





Abstract
The legume-rhizobium symbiosis is initiated through the activation of the Nodulation (Nod) factor-signaling cascade, leading to a rapid reprogramming of host cell developmental pathways. In this work, we combine transcriptome sequencing with molecular genetics and network analysis to quantify and categorize the transcriptional changes occurring in roots of Medicago truncatula from minutes to days after inoculation with Sinorhizobium medicae. To identify the nature of the inductive and regulatory cues, we employed mutants with absent or decreased Nod factor sensitivities (i.e. Nodulation factor perception and Lysine motif domain-containing receptor-like kinase3, respectively) and an ethylene (ET)-insensitive, Nod factor-hypersensitive mutant (sickle). This unique data set encompasses nine time points, allowing observation of the symbiotic regulation of diverse biological processes with high temporal resolution. Among the many outputs of the study is the early Nod factor-induced, ET-regulated expression of ET signaling and biosynthesis genes. Coupled with the observation of massive transcriptional derepression in the ET-insensitive background, these results suggest that Nod factor signaling activates ET production to attenuate its own signal. Promoter:β-glucuronidase fusions report ET biosynthesis both in root hairs responding to rhizobium as well as in meristematic tissue during nodule organogenesis and growth, indicating that ET signaling functions at multiple developmental stages during symbiosis. In addition, we identified thousands of novel candidate genes undergoing Nod factor-dependent, ET-regulated expression. We leveraged the power of this large data set to model Nod factor- and ET-regulated signaling networks using MERLIN, a regulatory network inference algorithm. These analyses predict key nodes regulating the biological process impacted by Nod factor perception. We have made these results available to the research community through a searchable online resource.
Most legumes and a few allied taxa are able to establish nitrogen-fixing symbioses with endophytic bacteria known generally as rhizobia. This mutualistic interaction represents a vital entry point for reduced nitrogen into the biosphere and is a property that underpins sustainable agriculture worldwide (Graham and Vance, 2003). The legume-rhizobium symbiosis is initiated by the plant’s recognition of specific bacterial lipochitooligosaccharides termed Nodulation (Nod) factors. Within minutes after Nod factor perception, target root hair cells exhibit biochemical and morphological responses that reprogram transcriptional and developmental outputs, culminating in a nitrogen-fixing symbiotic organ called the nodule. Myriad structural and functional alterations occur within the plant root in response to Nod factors, implicating a diverse repertoire of genes and proteins. The earliest known plant response to Nod factors involves depolarization of the root hair plasma membrane (Ehrhardt et al., 1992), a consequence of tip-focused ion fluxes (Felle et al., 1998). Such changes proceed and are likely causal to a transient cessation and subsequent reactivation of root hair polar growth, leading to the formation of a characteristic root hair curl (the shepherd’s crook), entrapping bacteria that will ultimately colonize the nodule organ. Bacteria enter the root hair following localized cell wall hydrolysis (Xie et al., 2012) within an inward-growing channel known as the infection thread. At the biochemical level, Nod factors elicit a complex series of changes, including ion fluxes at the root hair tip (Felle et al., 1998), periodic increases in intracellular calcium within and around the nucleus (Ehrhardt et al., 1996; de Ruijter et al., 1999; Wais et al., 2000), rearrangement of the root hair cytoskeleton (Cárdenas et al., 1998; Timmers et al., 1999), changes in phosphorylation profiles (Grimsrud et al., 2010), modification of membrane protein dynamics (Haney et al., 2011), and induction of cortical cell divisions (Ehrhardt et al., 1992; Timmers et al., 1999, and refs. therein). The perception of Nod factors and subsequent nodule development also modify the root transcriptional program, with gene induction observed as early as 3 h post inoculation (hpi; Pichon et al., 1992; Cook et al., 1995).
