





Plant-specialized metabolites account for arguably the largest and most diverse pool of natural products accessible to humans. Conferring the plants’ selective traits, such as UV defense, pathogen resistance, and enhanced nutrient uptake, these chemicals are crucial to a species’ viability. During biosynthesis of these compounds, a vast array of specialized enzymes catalyze diverse chemical modifications, with oxidation being one of the most predominant (Smanski et al., 2016; Dong et al., 2018). Considering these observations, it is not surprising that within plant genomes, two families of oxygenases are the most abundant: the cytochrome P450 monooxygenases (P450s) and the iron/2-oxoglutarate-dependent oxygenases (Fe/2OGs). These and other oxygenases represent the synthetic workhorses of plant-specialized metabolism and also play key roles in primary metabolism, cellular regulation, and fitness. Furthermore, the challenging and selective chemistry they catalyze cannot currently be matched by synthetic chemists. Since many plant natural products serve as valuable pharmaceuticals and commodity chemicals, plant oxygenases represent a promising toolset for synthetic biologists to manipulate plant traits or develop biocatalysts (Harvey et al., 2015). Here, we review families of plant oxygenases and their chemistry and suggest potential applications.
CYTOCHROME P450S
P450s (or CYPs) are hemoproteins that catalyze NADPH- and O2-dependent hydroxylation, demethylation, epoxidation, cyclization, and desaturation reactions, among others. P450s form a vast superfamily of proteins, the genes for which are found in bacteria, insects, fish, mammals, plants, and fungi. P450 nomenclature is based on amino acid sequence similarity, assigning proteins with greater than 40% sequence identity into the same family and proteins with greater than 55% sequence identity into the same subfamily (Nelson, 2006). Genomes of higher plants contain a large number of P450s. For example, the Arabidopsis (Arabidopsis thaliana) genome encodes 246 full-length P450s, accounting for approximately 1% of the Arabidopsis gene complement, and the Jatropha curcas (Barbados nut) genome is estimated to encode greater than 400 P450s (Schuler, 2015). Plant P450s have been shown to participate in a variety of pathways, including the biosynthesis of phenylpropanoids, alkaloids, terpenoids, lipids, cyanogenic glycosides, and glucosinolates, as well as plant growth regulators such as auxin, gibberellins, jasmonic acid, and brassinosteroids.
All eukaryotic P450s are strictly membrane bound and dependent upon partner flavin-dependent reductase proteins that provide reducing equivalents. Some also recruit cytochrome b5s as additional electron donors. These transmembrane domains make it difficult to obtain recombinant protein; therefore, it is challenging to obtain structural data for eukaryotic P450s. To date, a handful of human P450s and one plant member have been successfully crystallized by replacing the N-terminal signal peptide sequence to increase protein solubility. These structures are largely helical, with the heme buried in a solvent-exposed central cleft and an axial Cys residue additionally coordinating the iron cofactor (Fig. 1A). Oxygen activation generates the characteristic compound I intermediate (Fig. 1B) that catalyzes hydrogen atom transfer (HAT) from the substrate. While the mechanism of hydroxylation is largely understood (Fig. 1B; Box 1), control of the reaction outcome by nonhydroxylating members is a continuing field of study.
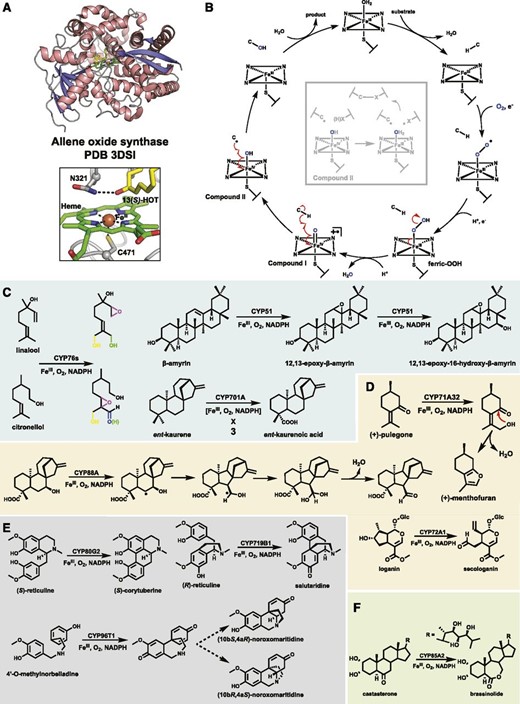
A, 3D structure of the plant P450 allene oxide synthase and zoomed-in view of the active site (PDB 3DSI). The heme scaffold is shown as green sticks, the ferric ion as an orange sphere, and a hydroxylated substrate mimic as yellow sticks. An active-site Asn residue helps tether the substrate above the heme. B, The mechanism of hydroxylation by the P450s invoking the potent compound I oxidant. A detailed discussion can be found in Box 1. The faded inset depicts an alternative nonhydroxylating pathway utilizing compound II for a second HAT step. C to F, Select plant P450 reactions discussed in the text that catalyze promiscuous hydroxylations (C), rearrangement coupled with hydroxylation (D), phenolic C-C bond coupling (E), and a Baeyer-Villiger-like oxidation (F). Possible independent reaction outcomes catalyzed by CYP76s are depicted by different colors. Dashed arrows represent spontaneous reactions.

