usually brought about by a randomly occurring LRR motif shortly downstream of the gene (Supplemental Table S5). In 29 of 30 random examples (Supplemental Table S4) of the remaining 405 cases, we observed a stop codon in the coding sequence interrupting the open reading frame in the transcript.
We believe that these cases with stop codons represent alleles of NLR genes that have recently been pseudogenized but that are still transcribed. An example supporting this theory is the Pm2 gene conferring resistance to Blumeria graminis, the causal agent of powdery mildew. The Pm2 gene was cloned from the wheat ‘Ulka’ (Sánchez-Martín et al., 2016), where it encodes a full-length NLR. The allele in cv Chinese Spring has a stretch of 12 nucleotides replaced by five other nucleotides, thus causing a frame shift leading to an early stop codon, but it is nonetheless still transcribed (Supplemental Fig. S2).
A Set of Validated NLR Gene Models in cv Chinese Spring
For the subsequent analyses that involve gene models, we proceeded with gene models from the IWGSC RefSeq v1.1. To select appropriate transcripts that encode complete NLRs, we used NLR-Parser to classify sequences and selected those containing a P-loop (Meyers et al., 1999), at least three consecutive motifs associated with the NB-ARC domain (motifs 1, 6, 4, 5, 10, 3, 12, and 2), and at least one LRR-associated motif (motif 9, 11, or 19). Of 4,983 transcripts (Supplemental Table S5) that either overlapped with an NLR locus or were classified as NLR associated by NLR-Parser, we identified 1,823 transcripts (corresponding to 1,560 genes) that encoded complete NLRs. In addition, we used ADR1 (AT1G33560) to search for homologs in the gene models. The four candidate ADR1 transcripts (TraesCS5D02G081800.1, TraesCS5B02G075900.1, TraesCS5A02G069600.1, and TraesCS5A02G069600.2) were added. Our final list of confirmed 1,827 NLR transcripts (Supplemental Table S6) is most likely an underestimate of the total NLR gene content. Manual curation would likely be required to obtain a more precise estimate of the total true NLR complement.
Phylogenetic Analysis of Wheat NLR Genes
NLRs constitute a large gene family, and only for a very few individual genes has a function been assigned. A phylogenetic analysis provides a means to order this large number of genes with respect to their sequence relationship and arrange them into subfamilies. We set out to establish this order and look for common features in various clades of the NLR gene family, which may hint at common functional attributes.
We extracted the NB-ARC domains from NLR protein sequences and calculated their phylogenetic relationships. For this analysis, we used the 1,827 NLR transcripts we identified in the IWGSC RefSeq v1.1 gene annotation rather than loci annotated by NLR-Annotator. The reasons are, first, an often-occurring intron within the NB-ARC domain complicating identification of the correct, complete open reading frame and subsequent translation without the support of transcript data. Second, in our subsequent analysis below, we intended to investigate integrated domains, which is not possible without gene models. Figure 3 shows the NLR phylogeny of cv Chinese Spring with clades highlighted that are discussed subsequently in this article. We also added known resistance genes to the tree. An interactive version of Figure 3 and related supplemental figures can be found at https://itol.embl.de/shared/steuernb.
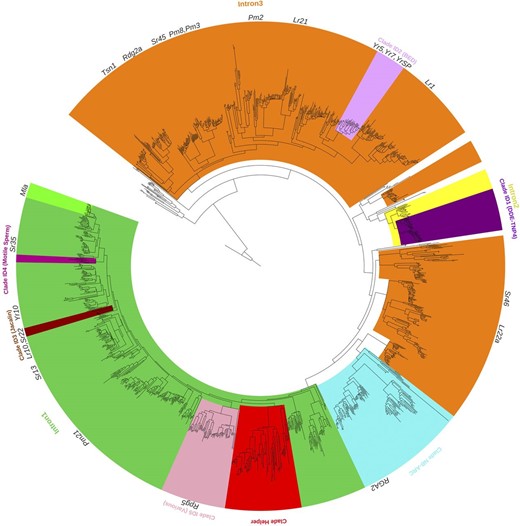
Phylogenetic tree of NLR genes in wheat. Cloned R genes were added to the tree. Specific clades are marked according to certain characteristics discussed in the main text: members of clades Intron1, Intron2, and Intron3 share characteristic intron/exon structure. Clades ID1 to ID5 are enriched for members with integrated domains. Members of clade NB-ARC have a truncated NB-ARC domain preceding a complete NB-ARC domain. Clade Helper is enriched for NLRs being hypothesized to be helpers. An interactive version of this figure is available at https://itol.embl.de/shared/steuernb.