Over the past decade, researchers have elucidated a Nod factor perception and signaling pathway in legumes and an overlapping mycorrhizal pathway in both legumes and nonlegumes (for review, see Venkateshwaran et al., 2013). Nod factor receptors, which include Lysine motif domain-containing receptor-like kinase3 (LYK3) and Nodulation factor perception (NFP) in Medicago truncatula and Nodulation factor receptor1 (NFR1) and NFR5 in Lotus japonicus, are plasma membrane-localized receptor-like kinase and kinase-like (RLK) proteins (Amor et al., 2003; Smit et al., 2007; Haney et al., 2011; Moling et al., 2014) consisting of an intracellular kinase domain and an extracellular region with two or three chitin-binding Lys motifs (LysM). LysM domains bind Nod factors through their chitin backbone (Petutschnig et al., 2010; Broghammer et al., 2012) and are implicated in Nod factor recognition specificity (Radutoiu et al., 2007). Nod factor receptor mutants in Medicago and Lotus spp. have subtly different phenotypes. While NFR1 and NFR5/NFP are essential for all symbiotic responses, lyk3 mutants (known as hair curling [hcl]) manifest excessive root hair curling, calcium spiking, and limited cortical cell division, but they exhibit an attenuated transcriptional response (El Yahyaoui et al., 2004; Mitra et al., 2004) and aberrant or absent infection threads (Catoira et al., 2001; Smit et al., 2007).
Downstream of the Nod factor receptors, a conserved set of genes initiate the host’s symbiotic responses to both symbiotic bacteria and mycorrhizal fungi. These include DOESN’T MAKE INFECTIONS1 (DMI1), DMI2, and DMI3, two nucleoporins, and the DMI3-interacting protein IPD3/CYCLOPS (Messinese et al., 2007; Yano et al., 2008). DMI2 (SYMRK in L. japonicus) encodes a Leu-rich repeat (LRR)-RLK required for calcium spiking and subsequent transmission of the Nod factor signal (Endre et al., 2002; Stracke et al., 2002). DMI1 (CASTOR and POLLUX in L. japonicus) encodes a nucleus-localized ion channel (Ané et al., 2004; Riely et al., 2007; Charpentier et al., 2008) that is also required for calcium spiking. DMI3 (CCaMK in L. japonicus), a calcium and calmodulin-dependent protein kinase, presumably decodes the nuclear calcium oscillations and activates a transcriptional cascade (Lévy et al., 2004; Mitra et al., 2004; Singh and Parniske, 2012). A diverse suite of symbiotic transcription factors (TFs), including the GRAS family proteins NODULATION-SIGNALING PATHWAY1 (NSP1) and NSP2, the NODULE INCEPTION1 protein, NUCLEAR FACTOR-Y A1 and B1 (NF-YA1 and NF-YB1), and several members of the APETALA2 (AP2)/ETHYLENE RESPONSE FACTOR (ERF) family of TFs, mediate symbiotic gene expression and are each essential for symbiotic infection and nodule organogenesis (Oldroyd et al., 2011; Venkateshwaran et al., 2013).
Given the complex developmental reprogramming that rhizobial infection and nodule organogenesis require, it is not surprising that these processes are tightly regulated by the plant. In particular, the plant hormone ethylene (ET) has long been recognized as a negative regulator of the legume-rhizobial symbiosis (Lee and LaRue, 1992). ET acts at multiple symbiotic stages during nodule initiation, and many of ET’s effects on symbiosis may be explained by negative regulation of Nod factor signaling, although the molecular mechanism(s) of such regulation is not understood. Thus, ET perception negatively regulates calcium spiking and root hair deformation (Oldroyd et al., 2001), infection thread persistence (Penmetsa and Cook, 1997), and primordium formation at sites opposite phloem poles (Heidstra et al., 1997; Penmetsa et al., 2003). M. truncatula ETHYLENE INSENSITIVE2 (EIN2) mutants (known as sickle [skl]) are not only insensitive to ET but are also hypersensitive to Nod factors (Oldroyd et al., 2001). Consequently, ET-insensitive skl mutants are hypernodulated by rhizobia, hyperinfected by mycorrhizal fungi, and display an increased susceptibility to root-infecting pathogens (Penmetsa et al., 2008).