Many characterized plant P450s catalyze hydroxylation (Schuler, 2015) of sp3 or sp2 C-H bonds, but their arsenal of catalysis is continually expanding as new plant natural product chemistry is discovered. P450 hydroxylases typically display strong preference toward a selective C-H bond of their substrate, but many have been shown to exhibit a general promiscuity, catalyzing a small percentage of altered regiospecific outcomes and/or activity upon related chemical scaffolds. For example, CYP76C subfamily P450s from Arabidopsis can install multiple hydroxyls into monoterpenol substrates (Fig. 1C; Höfer et al., 2014). Other P450s have dual functionality, such as Avena sativa (oat) CYP51H10 that hydroxylates C16 and epoxidates the C12-C13 bond of β-amyrin (Fig. 1C; Geisler et al., 2013) or ent-kaurene oxidase (CYP701A) that catalyzes three consecutive oxidation reactions to produce the GA precursor ent-kaurenoic acid (Fig. 1C; Helliwell et al., 2001; Mafu et al., 2016). In the latter reaction, the second turnover generates a dihydroxylated carbon center that dehydrates to an aldehyde prior to a third oxidation. In a similar manner, P450-dependent demethylation likely proceeds through hydroxylation, followed by decomposition to formaldehyde and the demethylated product (Kellner et al., 2015a). Another spontaneous rearrangement following hydroxylase activity by a P450 is thought to occur in (+)-menthofuran production by Mentha spicata (mint) CYP71A32 (Fig. 1D), wherein the newly installed hydroxyl group acts as a nucleophile (Bertea et al., 2001). In hydroxylation-derived reactions such as the latter, it is likely that additional protein residues aid in rearrangement.
Other P450s favor radical rearrangement of their substrate immediately following HAT but prior to hydroxylation. The ability to slow -OH rebound in these systems is not well understood but gives rise to unique P450 catalysis, for example, the ring contraction of ent-7-OH-kaurenoic acid (Fig. 1D) by CYP88A (Helliwell et al., 2001) and the oxidative ring opening of loganin to secologanin (Fig. 1D) by CYP72A1 or secologanin synthase (Irmler et al., 2000). In both systems, it is likely that radical rearrangement post HAT leads to -OH rebound at a different C atom than initially targeted that is ultimately dehydrated to give the final product (Hakamatsuka and Hashim, 1991; Yamamoto et al., 2000). Whereas these P450s delay hydroxylation, others completely suppress it by utilizing compound II for a second HAT, generating two substrate radicals that undergo recombination to form a new bond (Fig. 1B, inset). This strategy is utilized by P450s for olefin installation and the intra/intermolecular cyclization of C-C or C-O bonds, the latter being used extensively during plant benzylisoquinoline alkaloid biosynthesis (Kellner et al., 2015a, 2015b; Kilgore et al., 2016). The intramolecular C-C coupling of (S)-reticuline and (R)-reticuline is catalyzed by Coptis japonica CYP80G2 and Papaver somniferum CYP719B1, respectively (Fig. 1E; Kellner et al., 2015b). In both systems, HAT from hydroxyl groups on opposite rings leads to C-C bond formation via radical rearrangement (Mizutani and Sato, 2011). Subsequent enolization then rearomatizes, a step possibly facilitated by active-site residues. Both of these P450s must bind the phenolic rings such that the hydroxyls are primed for HAT, but the altered chirality likely brings different C atoms within close proximity for radical coupling. A more recently identified P450, CYP96T1 from daffodil (Narcissus pseudonarcissus), catalyzes a similar C-C coupling of 4′-O-methylnorbelladine to noroxomaritidine but produces an enantiomeric mixture (Fig. 1E), thought to arise from flexibility of the C-C coupled product prior to spontaneous N-C ring closure (Kilgore et al., 2016). The continued research on properties of P450s that enable targeting of an O-H bond and/or utilization of compound II for a second HAT step will yield valuable information for the rational design of atypical P450 reactivity.
Much of the P450’s catalytic capability extends from its ability to generate high-valent iron species that allow it to target otherwise unreactive C-H bonds. However, the ferric-peroxo species (Fig. 1B) is also thought to act as a nucleophile during the Baeyer-Villiger oxidation catalyzed by CYP85A2 in brassinosteroid biosynthesis (Fig. 1F; Nomura et al., 2005). In this system, the peroxo intermediate likely attacks the steroid backbone ketone and undergoes a Criegee-like rearrangement to produce the expanded ester-containing ring of the brassinosteroids. The examples described above represent only a subset of known plant P450 reactions, a list that is likely to expand in the future.
Plant P450s have become a focal point of synthetic biology research due to their abundance and the challenging oxidative chemistry they catalyze. Total synthetic routes to many clinically used plant metabolites are crippled by efficiency, selectivity, and cost, often at steps that plant oxygenases catalyze with ease (King-Smith et al., 2018). Chemo-enzymatic approaches using plant P450s expressed in Escherichia coli and yeast hosts have overcome some of these bottlenecks, enabling more efficient production of highly complex compounds. Among others, semisynthesis of the antimalarial drug artemisinin is a prominent example (Paddon and Keasling, 2014). Nevertheless, expression of plant P450s in nonplant hosts can be challenging, as they require coexpression of partner reductases, need to be localized to the endoplasmic reticulum membrane, and may produce numerous undesired products (O’Reilly et al., 2011; O’Connor, 2015). Additionally, some P450s have been shown to preferentially operate synergistically with other P450s (Quinlan et al., 2012) or in multienzyme metabolon complexes (Jørgensen et al., 2005; Ralston and Yu, 2006; Bassard et al., 2017), which must be considered when designing synthetic systems involving P450s. These challenges can be alleviated through the use of a heterologous plant host for which new strategies have been devised to achieve improved activity. One promising approach is to synthetically target P450s to the thylakoid membranes of the chloroplasts in planta (Nielsen et al., 2013). This not only insulates the engineered pathway from other metabolic pathways of the hosts but also obviates the dependency upon a partner reductase, as photosynthesis can provide the reducing equivalents required for catalysis (Lassen et al., 2014). This strategy, first tested in 2013, has since been used to reconstitute the entire dhurrin biosynthetic pathway via CYP79A1 (Gnanasekaran et al., 2016) and was recently adapted to produce artemisinic acid, the precursor to artemisinin, in tobacco (Nicotiana tabacum) leaves (Fuentes et al., 2016).