For the reasons mentioned above, we used gene models for our phylogenetic analysis. However, we also tested if we can use information from NLR-Annotator to construct phylogenetic trees as a function for genomes lacking a gene annotation. To this end, we used only the concatenated amino acid motifs within the NB-ARC domain that were identified by NLR-Annotator. The resulting tree (Supplemental Fig. S3) is similar to the tree based on whole NB-ARC protein sequences (Fig. 3). For example, clades with specific features that we identified (see sections below) were preserved in this tree based only on the concatenated NB-ARC motifs. To highlight the similarity between the two trees, we color-coded NLR loci in the concatenated NB-ARC domain tree corresponding to genes from several clades with distinct features. Apart from six outliers, all loci that clustered in the full-length NB-ARC domain tree (Fig. 3) also clustered in the concatenated NB-ARC motif tree (Supplemental Fig. S3). All six outliers originate from proteins with two complete NLRs, which are fused. With NLR-Annotator, those were separated as two loci, and the outliers show the phylogenetic position of the secondary NB-ARC domain in the protein.
Integrated Domains
Some NLR proteins differ from the canonical NLR structure by having an additional integrated domain(s). This structure has been demonstrated in other plant species (Cesari et al., 2014; Baggs et al., 2017) and has also previously been reported in wheat (Bailey et al., 2018). It has been proposed that these domains act as a decoy or bait (Cesari et al., 2014; Baggs et al., 2017), in which the gene from which the integrated sequence originates encodes for a protein that is a target of a pathogen effector(s). A detailed study of additional domains integrated into NLRs can potentially provide information about effector targets in plant-pathogen interactions.
We screened the NLR protein sequences for integrated domains. In total, we found that 129 of the 1,560 protein sequences carry integrated domains. The most prevalent domains were protein kinase (41 cases), DDE acidic triad-transposase (DDE_TNP_4; 26 cases), zinc finger (zf)-BED (named after the Drosophila proteins BEAF and DREF; 10 cases), Jacalin (eight cases), and Motile Sperm (seven cases; Supplemental Table S7).
We then associated integrated domains with the phylogeny of the NB-ARC domains of all NLRs. The clades with accumulated integrated domains are shown in Figure 3, and individual domains are displayed within the structure of NLRs in Supplemental Figure S4. In concurrence with previous studies (Bailey et al., 2018; Stein et al., 2018), we found that some clades of the phylogenetic tree are enriched for specific integrated domains. These are DDE_TNP_4 (clade ID1; Fig. 3), zf-BED (clade ID2; Fig. 3), Jacalin (clade ID3; Fig. 3), and Motile Sperm (clade ID4; Fig. 3). A fifth clade (clade ID5; Fig. 3) with 66 members is heavily enriched (47 proteins) for various integrated domains, which tend not to be in the above-mentioned specific clades (ID1–ID4), including protein kinase, Kelch, Grass, Exo70, and WRKY domains. The major type of integrated domain that is not exclusively found in a specific clade are the protein kinases.
Tandem NLRs
NLRs occurring in tandem can function together. They can be arranged in a head-to-head formation in which the genes are on opposite strands and the distance between gene starts is shorter than the distance between gene ends (Narusaka et al., 2009). We searched for this type of tandem NLR genes in the reference wheat genome. We found 52 NLR pairs with a position as described above and a maximum distance of 20 kb (Supplemental Fig. S5; Supplemental Table S8). Twenty-five pairs had one mate located in the red Helper clade. Fifteen of those pairs have the other mate located in clade ID5, which accumulates various integrated domains.
Intron-Exon Structure
We investigated introns in NLR genes and observed a large diversity in terms of length and number of introns. The average intron length is 1,197 bp but they can extend up to 24,586 bp, while the average intron number is two and ranges between zero and 23 (Supplemental Fig. S6). Another interesting feature is the position of introns. Several clades in the phylogeny (Fig. 3; Supplemental Fig. S6, orange clades) do not contain an intron in the coding region of the NB-ARC domain. Known R genes without an intron in the NB-ARC domain are Tsn1, Sr45, Pm2, Pm3, Pm8, Lr1, Lr21, Yr5, Yr7, and YrSP. A large number (608) of NLRs, however, have an intron right after the resistance nucleotide-binding site A (RNBS-A; motif 6) within the NB-ARC domain. All those genes are members of the green clade Intron1 in Figure 3 (and Supplemental Fig. S5). Only three exceptions in that clade were observed to not have this feature. Several cloned R genes, such as Sr22, Sr35, Lr10, and those in the Mla family (Sr33, Sr50, and SrTA1662), have this common feature. Another pattern of intron within the NB-ARC domain was observed in two distinct clades, where an intron is within the coding sequence of the RNBS-C (motif 10). One of the clades (Fig. 3, yellow clade Intron2), which includes those NLRs with an integrated DDE_TNP_4 domain, has 64 members. The other clade (Fig. 3, light blue clade NB-ARC) with an intron within the RNBS-C (motif 10) has 121 members.