Several array-based studies have characterized transcriptional changes occurring during nodule formation (El Yahyaoui et al., 2004; Mitra et al., 2004; Lohar et al., 2006; Benedito et al., 2008; Breakspear et al., 2014). Most studies analyzed relatively late time points and were limited by their incomplete representation of plant genes, reduced sensitivity, and inability to reliably discriminate close paralogs. Recent advances in next-generation sequencing technology have given a new dimension to transcriptome analyses, providing higher technical reproducibility and a wider dynamic range for the detection of changes in gene expression (Fu et al., 2009). Transcriptome sequencing (RNA-seq) has been applied to detect symbiosis-induced transcriptional changes in different legume species (Fu et al., 2009; Libault et al., 2010; Hayashi et al., 2012; Rose et al., 2012; Roux et al., 2014), but often at late developmental time points, at small scales, and with limited replication. Thus, the very early transcriptional changes (e.g. minutes to hours after inoculation) occurring upon the perception of Nod factors remain poorly documented.
In this work, we combine massive transcriptome sequencing with molecular genetics, promoter:reporter gene fusions, and network analysis to quantify, categorize, and spatially and temporally resolve transcriptional changes occurring in M. truncatula roots on the scale of minutes to days after inoculation with Sinorhizobium medicae. Our 2-fold aim was to identify symbiosis-specific genes whose transcription is activated by Nod factors and to characterize the ET biosynthesis and signaling pathways in M. truncatula in the context of early symbiotic development. The outcome is a deep analysis of the early transcriptional changes occurring in M. truncatula hours after inoculation, providing a detailed digital time course of gene expression and highlighting numerous novel genes coexpressed with known markers of symbiosis.
The rich collection of genetic and temporal variation in this large compendium also permits us to perform expression-based regulatory network inference to predict novel relationships between regulatory proteins and target genes as well as to predict candidate regulators of early symbiosis. The analysis reveals several distinct transcriptional outcomes that include ET responses nested within Nod factor responses and further nesting of other hormone pathways within the ET and Nod factor networks. The most dramatic of these is a large suite of Nod factor-activated genes that are either negatively or positively regulated by a Nod factor-dependent ET circuit, which we interpret as a negative feedback loop. Transcriptional profiling as well as promoter:GUS fusion studies reveal that ET biosynthesis is induced primarily in the epidermis during rhizobium infection and, subsequently, in the meristematic region in mature nodule tissues. Taken together, these results suggest that the spatiotemporal regulation of ET production is an important component of symbiotic regulation and nodule development.
RESULTS
Massive Transcriptional Reprogramming of Roots in Response to Rhizobium
With the goal of characterizing the host’s earliest transcriptional responses to bacterial signals at high temporal resolution, 5-d-old roots of M. truncatula were inoculated with S. medicae ABS7M (pXLGD4) at densities of 150 cells µL−1 (optical density at 600 nm = 0.001). Tissue was sampled at nine points over a 48-h time course, with the first postinoculation time point at 30 min. Live bacteria were used to conserve the biological context of Nod factor delivery and to account for the possibility that certain rhizobium-induced responses may be independent of Nod factors. Host genetics was used to infer the nature of the inciting ligand (e.g. Nod factor or potentially other bacterial factors), and for the purpose of presenting the data, we operationalize mutant transcription phenotypes as the products of Nod factor and ET ligands. Rather than using multiple alleles of individual genes, we use strong alleles of different genes acting in the same pathway. We attribute the symmetry of responses among mutants to their action in a common pathway, namely, Nod factor signaling, thereby reducing the possible confounding effect of idiosyncrasies of the particular genes and alleles under test. Wild-type M. truncatula was inoculated in parallel to nfp and lyk3 LysM receptor mutants that are either insensitive (nfp; Amor et al., 2003) or minimally sensitive (lyk3/hcl-1; Catoira et al., 2001) to Nod factors and to skl, which is an ET-insensitive mutant that exhibits excessive symbiotic development (Penmetsa and Cook, 1997) and is approximately 20-fold more sensitive to Nod factor than wild-type plants (Oldroyd et al., 2001).