The P450 scaffold itself also holds great potential for engineering novel catalysts, as demonstrated by pioneering work of the Arnold lab and others. Through high-throughput directed evolution approaches, bacterial P450-BM3 and others have been evolved extensively for a wide range of oxidations on a diverse set of substrates (Jung et al., 2011; Renata et al., 2015; Arnold, 2018). P450-BM3 mutants are being used in the chemo-enzymatic synthesis of plant metabolites such as nigaelladine A (Loskot et al., 2017), a norditerpenoid alkaloid from Nigella glandulifera (Chen et al., 2014). The P450-BM3 system was targeted because it is naturally fused to its partner reductase, a trait not observed for any known plant P450. However, successful fusion of plant P450s to partner reductases has enabled an increase in their catalytic efficiency (Didierjean et al., 2002; Schückel et al., 2012; Sadeghi and Gilardi, 2013). Directed evolution of P450s has also yielded novel activities and even some previously unobserved for any natural catalyst, such as carbene/nitrene insertions to generate cyclopropyl/butyl groups, aziridination, and sulfimidation, greatly expanding the utility of this enzyme scaffold (Coelho et al., 2013; Wang et al., 2014; Brandenberg et al., 2017; Chen et al., 2018a). Such exhaustive evolution of any plant P450 has yet to be explored, owing to their difficulty in heterologous expression, but their native promiscuity could yield improved access to a plethora of plant secondary metabolites. For example, researchers observed that only three amino acid variations were needed to efficiently alter P450 activity in carnosic acid biosynthesis (Scheler et al., 2016). Significant advancement in P450 structure and mechanistic control are necessary to expand such efforts in the future.

FE/2OGS
The second largest oxygenase family in plants is the Fe/2OGs, represented by ∼130 genes in Arabidopsis (Hagel and Facchini, 2018). The Fe/2OGs participate in many of the same plant pathways as P450s and also function in histone modification and DNA repair, roles conserved in all domains of life. These oxygenases utilize a nonheme ferrous cofactor and a 2OG cosubstrate for O2 activation to functionalize unreactive C-H bonds, concomitant with the production of CO2 and succinate coproducts. Plant Fe/2OGs mainly catalyze hydroxylation reactions, and other reactivities include C/N/O demethylation, olefin installation, C-/O,C bond coupling, and methoxy migration (Hagel and Facchini, 2018). Furthermore, bacterial Fe/2OGs catalyze C-N cyclization, epoxidation, halogenation, azidation, epimerization, and endoperoxdiation (Vaillancourt et al., 2005; Steffan et al., 2009; Chang et al., 2014, 2016; Matthews et al., 2014; Dunham et al., 2018). The wide range of catalytic functions, their cytosolic nature, and the absence of a required partner reductase make Fe/2OGs a valuable source of novel biocatalysts.
The mechanism and structure/function relationships of Fe/2OGs have been studied extensively and are largely understood for the common hydroxylation outcome (Fig. 2, A and B; Box 2; Bollinger et al., 2015). Structurally, they are defined by a conserved cupin or double-stranded β-helix, and the ferrous cofactor is coordinated by a conserved 2His-1Glu/Asp (HxD/ExnH) facial triad with the 2OG cosubstrate coordinated via its C1 carboxylate and C2 carbonyl (Fig. 2A). The substrate binds in the general vicinity such that its target carbon is positioned closest to the iron center. No plant Fe/2OG has been characterized biophysically in great depth, and only four x-ray crystal structures have been obtained (Wilmouth et al., 2002; Sun et al., 2015; Kluza et al., 2018). Besides a histone demethylase, the three other plant Fe/2OG structures are involved in plant specialized metabolism and share a highly similar secondary structure (∼2.5 Å root-mean-square deviation), despite disparity in substrates and reactivity. This conservation may extend to most specialized plant Fe/2OGs, suggesting these scaffolds could be more tractable for engineering purposes than their bacterial counterparts.
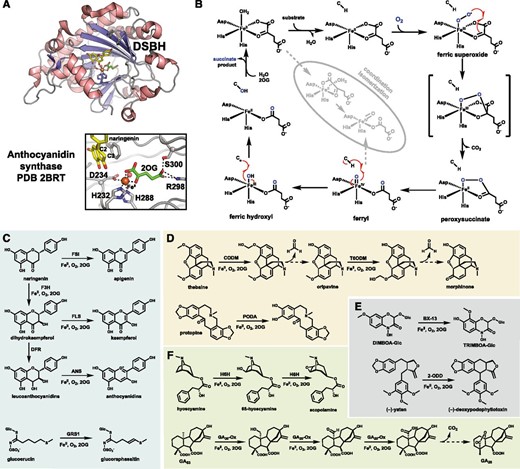
A, Overall double-stranded β-helix (DSBH) fold and active site of the plant Fe/2OG anthocyanidin synthase bound to substrate analog naringenin (PDB 2BRT). Naringenin is shown as yellow sticks, the 2OG cosubstrate as green sticks, the ferrous cofactor as an orange sphere, a water molecule as a red sphere, and the iron-binding facial triad residues and 2OG-stabilizing residues as gray sticks. B, The standing mechanistic proposal for Fe/2OG-dependent hydroxylation. A detailed discussion can be found in Box 2. The faded inset depicts possible routes to coordination relocation of the key ferryl intermediate to possibly suppress the canonical hydroxylation reactivity. C to F, Select plant Fe/2OGs described in the text that participate in flavonoid biosynthesis and catalyze olefin installation (C), catalyze demethylation or dealkylation (D), catalyze unique rearrangements (E), and catalyze iterative turnovers upon the same substrate (F). Dashed arrows represent spontaneous reactions. ANS, Anthocyanidin synthase; CODM, codeine O-demethylase; FLS, flavonol synthase; FSI, flavone synthase I; GA20ox, GA 20-oxidase; GRS1, glucoraphasatin synthase1; H6H, hyoscyamine-6β-hydroxylase; PODA, protopine O-dealkylase; T6ODM, thebaine-6 O-demethylase; DIMBOA, 2-(2,4-dihydroxy-7-methoxy-1,4-benzoxazin-3-one)-b-D-glucopyranose; TRIBOA, 2-(2,4,7- trihydroxy-8-methoxy-1,4-benzoxazin-3-one)-b-D-glucopyranose; 2-ODD, Phex30848.