A Clade with an Extended NB-ARC Domain
The 121 members of the clade NB-ARC (Fig. 3) almost exclusively (with only five exceptions) share a characteristic feature, in which the NB-ARC domain is preceded by a truncated NB-ARC domain (Supplemental Fig. S7). The truncation is after the motif RNBS-B (motif 5). A prominent member of this clade is Resistance gene analog2, which was found to be required for Lr10-mediated resistance (Loutre et al., 2009). Members of this clade have an intron separating the truncated and the complete NB-ARC domains. As already described in the previous section, members of this clade have an intron within RNBS-C (motif 10).
NLR Expression
In a previous study, we observed that in wheat the NLRs encoded by the stem rust resistance genes Sr22 and Sr45 were expressed at low levels in leaves of seedlings (∼30/60 Gb sequencing resulted in less than 20×/15× coverage of Sr22/Sr45, respectively; Steuernagel et al., 2016). To test whether this observation of low expression could be extended broadly to NLRs in wheat, we sequenced the transcriptomes of young leaves, generating more than 305 Gb of data in three samples. We also enriched the same samples for NLR genes (cDNA R gene enrichment sequencing [RenSeq]; Andolfo et al., 2014) to detect lowly expressed NLR genes. We considered expression of a gene detectable if more reads would map to it than necessary to cover the entire length fivefold. In the combined data sets, we found 1,074 NLR genes being detectable as transcribed. In the unenriched transcriptome data, 859 genes (80%) could be detected. Down-calculating those numbers (Fig. 4), we estimate that generating 50 Gb of wheat transcriptome data would allow for 55% of NLRs expressed in young leaves to be detected.
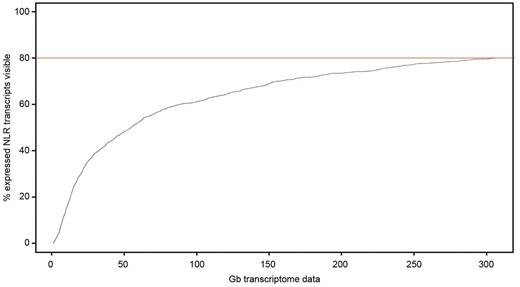
Estimated detectability of NLR transcripts in transcriptome studies. The percentage of expressed NLR transcripts (y axis) in leaves from the three-leaf stage is shown in relation to the amount of sequencing data (x axis) used as an input. A transcript is considered detectable if the combined length of mapped reads exceeds 5 times the length of the transcript. The red line indicates the maximum percentage of NLR transcripts detected with 305 Gb of RNA sequencing data.
Stress-Induced Expression of NLRs
There are more than 1,000 NLRs in the wheat genome, representing 1% to 2% of the total gene-coding capacity (International Wheat Genome Sequencing Consortium, 2018). However, the immunity conferred by NLRs comes at a metabolic cost, and overactivation of immune responses may give rise to stunted growth (Yi and Richards, 2009; Choulet et al., 2014). Therefore, we assume that NLR expression would be tightly regulated.
As an example, we checked whether NLR expression changes after biotic stress. Plants respond to conserved pathogen-associated molecular patterns (PAMPs) or microbe-associated molecular patterns such as chitin, a constituent of fungal cell walls, or flagellin from the bacterial flagellum. Molecular events that occur during PAMP-triggered immunity are highly conserved between dicots and monocots and include rapid production of reactive oxygen species, mitogen-activated protein kinase induction, and defense gene induction (Dangl et al., 2013). We hypothesized that, as a first layer of defense, the PAMP-triggered immune response might also include increased expression of NLRs to help prime the plant defense system for effector-triggered immunity. To test this hypothesis, we infiltrated young wheat plants (at the three-leaf stage) with two PAMPs, namely chitin (Kombrink et al., 2011) and flg22, a 22-amino acid peptide from flagellin (Takai et al., 2008), and then sequenced the transcriptomes of water-treated and PAMP-challenged plants at 30 min and 3 h after challenge. Three (non-NLR) genes that had previously been reported to be PAMP responsive (Schoonbeek et al., 2015) showed differential expression by reverse transcription quantitative PCR, confirming a responsivenes to PAMP treatment (Supplemental Fig. S8).
Within our set of 628 NLR genes for which we could obtain expression data (Supplemental File S1), nearly half were differentially expressed. In total, 266 genes (257 in the case of chitin and 194 in the case of flg22) were found to be at least twofold stronger expressed than before treatment. Moreover, the expression of 243 (234 in the case of chitin and 166 in the case of flg22) of those genes were reduced again to resting state levels at 180 min after treatment. We visualized this general tendency by plotting expression values of NLR genes in ternary plots where the dot size and color additionally depict the maximum expression found at any time point (Fig. 5).