To correlate gene expression at specific time points with early symbiotic development, morphological changes occurring in roots and root hairs were monitored for 3 d following inoculation. S. medicae ABS7M (pXLGD4) constitutively expresses the lacZ (for β-galactosidase, EC 3.2.1.23) gene, allowing the observation of the bacteria throughout the infection process. Host responses were first evident at 3 hpi, when root hairs began to swell, followed by asymmetric tip growth and tip branching between 3 and 6 hpi (Fig. 1). We observed infrequent root hair curls at 12 hpi, which became significantly more prevalent by 24 hpi, the stage at which blue-stained bacterial microcolonies were initially observed within root hair curls. Bacterial entry was first evident as infection threads contained within root hairs at 36 hpi, reaching the base of infected trichoblast cells by 48 hpi. By 72 h, nascent nodule primordia had developed, at which time successful infection threads had reached dividing cells within the root inner cortex.
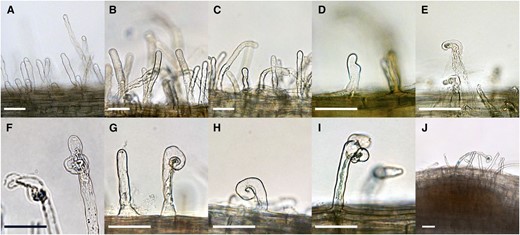
Root responses and infection development during the experimental time course. Roots of 5-d-old M. truncatula seedlings were inoculated with S. medicae ABS7M constitutively expressing the lacZ gene. Morphological features, including root hair deformations and inner cortical cell divisions, were monitored in the susceptible zone proximal to the root tip across time. A, Uninoculated roots. B, Roots at 0.5 hpi. C, Roots at 1 hpi. D, Roots at 3 hpi. E, Roots at 6 hpi. F, Roots at 12 hpi, G, Roots at 24 hpi. H, Roots at 36 hpi. I, Roots at 48 hpi. J, Roots at 72 hpi. Scale bars = 50 μm.
RNA was isolated, reverse transcribed, and sequenced from four biological replicates of whole roots harvested from noninoculated plants and at eight time points between 30 min and 48 hpi. The selected time points encompass host responses from Nod factor perception through root hair infection and cortical cell division. A total of 144 complementary DNA libraries were sequenced to an average depth of 15 million reads (Supplemental Fig. S1), for a total of 220 Gb of trimmed data. After employing quality filters, 69,237 annotated genes matched one or more reads, among which 46,720 were associated with a minimum of 30 independent reads. Following normalization (Robinson and Oshlack, 2010) and the imposition of a conservative 0.001 false discovery rate (FDR) cutoff (Supplemental Fig. S2), 10,985 genes were identified as differentially expressed between genotypes using the statistical package edgeR (Supplemental Table S1). Among these, 3,631 were differentially expressed between cv Jemalong A17 and nfp, 1,996 between cv Jemalong A17 and lyk3, and the vast majority, 8,760, between cv Jemalong A17 and skl (Supplemental Fig. S3).
We developed a Web interface to make the results of our experiments more easily accessible. The entirety of our expression data and the accompanying MERLIN network analyses (see below) can be queried and downloaded at http://pages.discovery.wisc.edu/∼sroy/medicago_symbiosis_transcriptome/query.php.
Hierarchical Clustering Highlights Nod Factor- and ET-Dependent Transcriptional Changes and Identifies Symbiosis-Related Genes
Model-based hierarchical clustering was used to extract the main patterns of gene expression among the four genotypes (Fig. 2). MCLUST version 3 (Fraley and Raftery, 2006) resolved the 10,985 differentially expressed genes into 11 groups (G0–G10) based on their expression profiles across samples (Fig. 2A). To compare the trends in gene expression of each group as a function of plant genotype, expression values were normalized to those observed in noninoculated wild-type plants. Figure 2B presents an overview of expression for each of the 11 groups, depicted graphically as the aggregate average expression value for each genotype and time point.