The specialization of plant Fe/2OGs is prominently displayed by those involved in flavonoid biosynthesis (Fig. 2C). The flavonoid precursor naringenin acts as the substrate for both flavanone-3-hydroxylase (F3H) and flavone synthase I; the former hydroxylates C3, whereas the latter desaturates along the C2-C3 bond to yield apigenin. The F3H reaction product dihydrokaempferol serves as the substrate for flavonol synthase (FLS) that also desaturates the C2-C3 bond to yield kaempferol. Alternatively, the ketone of dihydrokaempferol can be reduced by the NADPH-dependent reductase DFR to give various leucoanthocyanidins that are desaturated once again along the C2-C3 bond by a fourth Fe/2OG, anthocyanidin synthase (Saito et al., 1999). This desaturation triggers spontaneous dehydration of C4 to produce the oxinium-containing anthocyanidins leading to the vibrant pigments of many flowering plants. These four Fe/2OGs are an excellent group of related enzymes from which structural features dictating substrate promiscuity and specificity can be learned. Structures of anthocyanidin synthase bound to narigenin or dihydrokaempferol (Wilmouth et al., 2002; Welford et al., 2005) suggest that these Fe/2OGs, assuming a roughly similar binding mode, initially target C3. While this is in line with hydroxylation by F3H, desaturation via initial targeting of C3 would not correlate to standing and recently supported mechanistic proposals of either an adjacent-heteroatom-assisted or dual HAT pathway (Dunham et al., 2018), the latter of which would require the C2-H bond to be directed toward the iron center. Further analysis of these Fe/2OGs may reveal a third desaturation strategy. It is worth noting that an other known plant Fe/2OG catalyzing desaturation, glucoraphasatin synthase1 from Raphanus raphanistrum subsp. sativus (radish; Fig. 2C), installs an alkene adjacent to an S atom of its glucoerucin substrate, consistent with the proposed heteroatom-assisted mechanism (Kakizaki et al., 2017; Dunham et al., 2018).
Other plant Fe/2OGs have repurposed their hydroxylase activity to trigger rearrangement similar to P450 reactions. For the N-Lys demethylases involved in histone regulation, these rearrangements occur spontaneously, as the charged amino group is an inherently good leaving group (Tsukada et al., 2006; Chen et al., 2011). In other cases, such as codeine/thebaine-6 O-demethylase and protopine O-dealkylase (Fig. 2D; Farrow and Facchini, 2013), assistance from surrounding active-site residues for proton donation is likely. These Fe/2OGs are the first members shown to catalyze O-demethylation, and the dealkylation activity of protopine O-dealkylase was previously observed only for P450s (Farrow and Facchini, 2013). Other rearrangements are catalyzed by the Fe/2OGs Phex30848 (or 2-ODD) and BX13 that catalyze the C-C bond formation of (−)-yatein (Lau and Sattely, 2015) and an odd methoxy migration in benzoxazinoid biosynthesis (Handrick et al., 2016), respectively (Fig. 2E). The exact mechanisms remain unclear but could invoke rearrangement of thecarbon radical post HAT instead of an -OH-modified product. Other Fe/2OGs can catalyze additional oxidations on their initially hydroxylated products. Identified as the main contributor to the famous dwarf phenotype characterized by reduced levels of the growth hormone GA in an Oryza sativa (rice) mutant (Sasaki et al., 2002), GA 20-oxidase catalyzes three consecutive oxidations of the C20 methyl of GA53 to produce GA20 (Fig. 2F). GA 20-oxidase converts this methyl group to a hydroxyl, formal, and carboxyl group consecutively, of which the final reaction triggers rearrangement, installing the characteristic carboxyl bridge. Another dual-function Fe/2OG is the epoxide-forming hyoscyamine-6β-hydroxylase in Solanaceae plants that first hydroxylates the C6 position prior to C-O ring closure with C7 in two independent turnovers (Ushimaru et al., 2018; Fig. 2F). Hyoscyamine-6β-hydroxylase differs from other epoxidizing Fe/2OGs that directly target alkenes in a single turnover (Chang et al., 2016) and is more similar to other C-O-cyclizing members such as clavaminate synthase (Zhou et al., 2001) and the loline oxidase LolO (Pan et al., 2018). Additional studies are required to fully understand the intricacies of the reaction outcome in this enzyme family.
Although not as heavily studied or utilized as biocatalysts compared with P450s, Fe/2OGs could serve as valuable synthetic biology tools for engineering purposes. Their involvement in numerous plant hormone and defense pathways makes them good targets for altering certain plant traits, as demonstrated by the generation of pathogen resistance Solanum tuberosum (potato) through CRISPR targeting of the DOWNY MILDEW RESISTANCE6 gene, an Fe/2OG involved in plant defense signaling (Zeilmaker et al., 2015). Specialized plant Fe/2OGs in particular could be useful due to the wide array of substrate chemical scaffolds they accommodate and a potentially conserved secondary structure. For example, a recent review proposed a plausible chemo-enzymatic synthesis for natural and modified vinblastine/vincristine alkaloids through the use of desacetoxyvindoline-4-hydroxylase from Catharanthus roseus (Madagascar periwinkle; King-Smith et al., 2018). Such approaches using bacterial Fe/2OGs for natural product synthesis have already been demonstrated (Zhang et al., 2018; Zwick and Renata, 2018). Moreover, Fe/2OGs could be engineered to produce novel halogenated natural products in planta, a strategy exemplified by engineering chlorinated monoterpene indole alkaloid production in Madagascar periwinkle using bacterial FAD-dependent halogenases (Runguphan et al., 2010). It was recently demonstrated that a bacterial Fe/2OG amino acid hydroxylase could be modified to catalyze halogenation of its native substrate, albeit at reduced efficiency (Mitchell et al., 2017a). Theoretically, plant Fe/2OG hydroxylases could be engineered to produce novel halogenated compounds as well. Researchers have also recently developed a promising chemical handle upon Fe/2OG activity in vivo through the use of a modified or bumped N-oxalylglycine (NOG) chemical inhibitor (Sudhamalla et al., 2018). NOG is a structural analog of 2OG that nonspecifically inhibits Fe/2OG activity. By expanding the active site of an Fe/2OG protein involved in nucleotide repair via rational design, it was demonstrated that NOG harboring small aliphatic groups at the C4 position could selectively inhibit only the engineered enzyme while wild-type activity was unaffected. This approach could be applied to transgenic plants to selectively regulate artificially engineered Fe/2OG-gated pathways.