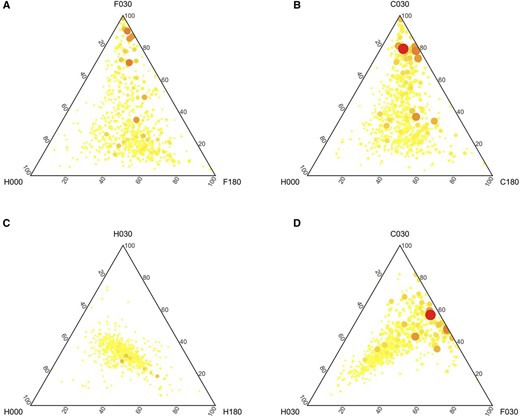
Expression of NLRs following PAMP treatment. Ternary diagrams show the relative gene expression between samples before treatment (000), after 30 min (030), and after 180 min (180), where A represents flg22 (F) treatment, B represents chitin (C) treatment, and C represents mock (H) treatment. The ternary diagram (D) shows the difference for mock, flg22, and chitin 30 min after treatment. Color (from yellow to red) and dot size are proportional to the maximum expression value for the respective gene.
Most NLR genes that showed differential expression after chitin treatment also showed differential expression after treatment with flagellin (Supplemental Fig. S9A). In particular, when we relaxed the threshold of twofold overexpression, we noted that treatment with either PAMP caused up-regulation of the same genes (observed in 255 cases out of 266 differentially expressed genes; Supplemental Fig. S9B).
The up-regulated NLR genes were not associated with specific phylogenetic clades.
DISCUSSION
In this study, we present the tool NLR-Annotator for de novo annotation of NLR loci. Our tool is distinguished from standard gene annotation in that it does not require transcript support to identify NLR-associated loci. NLR-Annotator is therefore a powerful tool for the identification of potential R genes that could be used in breeding for resistance to wheat pathogens. Very often, however, the functional copy of an R gene is not present in the accession that was chosen for a reference genome. The positive selection imposed on NLRs often results in extensive accessional sequence and copy number variation at NLR loci (Noël et al., 1999; Kuang et al., 2004; Chavan et al., 2015; Seo et al., 2016). Thus, the corresponding NLR homolog in the wheat reference accession, cv Chinese Spring, would not be considered by a standard gene annotation. NLR-Annotator, on the other hand, is not limited to functional genes. In our analysis of cv Chinese Spring, we found 3,400 loci, while only 1,560 complete NLRs could be confirmed through gene annotation in IWGSC RefSeq v1.1. Within the additional loci identified by NLR-Annotator there might be some NLRs with potential for function. However, we speculate that most of these are nonfunctional pseudogenes. Nevertheless, it is important to define these loci in the reference accession cv Chinese Spring because these genes may have functional alleles in other accessions. A case in hand concerns Pm2 (from cv Ulka), which in cv Chinese Spring has an out-of-frame insertion/deletion leading to an early stop codon. The corresponding locus in cv Chinese Spring is still transcribed at a detectable level (Supplemental Fig. S2). The frameshift interrupting the open reading frame led to two remaining open reading frames, still large enough to have been annotated as separate genes.
In some cases (e.g. transcript studies or inspection of integrated domains), an analysis of NLRs requires functional genes. Previous work on describing NLRs in earlier versions of the cv Chinese Spring genome have searched within the genome-wide gene annotation for NLRs. This led to fewer NLRs being identified, for example 16 full-length NLRs in the 2014 cv Chinese Spring survey sequence (International Wheat Genome Sequencing Consortium, 2014; Clavijo et al., 2017) and 1,174 full-length NLRs in the 2017 cv Chinese Spring W2RAP assembly (Clavijo et al., 2017). Our study benefitted from an improved general annotation of wheat NLRs made possible by supplying the IWGSC RefSeq v1.1 annotation pipeline with cDNA data enriched for NLR transcripts through NLR enrichment sequencing (International Wheat Genome Sequencing Consortium, 2018), and second, we developed NLR-Annotator to identify NLRs independently of gene annotation.
To improve the annotation further would require manual annotation locus-by-locus, in each case taking into account existing gene annotations, mapped transcript data from different sources, and our NLR-Annotator loci. All these data would have to be integrated and inspected to provide bona fide gene models. Given the diversity of NLR complements in different wheat accessions (Thind et al., 2017), the amount of work required for this effort is considerable. For practical applications such as R gene cloning, our automated NLR locus discovery will, in most cases, likely be sufficient to identify a candidate gene, which can be annotated ad hoc in the relevant accessions. For studies requiring annotated genes, we have to settle for a subset of the actual NLR complement.