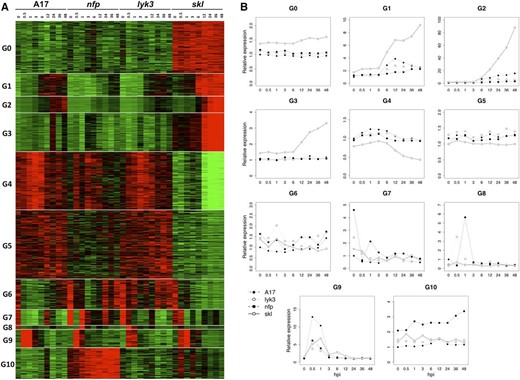
Identification of 11 coexpression groups. A, Heat map representing the expression of 10,985 differentially expressed genes identified in the RNA-seq experiment, where the average Trimmed Mean of M component (TMM) count values of a given gene across all samples was used as a normalization factor. The vertical axis organizes genes according to coexpression. The horizontal axis is a genotype-specific time course. Samples with gene TMM count values larger than the average TMM count for that gene are represented in red, while samples with lower counts than the average are represented in green. Whenever transcript values are close to the average value, samples are colored in black. B, Line graphs showing the average relative expression value of all genes in that group. TMM count values of any given gene were normalized by the count value of that particular gene at 0 hpi in cv Jemalong A17. A similar methodology was used to generate all expression figures in this work.
Rhizobial inoculation and host genotype both profoundly impacted transcriptional responses, as indicated by the distinct patterns of gene expression among the four genotypes in each group. Several groups of genes were most heavily influenced by ET perception (i.e. G0 and G4), Nod factor perception (i.e. G1–G3), or in response to bacterial inoculation independent of genes for Nod factor or ET perception (G9). Among these, the largest group of genes included those whose transcriptional induction required the Nod factor perception pathway and whose expression was hyperactivated in the ET-insensitive background of skl, namely, groups G1, G2, and G3. Based on their genetic dependencies, we designate this large set of G1 to G3 genes as Nod factor-induced, ET-repressed transcripts. Using this same logic, we note that a subset of genes in G4 also require the Nod factor pathway for induction, but for these, ET perception is required as a positive factor for gene induction (discussed further below). Thus, the vast majority of Nod factor-activated genes, including all genes in G1, G2, and G3 and a subset of genes in G4, are also strongly modulated by ET. The average expression of G1 and G2 genes in wild-type M. truncatula increased starting 3 to 6 hpi, and the magnitude of this increase was significantly amplified in the skl mutant. Mutations in NFP and LYK3 differentially impacted the expression of G1 and G2 genes, such that NFP was typically essential for gene induction, while transcriptional activation was evident in lyk3, but at significantly lower levels than observed in the wild type. Induction of G3 genes also required NFP and LYK3, although their up-regulation was significantly lower and later compared with most G1 and G2 genes, first evident at 24 hpi in the wild type (Fig. 1A) and at 12 hpi in skl.
Legume roots undergo profound physiological and developmental changes in response to rhizobium. We utilized Pathway Studio 9 and ResNet Plant 4 (Nikitin et al., 2003) to identify overrepresented groups of genes functioning in common signaling pathways and biological processes correlated with such changes (Supplemental Table S2). For each gene, the best BLASTn match in Arabidopsis (Arabidopsis thaliana) was identified and classified into the Gene Ontology (GO) categories of biological process and molecular function. A hypergeometric test was used to identify enriched GO terms within each cluster at P < 0.001. Overrepresented GO terms in G1 include biotic and abiotic stress response as well as polysaccharide and hormone biosynthesis, pollen tube growth, and receptor kinase activity. We emphasize that GO terms for biological process are best treated as analogies until proven otherwise; for example, pollen tube growth might or might not indicate Nod factor-induced changes to polar root hair growth, and response to stress does not necessarily indicate a defense reaction. G2 is enriched for TFs, kinases, and genes implicated in plant hormone biosynthesis and signaling, while G3 is enriched for genes implicated in cell division, DNA replication and repair, and transcription. Genes clustered in G0 and G4 possess expression profiles heavily influenced by ET. G0 genes exhibit constitutively high levels in the ET-insensitive mutant skl, while the average expression profile of G4 genes involves down-regulation in skl starting approximately 6 hpi (Fig. 2B). G0 is enriched for genes implicated in RNA processing as well as DNA synthesis and chromatin organization, while G4 contains numerous genes implicated in biotic and/or abiotic stress and cell wall organization. The average expression level of genes in group G5 is lower in skl relative to the other genotypes, but in contrast to G4, the differences in transcript abundance are relatively constant with time and are of lower magnitude. Interestingly, genes involved in plant defense are enriched in G5 as well as in G6 and G10, with the genes in G6 tending toward higher transcript abundance in nfp and lyk3 backgrounds compared with wild-type plants, while conversely, genes in G10 have higher expression in the nfp background.