FLAVIN MONOOXYGENASES
Flavin-dependent monooxygenases (FMOs) utilize an organic FAD cofactor and O2 for oxidation. Their reactions are also dependent on reducing equivalents from a NADPH cosubstrate, and, in contrast to metalloenzymes capable of accessing high-valent oxo species to target unreactive C-H bonds, FMO activity is restricted to the targeting of nucleophilic or electrophilic substrates. Currently, only three sets of plant FMOs are known, the FMO1s, glucosinolate-S-oxidases (GS-Oxs), and YUCCAs, all with highly specific biochemical functions (Schlaich, 2007). This is in stark contrast to homologous human FMOs that lack specificity and play a general role in xenobiotic metabolism (Lattard et al., 2004). Bacterial FMOs are vastly more diversified than both aforementioned sets, with known members capable of halogenation, oxidative rearrangements, and more (Huijbers et al., 2014). The term FMO can be applied across many classes of FAD/O2-utilizing enzymes, most of which have not been identified in plant genomes. Plant FMOs are defined by a double Rossmann fold that simultaneously binds FAD and NADPH via conserved motifs in the N/C termini (Fig. 3A; Huijbers et al., 2014). Reduction of the FAD cofactor by NADPH allows for the activation of O2 to produce the common hydroperoxyflavin or C4α(OOH)FAD intermediate for catalysis, observed in all characterized FMOs (Fig. 3B). Structures of bacterial FMOs have also suggested a second role of the bound NADP+ in stabilizing the reactive intermediate, which needs to be investigated further (Alfieri et al., 2008).
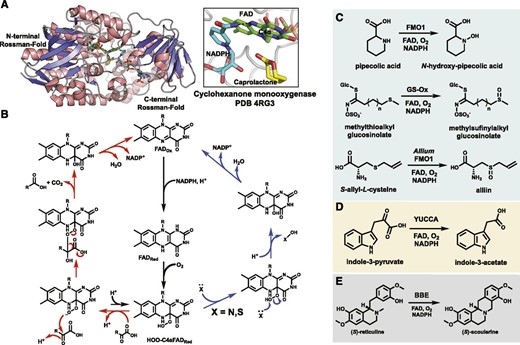
A, The double Rossmann fold and active site of a bacterial cyclohexanone monooxygenase bound to its caprolactone product (PDB 4RG3). No plant FMO has been crystallized, but this bacterial Baeyer-Villiger monooxygenase (BVMO) is predicted to share an overall secondary structure. The FAD cofactor is shown as green sticks, the NADPH cosubstrate as cyan sticks, the product as yellow sticks, and the C4α atom that reacts with O2 is labeled. B, The proposed mechanisms for plant FMO activity utilizing the C4α(OOH)FAD intermediate as an electrophile (blue path) or nucleophile (red path). C to E, Select plant FMO reactions discussed in the text that catalyze hetero-atom oxidation (C), oxidative decarboxylation (D), and a C-C coupling reaction catalyzed by the plant FAD-dependent oxidase BBE (E).
Among the three classes of plant FMOs, two catalyze heteroatom oxidation and the other catalyzes oxidative decarboxylation. Arabidopsis FMO1 was first identified as an enzyme linked to plant systemic acquired resistance (Mishina and Zeier, 2006), but its function was only resolved recently. Two groups independently discovered that FMO1 is responsible for the N-hydroxylation of pipecolic acid (Fig. 3C), producing the mobile signal molecule of the systemic acquired resistance pathway (Chen et al., 2018b; Hartmann et al., 2018). This reaction likely proceeds via nucleophilic attack upon the C4α(OOH)FAD intermediate and is shared by the GS-Oxs (Hansen et al., 2007; Kong et al., 2016). GS-Oxs catalyze the S-oxidation of thiol-glucosinolates to their respective sulfinyls, with homologs participating in alliin biosynthesis in Allium sativum (garlic; Fig. 3C; Yoshimoto et al., 2015). The GS-Oxs are highly selective toward glycosylated substrates (Hansen et al., 2007), further highlighting the distinction compared with the promiscuous human FMOs. The YUCCA family FMOs catalyze the oxidative decarboxylation of indole pyruvate to indole-3-acetic acid (IAA; Fig. 3D), the primary plant growth hormone (Mashiguchi et al., 2011). YUCCAs were initially proposed to catalyze the N-hydroxylation reaction earlier in the IAA pathway (Zhao et al., 2001), but follow-up studies clarified their bona fide activity (Mashiguchi et al., 2011; Stepanova et al., 2011). This reaction proceeds via nucleophilic attack by the C4α(OOH)FAD on the α-keto group and subsequent decarboxylation with O-O bond cleavage to produce IAA (Dai et al., 2013). Detection of a short-lived C4α(OOH)FAD species is consistent with its nucleophilic role compared with other FMOs (Dai et al., 2013). The YUCCAs are chemically similar to the BVMOs found in bacteria that oxidize linear or cyclic ketones to new ester compounds via rearrangement of a Criegee intermediate between the substrate and peroxy-flavin (Mascotti et al., 2015). No BVMO has been identified in plants, but these bacterial enzymes share conserved sequence and secondary structure features with plant and human FMOs, suggesting that plant FMOs could potentially be engineered to BVMO-like enzymes (Huijbers et al., 2014). In fact, a human FMO was recently shown to catalyze a BVMO-like reaction (Fiorentini et al., 2017). There are also Baeyer-Villiger-like oxidations of plant metabolites with unknown biosynthetic routes, such as the degradation of (+)-camphor in Salvia officinalis (sage; Croteau et al., 1984). The BVMOs have been used in numerous chemo-enzymatic applications, and their selectivity has also been successfully engineered (van Beek et al., 2012; Li et al., 2018a; Woo et al., 2018).