We set out to investigate the sequence diversity relationships within the NLR gene family in cv Chinese Spring. It has been well documented that in NLRs, the NB-ARC domain is far more conserved than the LRRs. Therefore, phylogenetic studies compare the NB-ARC domains to explore and depict the sequence relationship among NLRs (Jupe et al., 2012; Andolfo et al., 2014; Bailey et al., 2018). Extracting NLR domains from NLR loci defined by NLR-Annotator is complicated by the presence of introns within domains, the boundaries of which can only be defined properly using transcript data. In addition, we were interested in cataloging the occurrence and distribution of integrated domains in the NLRs. This can only be done on annotated genes. Therefore, in our subsequent analysis, we used the 1,540 annotated NLRs to compute a phylogenetic tree.
With the reference sequence, we studied the phylogeny, integrated domains, and helper NLRs. This has been recently described in detail by Bailey et al. (2018) based on an analysis of the W2RAP assembly of wheat. In brief, we found that the new assembly supports the observations made in the study by Bailey et al. (2018). One additional feature that we discovered is a clade of NLRs that contain elongated NB-ARC domains (clade NB-ARC in Fig. 3). The elongated part of the NB-ARC domain precedes the canonical NB-ARC domain and consists predominantly of motifs 6, 4, and 5, often preceded by motif 1 (Supplemental Figs. S1 and S7). We do not know the function of this elongated NB-ARC domain. However, a member of this clade is RGA2 which has previously been described to be essential for Lr10 function (Loutre et al., 2009).
To expand the previous phylogenetic analysis to include the entire set of all 3,400 NLR loci, we developed a new feature in NLR-Annotator. By reducing the NB-ARC domains to only be represented by the even more conserved motifs (Supplemental Fig. S1), we can be sure to exclude introns from the phylogenetic analyses and can also exploit the a priori knowledge of the positions of the motifs within the NB-ARC domain to avoid an external multiple alignment. This then permits using the alignment from NLR-Annotator directly as an input to compute a phylogenetic tree. The resulting phylogeny (Supplemental Fig. S3) was validated by visualizing leaves that correspond to genes from specific clades in the phylogeny derived from gene models (Fig. 3). The phylogenetic relationship of genes was maintained for corresponding loci in the tree generated from NLR-Annotator. One example, however, was found in the Mla region, where a locus was associated with a neighboring subclade rather than the other loci of the Mla region.
Finally, with the IWGSC RefSeq v1.0 gene annotation v1.1, we performed NLR expression profiling. We found that many NLRs are expressed at low levels, requiring either extremely deep sequencing or target enrichment to be detected. We also looked at NLR induction in response to PAMPs and found that a subset of NLRs are triggered following PAMP exposure. The transcriptional response was measurable within 30 min and had returned to base level after 3 h. This response shows the tight regulation of the NLR immune system and may reflect the fine-tuning between the cost of defense and resource allocation for growth and reproduction.
CONCLUSION
In this study, we introduce NLR-Annotator to detect NLRs in a genome sequence. The power and novelty of this tool is that it is completely independent of a preceding gene annotation, thus enabling rapid ab initio interrogation of the NLR complement of a genome without reliance on gene expression or other limitations of gene prediction pipelines. We then used NLR-Annotator to catalog NLRs in the reference sequence of wheat cv Chinese Spring and extended our analysis to comprehensively characterize this complex multigene family. We predict that this tool will also facilitate the cloning of functional R genes from plant genomes by positioning NLR loci within mapped intervals, as demonstrated here for leaf and stem rust, two major diseases of wheat.
MATERIALS AND METHODS
NLR-Annotator
The NLR-Annotator pipeline is divided into three steps: (1) dissection of genomic input sequence into overlapping fragments; (2) NLR-Parser, which creates an xml-based interface file; and (3) NLR-Annotator, which uses the xml file as input, annotates NLR loci, and generates output files based on coordinates and orientation of the initial input genomic sequence. All three programs are implemented in Java 1.5. Source code, executable jar files, and further documentation have been published on GitHub (https://github.com/steuernb/NLR-Annotator). The version of NLR-Parser used here has been published previously along with the MutRenSeq (Steuernagel et al., 2016) pipeline and uses the MEME suite (Bailey and Gribskov, 1998). All genomes annotated in this publication were dissected into fragments of 20 kb length overlapping by 5 kb. Manual investigation of questionable Arabidopsis (Arabidopsis thaliana) protein sequences was performed using SMART (Letunic et al., 2015).
Testing NLR-Annotator on TAIR10
We classified protein sequences from Arabidopsis (TAIR10) as NLR proteins using HMMER (Eddy, 2011) version HMMER-3.1b1 and models from Pfam-A (https://pfam.xfam.org/) version 31.0. Proteins having an NB-ARC domain with an E value < 1E-5 were used for further processing.