Prior molecular and genetic studies identified genes that are rapidly induced following treatment with either rhizobium or isolated Nod factors. Several of these genes mediate host responses in a Nod factor-dependent manner, whereas others are common transcriptional markers reporting Nod factor perception. Notably, 30 of these classical symbiosis-related genes were differentially expressed in our data set, typically contained within G1, G2, and G3. Among these are the symbiosis markers Early Nodulin11 (ENOD11) and Rhizobium-induced peroxidase1 as well as the early TFs NSP2, Ethylene response factor required for Nodulation1 (ERN1), NF-YA1, and Rhizobium-directed polar growth (Fig. 3; Supplemental Table S3). While the intensity and kinetics of induction for these classical symbiotic genes varied in the wild type, their induction was consistently accentuated in skl and manifested a weak or absent response in one or both of the Nod factor receptor mutants, profiles consistent with their symbiotic designation. By analogy to the coexpressed classical symbiosis genes, we conclude that the other 2,356 genes in G1, G2, and G3 likely also participate in symbiotic processes.
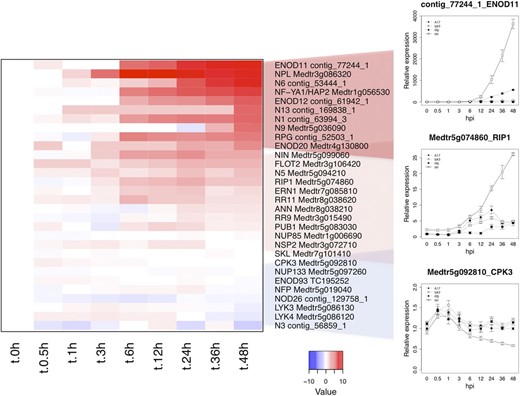
RNA-seq expression graphs of known symbiosis-related genes. The heat map represents relative log-transformed expression values (average TMM counts normalized to cv Jemalong A17 at 0 hpi) of genes previously shown to function in Nod factor signaling or to be required for nodule development. Values in the line graphs show average TMM counts normalized to cv Jemalong A17 at 0 hpi. Error bars represent se calculated from four independent biological replicates.
The massive induction of gene expression in skl may be a consequence of a deregulated response to Nod factors; alternatively, it may indicate a Nod factor-independent, ET-repressed response. If the accentuated transcriptional response in skl requires Nod factors, then mutations in the Nod factor signaling pathway should be epistatic to skl-dependent changes in gene expression. To test this hypothesis, we generated a homozygous dmi1-1 skl double mutant. dmi1-1 mutants undergo early ion fluxes and root hair swelling in response to Nod factors but lack root hair curling and rhizobial infection and fail to activate Nod factor-elicited calcium oscillations, ENOD expression, or cortical cell divisions and, consequently, do not nodulate (Catoira et al., 2000; Ané et al., 2004). We inoculated 5-d-old wild-type, dmi1-1, skl, and dmi1-1 skl double mutant seedlings with S. medicae ABS7M (pXLGD4). Transcription of the marker genes ENOD11, NIN, and ERN1 was analyzed by means of quantitative real-time PCR (qPCR) at intervals over 48 h (Supplemental Fig. S4). Consistent with RNA-seq data, we observed induction in response to rhizobium in the wild type, elevated expression in skl, and absent induction in the Nod factor signaling mutant dmi1-1. Induction was also absent in the dmi1-1 skl double mutant, indicating that ET-dependent negative regulation is dependent on Nod factor perception.