Other notable classes of FAD-dependent oxidases found in plants include the berberine-bridging enzymes (BBEs) and FAD-epoxidases. BBE, or (s)-reticuline oxidase from Eschscholzia californica (California poppy), catalyzes a C-C coupling reaction (Fig. 3E), producing H2O2 as a by-product, and utilizes a bicovalently tethered FAD cofactor (Daniel et al., 2017). Other members participate in similar cyclizations during cannabinoid metabolism (Daniel et al., 2017). FAD epoxidases specifically target alkenes of carotenoids or the triterpenoid precursor squalene and have a different source of reducing equivalents from other FMOs (Büch et al., 1995; Han et al., 2010). It is also worth mentioning the bacterial flavo-halogenases that generate a reactive hypohalous ion to chlorinate and brominate electron-rich substrates (van Pée and Patallo, 2006; Payne et al., 2016). Improvement of the aforementioned in planta halogenation of indole alkaloids (Runguphan et al., 2010) could lead to promising new therapeutics, as it is estimated that one-third of the drugs in clinical trials are halogenated (Fraley and Sherman, 2018), and the presence of halogens can significantly enhance drug potency (Imai et al., 2008; Gillis et al., 2015).
CAROTENOID CLEAVAGE DIOXYGENASES
First identified in Zea mays (maize; Schwartz et al., 1997), the carotenoid cleavage dioxygenases (CCDs) are a specialized class of non-heme-iron-dependent enzymes that cleave the electron-rich alkene chain of various carotenoids and apocarotenoids. While β-carotenes are important antioxidants and pigments themselves, CCD activity produces the precursors for the important signaling hormones strigolactone and abscisic acid, along with many notable fragrant compounds. The identification of plant CCDs has since revealed mammalian homologs involved in retinal production and bacterial homologs capable of metabolizing noncarotenoid substrates. In plants, CCD1, CCD4, CCD7, and CCD8 and the neoxanthin cleavage enzymes are universally conserved (Auldridge et al., 2006; Harrison and Bugg, 2014), the latter of which specifically target epoxidized carotenoids during the biosynthesis of abscisic acid, a key regulator of drought tolerance and seed dormancy (Fig. 4C). A number of specialized plant CCDs have also been identified, such as CCD2 from Crocus sativus, which cleaves zeaxanthin during corcin biosynthesis (Frusciante et al., 2014), and lycopene cleavage dioxygenase from Bixa orellana, which is capable of cleaving the β-carotenoid precursor (Satpathy et al., 2010).
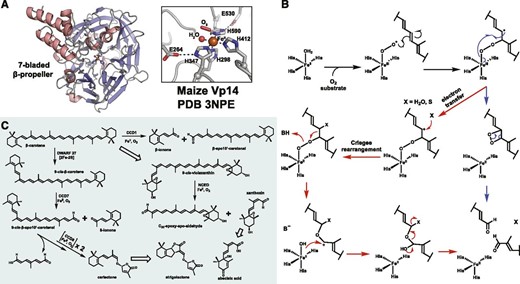
A, The seven-bladed β-propeller structure and active site of iron-bound Vp14, a plant CCD from maize. The four iron-coordinating His and the conserved second-sphere Glu residues are shown as gray sticks, the ferrous cofactor as an orange sphere, and a water molecule as a red sphere. An end-on coordinating O2 is modeled and shown as a red stick, but the accuracy of this model is suspect due to low resolution of the structure (∼3.1 Å). Structures of bacterial CCDs (sub-2 Å resolution) contain clear water molecules modeled at said location. B, Mechanistic proposals for CCD dioxygenation invoking either a dioxetane (blue path) or Criegee-like (red path) rearrangement. These two possibilities are explained further in Box 3. C, Schematic of conserved CCD reactions catalyzed in higher plants. Hollow arrows represent multiple and/or non-CCD enzyme reactions. DWARF27 is a 2Fe-2S-cluster-dependent carotenoid isomerase. NCED, Neoxanthin cleavage enzyme.
The cleavage activity of CCDs is catalyzed by a single ferrous cofactor and O2 molecule, drawing all four reducing equivalents from the primary substrate. Structural characterization and sequence comparison of CCDs revealed a conserved seven-bladed β-propeller architecture housing a 4-His coordination motif (Fig. 4A; Messing et al., 2010; Sui et al., 2013), also present in the bacterial lignostilbene dioxygenases that catalyze similar oxidative cleavage reactions (Bugg et al., 2011). Three second-sphere Glu residues that form hydrogen bonds with the His residues are also conserved (Sui et al., 2013; Priya and Siva, 2015). The exact mechanism for CCD-catalyzed cleavage remains in question, but the current proposal invokes the action of a Fe(III)-superoxide species (Fig. 4B; Box 3). Limited biochemical and structural studies of the plant CCDs have been reported to date.
The specialization of plant CCDs is exemplified by members CCD7 and CCD8 that act sequentially to produce carlactone, the direct precursor to strigolactone, a more recently identified plant hormone that controls root and shoot branching. Similar to CCD1 and CCD4 (Harrison and Bugg, 2014), CCD7 catalyzes the selective 9/10 cleavage of β-carotene; however, it specifically recognizes 9-cis-β-carotene (Fig. 4C) and displays no activity upon the more abundant trans-conformer (Bruno et al., 2014). Its 9-cis-β-apo-10′-carotenal product then serves as the substrate for CCD8, the most chemically unique plant CCD, catalyzing sequential dioxygenations to produce carlactone (Fig. 4C; Harrison et al., 2015). The first cleavage reaction of CCD8 is likely akin to others, but the second results in a unique rearrangement to produce the enol ether of carlactone. CCD8 seems to not release its initial cleavage product, an observation consistent with the recent proposal involving a covalent enzyme-product complex (Harrison et al., 2015). Alternatively, this could be achieved via direct coordination to the iron cofactor. Pre-steady-state kinetic analysis of CCD8 under submolar O2 concentrations and anaerobic crystallographic experiments would help decipher this mechanism. For other CCDs, substrate positioning seems to be the major factor dictating reaction outcome. Engineering of CCD activity could be used for the production of high-value rose (Rosa spp.) ketones, such as ionones and damascenones, used in fragrances and oils, but it will require improved structural and mechanistic knowledge to efficiently alter reactivity.