To compare NLR loci from NLR-Annotator with NLR protein sequences, we extracted gene positions from TAIR10 gene annotation (ftp://ftp.arabidopsis.org/home/tair/Genes/TAIR10_genome_release/TAIR10_gff3/TAIR10_GFF3_genes.gff) and searched for overlapping intervals.
R Gene Intervals
Coding sequences of cloned R genes were aligned to pseudochromosomes of wheat (Triticum aestivum) ‘Chinese Spring’ using BLASTn (Zhang et al., 2000) and default parameters. To align marker sequences extracted from the literature to the pseudochromosomes, we used BLASTn and the parameter –task blastn to amend for short input sequences. In uncertain cases, matching positions were validated using DOTTER (Sonnhammer and Durbin, 1995).
Comparison of NLR Loci with Gene Annotation
Comparison of NLR loci (generated from NLR-Annotator) with gene models from IWGSC gene annotation RefSeq v1.1, was done using the Java program CalculateNLRGenes.java (https://github.com/steuernb/wheat_nlr). Overlaps were calculated using the positions of gene transcripts. An overlap was only considered if both the locus and the transcript were on the same strand.
Selection of NLR Genes from Gene Annotation
To identify NLRs in the RefSeq v1.1 gene annotation, NLR-Parser was run on coding sequences of gene models. Tab-separated output from NLR-Parser was generated using output option –o. The last column in that table is a list of identified motifs (Jupe et al., 2012). Genes were selected by the presence of motif 1 (P-loop), the presence of either motif 9, 11, or 19 (LRR motifs), and at least one of the following consecutive motif combinations: 1, 6, 4; 6, 4, 5; 4, 5, 10; 5, 10, 3; 10, 3, 12; or 3, 12, 2. Motifs detected in reverse frames of sequences were discarded (one case, TraesCS3D01G521000LC). Examples from cases with discrepancies between NLR-Annotator and gene annotation were manually inspected using Web Apollo (Lee et al., 2013).
Phylogenetic Analysis
Protein sequences from NLR genes as well as from known R genes were screened for the NB-ARC domain using HMMER v. 3.1b1 (Eddy, 2011) and Pfam-A v. 27.0 (http://pfam.xfam.org/). The command line call was hmmscan–domtblout outputfile Pfam-A.hmm inputfile. Intervals within each sequence were defined based on the presence of motifs 1, 6, 4, 5, 10, 3, 12, and 2. An interval has to start with motif 1; other motifs may be absent but, if present, the order is not allowed to be changed. For each protein sequence, the largest interval including 20 flanking amino acids was used as the NB-ARC domain. A multiple alignment of NB-ARC domain sequences was generated using Clustal Omega (Sievers et al., 2011). A phylogenetic tree was generated using FastTree (Price et al., 2010). The tree was visualized using iTOL (Letunic and Bork, 2016).
Integrated Domain Analysis
Protein sequences of NLRs were screened for domains using HMMER v. 3.1b1 (Eddy, 2011) and Pfam-A v. 27.0 (http://pfam.xfam.org/). Domains with an E value < 1E-5 were regarded and filtered for domains usually overlapping with the NB-ARC domain (NACHT and AAA) as well as the LRR domain. Java source code was deposited on GitHub (https://github.com/steuernb/wheat_nlr).
Tandem NLR Analysis
General feature format files of both high- and low-confidence genes from the IWGSC RefSeq annotation v1.1 were screened for pairs of NLR genes that were on reverse complementary strands, at a distance of less than 20 kb, and in a head-to-head relation (i.e. the distance between gene starts is shorter than the distance between gene ends). Java source code was deposited on GitHub (https://github.com/steuernb/wheat_nlr).
Intron-Exon Structure
Intron-exon structure was derived from general feature format files of both high- and low-confidence genes from the IWGSC RefSeq annotation v1.1. Script for generating the file adding information to iTOL is deposited on GitHub (https://github.com/steuernb/wheat_nlr).
Growing cv Chinese Spring and Extracting RNA
Plants of wheat ‘Chinese Spring’ (CS42, accession Dv418) were grown in the greenhouse. The tissues were taken from different parts of the plants at various growth stages. The total RNA was extracted with the RNeasy Plant Mini Kit (Qiagen catalog no. 74904) following the company’s protocol. The total RNA samples were then digested with DNase I (Roche 11284932001) using the company’s protocol. After the DNA digestion, the total RNA samples were purified with Agencourt AMPure XP beads (part no. A63881) at 0.5× rate.