We also observed Nod factor pathway-independent effects of ET signaling. In particular, gene expression in G2 was elevated in the ET-insensitive background prior to Nod factor perception. The average normalized expression of the G2 genes in uninoculated nfp and lyk3 mutants (1.19 ± 0.04 and 1.1 ± 0.04, respectively) was comparable to that in wild-type cv Jemalong A17 (1). However, the average normalized value of G2 gene expression in uninoculated skl was nearly twice that of cv Jemalong A17 (1.85 ± 0.09). To account for the possibility of a normalization artifact, we quantified expression levels in skl compared with the wild type for seven G2 genes using qPCR, namely, ERN1, NIN, ENOD11, NF-YA1, 1-aminocyclopropane-1-carboxylic acid synthase4 (ACS4), ACS7, and ACS8 (see Fig. 7B; Supplemental Fig. S4; Laporte et al., 2014). Although the level of basal expression in the skl background varied depending on the gene, we observed that five of the seven genes differed significantly in their expression in skl relative to the wild type, with a 1.4- to 5.8-fold increase in transcript abundance. This suggests that ET may negatively regulate Nod factor-responsive genes in the absence of rhizobium and that the skl mutant may be predisposed to an accentuated symbiotic response by the increased baseline abundance of symbiotic signaling proteins.
Nod Factors Elicit a Massive Reprogramming of the Host Signaling Pathways within the First 24 h after Rhizobium Inoculation
Early symbiotic reprogramming is likely executed by TFs and kinases, which are abundant in groups G1, G2, and G3. Sixty-four of the genes from these groups contain conserved DNA-binding domains, suggesting that they contribute to the regulation of gene expression. Notably, the well-characterized symbiosis TFs NIN, ERN1, and NSP2 exhibit induction by 3 hpi and, thus, are among the earliest responding G2 genes (Fig. 4A). Their rapid up-regulation reflects their importance in effecting subsequent symbiotic gene expression and their absolute requirement for nodulation (Kaló et al., 2005; Andriankaja et al., 2007; Marsh et al., 2007). Groups G1, G2, and G3 contained an additional 20 TFs that are coinduced along with NIN, ERN1, and NSP2 by 3 hpi (Fig. 4A), suggesting that these uncharacterized genes also function to direct the earliest changes in gene expression.
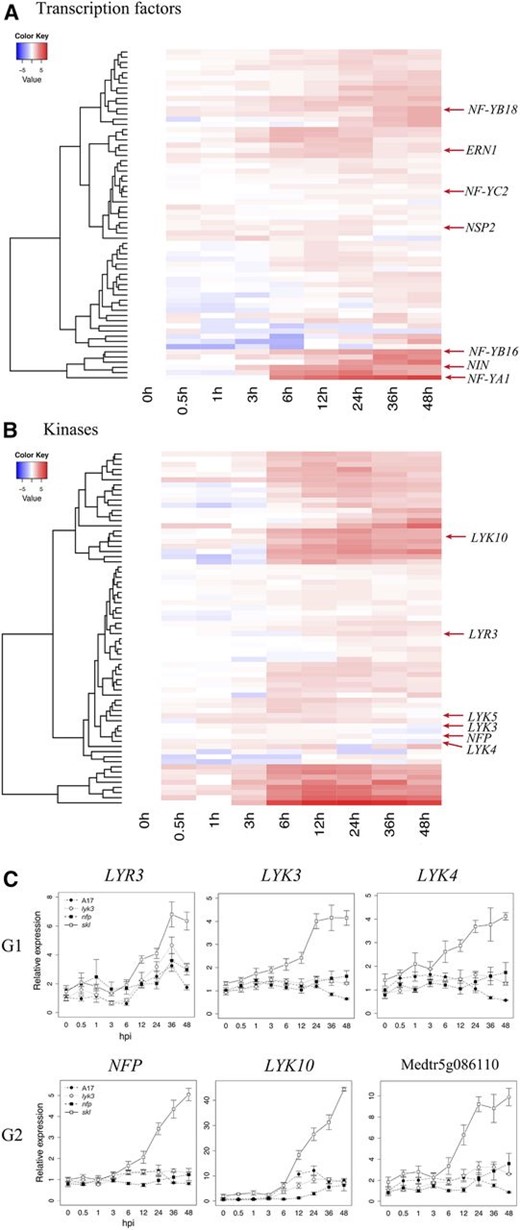
Early symbiotic induction of kinases and TFs in M. truncatula ‘Jemalong A17’ at various times after inoculation. A and B, The heat maps depict the expression patterns of differentially expressed TFs (A) and kinases (B) contained within expression groups G1 to G3 (Fig. 2). C, Differentially expressed members of the LysM family of plant receptors and receptor-like kinases. Heat-map values represent log-transformed relative expression values (average TMM counts normalized to cv Jemalong A17 at 0 hpi). Line graph values correspond to average TMM counts normalized to cv Jemalong A17 at 0 hpi. NFP corresponds to IMGA 3.5 version 4 gene Medtr5g019040; LYR3, Medtr5g019050; LYK10, Medtr5g033490; LYK3, Medtr5g086130; LYK4, Medtr5g086120; and Medtr5g086110. The vertical axis dendrogram organizes genes according to coexpression. The horizontal axis is a time course for wild-type cv Jemalong A17. Error bars represent se calculated from four independent biological replicates.