EXTRADIOL-CLEAVING DIOXYGENASES
Similar in reactivity to the CCDs, the extradiol-cleaving dioxygenases (ECDs) are iron-dependent enzymes that catalyze oxidative cleavage of C-C bonds. However, ECDs selectively target catechols to cleave the adjacent phenolic bonds, hence the name extradiol (Lipscomb, 2008). These enzymes contain a single ferrous cofactor coordinated via two His and one Asp residues, with their diol substrates occupying two other coordination sites (Lipscomb, 2008). In plants, there are a number of annotated ECDs, but few members have been functionally characterized. DOPA 4,5-dioxygenase (DODA) is responsible for the production of betalamic acid, the betalain pigment precursor, via cleavage of l-DOPA (Fig. 5A; Christinet et al., 2004; Chung et al., 2015), LigB from Arabidopsis cleaves caffealdehyde (Fig. 5A; Weng et al., 2012), and a third is proposed to be involved in stizolob(in)ic acid biosynthesis (Saito and Komamine, 1976). The 4,5 cleavage reaction of DODA produces 4,5-seco-DOPA that spontaneously rearranges to betalamic acid with high enantiomeric selectivity in both in planta and in vitro assays, suggesting that DODA may also play an additional role (Christinet et al., 2004). Bacterial ECDs catalyze a wide range of catechol cleavage reactions, some of which have been expressed in Medicago sativa (alfalfa) for bioremediation (Wang et al., 2015). It is possible that some of the uncharacterized plant ECDs have additional biosynthetic roles.
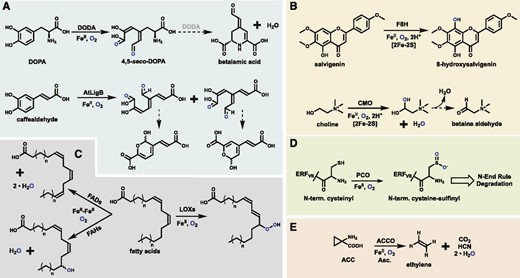
The representative reactions catalyzed by plant ECDs (A), RSOs (B), the LOXs and FaD/FaHs (C), PCOs (D), and ACCO (E). The role of DODA during the spontaneous formation of betalamic acid remains in question. Molecular oxygen atoms are shown in blue for tracing purposes. Dashed arrows represent spontaneous rearrangements, and hollow arrows represent multistep pathways. CMO, Choline monooxygenase.
RIESKE-TYPE OXYGENASES
Rieske-type oxygenases (RSOs) represent a small family of non-heme-iron-dependent oxygenases in plants. These enzymes contain separate ferrous- and 2Fe-2S-cluster-binding domains, the latter of which transfers electrons to the iron center via a bridging Asp residue during the catalytic mechanism for oxygen activation (Ferraro et al., 2005; Barry and Challis, 2013). Crystallized RSOs from bacteria revealed functional trimers with these domains juxtaposed. All higher plants possess a set of RSOs involved in chlorophyll degradation (Gray et al., 2004), and the only other known plant RSO is choline monooxygenase (Rathinasabapathi et al., 1997), which produces the osmoprotectant Gly-betaine (Fig. 5B; Hibino et al., 2002). RSOs are more abundant in bacterial genomes catalyzing known monohydroxylations and dihydroxylations, demethylations, N oxidation, and even C-C bond coupling (Barry and Challis, 2013), highlighting the synthetic potential of the RSO scaffold. A recent study identified a novel RSO from Ocimum basilicum (basil) trichomes that functions as a flavone-8-hydroxylase with strong selectivity toward salvigenin (Fig. 5B; Berim et al., 2014). Additional uncharacterized RSO homologs found in plant genomes may be proven to operate in specialized metabolism as well.
FATTY ACID OXYGENASES
The oxygenation of fatty acids is utilized by plants for the production of signaling and defense compounds. All higher plants possess two classes of oxygenases that target polysaturated or unsaturated fatty acids. The lipoxygenases (LOXs) are mononuclear nonheme enzymes that catalyze the insertion of molecular oxygen into unsaturated bonds to form hydroperoxo units (Fig. 5C; Baysal and Demirdöven, 2007). These compounds are then cleaved during production of various plant volatiles and oxylipins, such as the hormone jasmonic acid (Vick and Zimmerman, 1984; Fuller et al., 2001; Baysal and Demirdöven, 2007). LOXs are abundant in both plants and animals, utilizing three His residues and their C-terminal carboxyl group to coordinate an iron cofactor. This assembly differs from the di-iron cofactor used by the fatty acid hydroxylases (FaHs; Broun et al., 1998). Di-iron enzymes constitute an abundant superfamily that operates in critical biological processes in bacteria, such as ribonucleotide reduction and methane oxygenation, and catalyzes many monooxygenation or dioxygenation reactions such as N-hydroxylation in chloramphenicol biosynthesis (Jasniewski and Que, 2018; Komor et al., 2018). FaHs have only been found in select plant species but are closely related to the fatty acid desaturases (FaDs) present in nearly all higher plants (Fig. 5C; Broadwater et al., 2002). Sequence comparison of FaDs and FaHs revealed only seven class-specific variations (Broun et al., 1998). Subsequent mutagenesis studies showed that the desaturation:hydroxylation activity partition of Arabidopsis FAD2 and several FaHs could be manipulated with as few as a single amino acid change (Broun et al., 1998; Broadwater et al., 2002). Manipulation of plant LOXs and FaD/FaHs could be used to gain access to a range of high-value fatty acid derivatives and to manipulate plant growth and development.