Enrichment of cDNA
cDNA for targeted enrichment was prepared with the KAPA Stranded mRNA-Seq Kit (F. Hoffmann-La Roche) following the manufacturer’s protocol with minor modifications. Briefly, 5 µg of total RNA was used to capture mRNA with magnetic oligo(dT) beads, followed by fragmentation for 6 min at 85°C to generate 300- to 400-nucleotide fragment sizes. First and second strands were synthesized according to the manufacturer’s protocol. No A-tailing step was performed, and an adapter (100 nm final concentration) from the NEBNext Ultra DNA Library Prep Kit for Illumina (New England Biolabs) was used for ligation. After ligation, 3 µL of USER enzyme (New England Biolabs) were added to the reaction and incubated at 37°C for 30 min. Postligation purifications and amplification (eight cycles) were performed according to the manufacturer’s protocol, only substituting KAPA Library Amplification Primer Mix (10×) with NEBNext Universal PCR Primer for Illumina and NEBNext Index primers for Illumina (2 µm final concentrations each). Amplified libraries were purified using Agencourt AMPure XP beads (Beckman Coulter) with a 1:0.65 ratio of amplified cDNA to beads. Individual libraries were quantified with the Quant-iT PicoGreen dsDNA Assay Kit (Thermo Fisher Scientific), and equimolar amounts were used for targeted enrichment with a custom MYcroarray MYbaits kit (Arbor Biosciences) and the corresponding protocol. Enriched libraries were sequenced on an Illumina HiSeq 2500. Raw data were deposited in the Sequence Read Archive under study identifier PRJEB23081. Sequences for the bait library (TV2) are available at https://github.com/steuernb/MutantHunter/blob/master/Triticea_RenSeq_Baits_V2.fasta.
Elicitation with PAMPs
The protocol was modified from Schoonbeek et al. (2015). The ‘Chinese Spring’ wheat plants were grown for 3 weeks in a growth cabinet under a 16/8-h day/night regime at 23°C/18°C. For each biological repetition, three strips (2 cm) were cut from leaves 2 and 3, placed in a 2-mL tube with sterile water, and vacuum infiltrated three times for 1 min. The following day, water was removed and replaced by fresh water or PAMPs dissolved in water at 1 g L−1 for chitin (Nacosy, Yaizu Suisankagaku Industry) or 500 nm for flg22 (Peptron). Samples were drained and flash frozen in liquid nitrogen after 30 or 180 min prior to pulverization with two stainless steel balls in a Geno/Grinder (SPEX). RNA was extracted using the RNAeasy plant kit (Qiagen), the concentration was determined on a NanoDrop 8000 spectrophotometer (Thermo Fisher Scientific), and quality was assessed with an RNA 6000 Nano chip on a Bioanalyzer 2100 (Agilent Technologies). After removal of genomic DNA with the TURBO DNA-free Kit (Thermo Fisher Scientific), 1 µg of RNA was converted to cDNA with SuperScript IV (Thermo Fisher Scientific), and expression of PAMP-inducible genes (Schoonbeek et al., 2015) was verified by quantitative PCR (Supplemental Fig. S8) using 0.4 µL of cDNA per 16-µL reaction with PCR SYBR Green JumpStart Taq ReadyMix (Sigma) on a LightCycler 480 (Roche Life Science). Gene expression of three biological replicates is expressed as log2 relative to the expression level at time 0, after infiltration and overnight incubation but before addition of fresh water or PAMP solutions. Expression was normalized to EF-1α (Elongation factor 1α, M90077; with primers 5′-ATGATTCCCACCAAGCCCAT-3′ and 5′-ACACCAACAGCCACAGTTTGC-3′). Genes tested were Ror2 (syntaxin-121 homologous to barley [Hordeum vulgare] required for mlo-specified resistance2, AK454359.1; 5′-TCGTGCTCAAGAACACCAAC-3′ and 5′-AATCGAGTGGCTCAACGAAC-3′), CMPG1-like (RING-type E3 ubiquitin transferase homologous to maize [Zea mays] immediate-early fungal elicitor protein CMPG1, TraesCS4B02G228700; 5′-GGACGCAACCAAGGAGAAGA-3′ and 5′-TTGAGCCCTCTGAAGTCCAT-3′), and pub23 (E3 ubiquitin-protein ligase homologous to barley and Arabidopsis PUB23, Traes_3B_0B86BCF93; 5′-CGTTCATCAGAATGCTCAGCTG-3′ and 5′-TTCTCTTTTGTAGGCACGAACCA-3′).
Expression Data Analysis
The transcript analysis pipeline (calculation of read counts and transcripts per million) is based on Kallisto (Bray et al., 2016) and is common with the global wheat study by Ramírez-González et al. (2018). Complete transcript lists were filtered for NLR genes as defined above. To estimate the detectability of NLR transcripts, we combined Kallisto read counts from three-leaf stage replicates. We considered a transcript to be expressed if, in total RNA or in RenSeq cDNA, the combined length of mapped reads (i.e. a read length of 150 multiplied by the sum of counts from all replicates) exceeded 5 times the length of the transcript itself. Testing the same criterion using only read counts based on 305 Gb of total RNA input data, we found 80% of NLRs. We then gradually reduced Kallisto read counts proportionally to a reduced input data to estimate the percentage of NLRs detected with less input data.
For the differential gene expression analysis, we preprocessed count values obtained with Kallisto using Degust (doi/10.5281/zenodo.3258932), loading only genes that had a read count of more than 10 in at least one sample. Data were normalized using the method Voom/Limma.
For ternary plots, data were preprocessed with the Java program (PAMP_RnaSeq.java) and plotted using the R library ggtern (http://www.ggtern.com/). The detailed R script can be found on GitHub (https://github.com/steuernb/wheat_nlr).
Accession Numbers
The data sets generated and/or analyzed during this study are available in the European Bioinformatics Institute (EMBL-EBI) short read archive under study numbers PRJEB23081 and PRJEB23056. Please see Supplemental Tables S5, S6, and S8 for accession numbers.
Supplemental Data
The following supplemental materials are available.
Supplemental Figure S1. Consensus structure of an NLR with a CC domain and the amino acid motifs that can be used to identify structures within.
Supplemental Figure S2. Example of problems with the NLR gene annotation in RefSeq v1.0.
Supplemental Figure S3. Phylogenetic tree of NLR loci based on concatenated motif sequences of the NB-ARC domain.
Supplemental Figure S4. Motifs and integrated domains with the phylogeny of NLRs in wheat.
Supplemental Figure S5. Paired-NLR candidates.
Supplemental Figure S6. Introns in NLRs.
Supplemental Figure S7. Consensus structures of NLRs from the clade NB-ARC.
Supplemental Figure S8. Expression of known PAMP-inducible genes in RNA samples.
Supplemental Figure S9. Overlap of differentially expressed genes after flagellin and chitin treatments.
Supplemental Table S1. Cloned resistance genes in Triticeae.
Supplemental Table S2. Number of NLR loci found in exemplary plant genomes.
Supplemental Table S3. Number of NLR loci per wheat chromosome.
Supplemental Table S4. Candidate NLR loci in Sr and Lr disease resistance gene map intervals projected onto cv Chinese Spring.
Supplemental Table S5. NLR loci and overlapping genes from RefSeq annotation v1.1.
Supplemental Table S6. List of transcripts that were either classified as NLR associated by NLR-Parser, overlapping with an NLR locus, or found by homology search with ADR1.
Supplemental Table S7. Domains found in NLR genes.
Supplemental Table S8. Paired NLR candidates.
Supplemental File S1. Gene expression in response to PAMP treatment of all genes.
ACKNOWLEDGMENTS
We thank the International Wheat Genome Sequencing Consortium for early access to the RefSeq v1.0 of cv Chinese Spring, our colleague Yajuan Yue and John Innes Centre Horticultural Services for plant husbandry, and the Norwich BioScience Institutes Computing Infrastructure for Science group for high-performance computing maintenance. We thank David Swarbreck and Gemy Kaithakottil for technical support with Web Apollo. We thank Tobin Florio (www.flozbox-science.com) for the artwork in Figure 1.
LITERATURE CITED
Author notes
This work was supported by the Biotechnology and Biological Sciences Research Council (grant nos. BB/L011794/1, PRR-CROP BB/G024960/1, BB/M011216/1, BB/P016855/1, and BB/P012574/1), the 2Blades Foundation (Wheat Stem Rust), the Betty and Gordon Moore Foundation (Angiosperm Immune Receptor), and the Gatsby Foundation (The Sainsbury Laboratory).
Articles can be viewed without a subscription.
Senior author.
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (www.plantphysiol.org) is: Brande B.H. Wulff ([email protected]).
General concept and design of studies: B.S., K.W., J.D.G.J., B.K., S.G.K., and B.B.H.W.; design and testing of NLR-Annotator: B.S. and K.W.; resistance gene mapping to RefSeq: B.S. and S.G.K.; analysis of integrated domains and NLR pairs: B.S., E.B., K.V.K., K.W., G.Y., C.U., and B.B.H.W.; experimental concept and design of PAMP-triggered NLR expression: B.S., H.-J.S., C.J.R., and B.B.H.W.; tissue collection and RNA extraction: H.-J.S. and G.Y.; cDNA RenSeq: K.W. and A.I.W.; bioinformatics analysis and figures: B.S., R.H.R.-G., and I.Y.; article draft: B.S. and B.B.H.W.; all authors read and approved the final article.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}