CYS DIOXYGENASES
Cys dioxygenases (CDOs) are mononuclear non-heme-iron-dependent enzymes that couple the reduction of O2 to the oxidation of a Cys thiol to sulfinic acid. Bacterial and animal CDOs target free Cys in taurine biosynthesis (Dominy et al., 2006; Stipanuk et al., 2009), but plant CDOs (PCOs) target N-terminal cysteinyl residues of select plant peptides (Weits et al., 2014). As these sequences vary significantly, it remains to be determined whether PCOs utilize a similar active-site architecture to their mammalian and bacterial homologs. Briefly, CDOs utilize a cupin fold with a three-His iron motif to which the l-Cys substrate coordinates via its thiol and amino groups (McCoy et al., 2006; Simmons et al., 2006). Mammalian CDOs also contain a conserved Cys(S)-Tyr(meta-C) cross-link in the active site (McCoy et al., 2006; Li et al., 2018b), believed to help stabilize reactive Fe-oxo species (Ye et al., 2007). PCOs, first identified in Arabidopsis, are linked to the regulation of hypoxic response and were recently shown to specifically oxidize the N termini of class VII Ethylene Response Factors (ERF-VIIs; Fig. 5D; Weits et al., 2014; White et al., 2017). During periods of low cellular oxygen, such as flooding, the ERF-VIIs trigger up-regulation of anaerobic genes, enabling the plant to enter a generally anaerobic state (Bailey-Serres et al., 2012). Under normal O2 levels, the ERF-VIIs are oxidized by PCOs, which initiates their degradation via the N-end rule pathway, suggestingthat the PCOs act as the primary O2 sensor in vivo (Licausi et al., 2011; White et al., 2017). Flashman and coworkers recently provided support for this notion through the kinetic characterization of all five Arabidopsis PCOs, which displayed apparent K m values toward O2 within the physiological range (White et al., 2018). These findings and the in vivo role of PCOs are analogous to the regulation of the hypoxia-inducible transcription factors in animals by a set of Fe/2OGs, the hypoxia-inducible transcription factor hydroxylases, or PHDs, which display an even stronger O2 dependence (Ehrismann et al., 2007; Tarhonskaya et al., 2014). Arabidopsis PCOs were also shown to possess unique substrate preferences, varied enzymatic rates, and O2 dependence, suggesting nonredundant roles in vivo (White et al., 2018). Suppression of PCO activity could be used to increase the stability and abundance of ERF-VIIs, leading to improved flood tolerance and crop recovery (Licausi et al., 2010; Gibbs et al., 2011).

AMINOCYCLOPROPANECARBOXYLATE OXIDASE
Found in all higher plants, aminocyclopropanecarboxylate oxidase (ACCO) catalyzes the final step of ethylene biosynthesis via the Yang cycle (Fig. 5E; McKeon et al., 1995). Produced in nearly all plant tissues, ethylene is a major plant hormone controlling fruit ripening, senescence, and other aspects of plant growth (Iqbal et al., 2017). Manipulation of ethylene levels, both exogenously and within the plant, is routinely exercised to hasten ripening and extend shelf life. Tissue-specific regulation of ACCO activity is a powerful strategy to modulate many aspects of plant physiology (Tonutti et al., 1997; Ben-Amor et al., 1999; Lemus et al., 2007). ACCO utilizes a ferrous cofactor and O2 to produce one equivalent of ethylene, CO2, and cyanide from ACC (Rocklin et al., 2004). This reaction is also dependent upon ascorbate, presumably providing reducing equivalents, and an apparent requirement for CO2 or bicarbonate that remains to be understood. ACCO is structurally related to the Fe/2OGs, but its mechanism is not 2OG dependent (Hagel and Facchini, 2018). ACC binds in a pocket analogous to that of 2OG and coordinates in a bidentate manner via its amino and carboxyl groups. HAT from C2 by an Fe(IV)-oxo species is believed to initiate breakdown to ethylene and cyanoformate, the latter of which is shunted away from the active site before it decomposes (Murphy et al., 2014). Interestingly, bacteria also use an Fe/2OG-like enzyme for ethylene production, known as the ethylene-forming enzyme, which directly oxidizes the iron-bound 2OG molecule to ethylene and two molecules of CO2 (Martinez and Hausinger, 2016; Martinez et al., 2017; Zhang et al., 2017). Ethylene-forming enzymes have garnered much interest as a sustainable source for bioethylene production, as their 2OG substrate is cheap and it does not produce a toxic by-product like cyanide compared with ACCO.
CONCLUSION
Although the enzyme families discussed here do not include all plant oxygenases, those chosen provide a representative picture of the diverse chemistry and catalytic strategies utilized by oxygenases in plant metabolism. While many of the reaction outcomes are chemically redundant (i.e. hydroxylation), each system has its own advantages regarding their adaptability, selectivity, and/or accessibility as potential synthetic biocatalysts. P450s have garnered much of the attention due to their abundance, plasticity, and established engineering efforts. However, the influx of plant genomic data has helped illuminate the activity and role of numerous other, less studied oxygenases in plant metabolism. Many of these oxygenase families have proven to be useful biocatalysts in bacterial systems, highlighting their synthetic potential. Similar endeavors using plant catalysts have presented many difficulties, as many plant enzymes are unstable in vitro, harbor suboptimal activity, and/or require additional partnering enzymes. Establishment of in vivo optimization techniques, such as directed evolution, in plant cells may ultimately circumvent this issue and allow the rapid generation of novel plant catalysts. A continued effort to enhance basic structural and biochemical knowledge of each oxygenase family should be emphasized to lay the foundation for future rational design of enzyme activity. If such strategies can be routinely accessed, the presence of oxygenases throughout plant genomes will give synthetic biologists access to nearly all facets of plant biochemistry and physiology.
LITERATURE CITED
Author notes
This work was supported by grants from the Edward N. and Della L. Thome Memorial Foundation, the Family Larsson-Rosenquist Foundation, and the National Science Foundation (grant no. 1709616). J.-K.W. was supported by the Arnold and Mabel Beckman Foundation, Pew Charitable Trusts (grant no. 27345), and the Kinship Foundation (grant no. 15-SSP-162).
Author for contact: [email protected].
Senior author.
A.J.M. and J.-K.W. wrote the article.
Articles can be viewed without a subscription.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}