Members of the heterotrimeric, nucleosome-like NF-Y family of TFs are targets of the NIN protein (Soyano et al., 2013) and mediate early infection and nodule development processes (Petroni et al., 2012; Laloum et al., 2013, 2014). NF-YA subunits bind to conserved CCAAT motifs in promoters, where they recruit the histone 2A/2B-like NF-YC and NF-YB subunits (Nardini et al., 2013), which also bind DNA, albeit nonspecifically. NF-YA1 and NF-YA2 are paralogs that are essential for both infection and nodule meristematic persistence in M. truncatula (Combier et al., 2006; Laloum et al., 2014; Laporte et al., 2014), while silencing of NF-YC1 leads to the arrest of nodule development and infection in common bean (Phaseolus vulgaris; Zanetti et al., 2010). In addition to NF-YA1, we observed that two B-type NF-Ys (NF-YB16 and NF-YB18) exhibit a Nod factor-induced and ET-repressed transcriptional response by 6 hpi (Fig. 4A). A third, C-type subunit (NF-YC2) with high similarity to the common bean gene NF-YC1 was also differentially expressed. The concurrent expression of these subunits suggests that they may form a common NF-Y complex that controls early symbiotic responses.
Protein kinases play a key role in the transduction and amplification of diverse intracellular and extracellular signals, and our transcriptional analysis reveals a massive, early shift in the expression of diverse protein kinases in response to rhizobium inoculation. Ten percent of all G1, G2, and G3 genes induced upon inoculation are annotated as protein kinases, most of which are predicted transmembrane receptor kinases and RLKs. Sixty-nine genes contained a conserved kinase domain (National Center for Biotechnology Information [NCBI] conserved domains), and approximately half of these were up-regulated within 6 h of inoculation (Fig. 4B). In agreement with previous proteomic analyses (Rose et al., 2012), these data indicate a rapid shift in the phosphoproteome, particularly in membrane-associated proteins, suggesting a coordinated response to extracellular ligands or stimuli. Among the transcriptionally regulated protein kinases, several are known to be involved in Nod factor signaling, including the putative Nod factor receptors NFP and LYK3. NFP and LYK3 are members of the larger family of LysM receptor-like genes, and indeed, a total of six LysM receptors are differentially expressed at early stages in our data set. Three G2 genes, NFP, LYK10 (Medtr5g033490), and Medtr5g086110, exhibited hyperaccumulation in skl and higher expression in the wild type relative to nfp (Fig. 4C). Interestingly, Medtr5g086110 encodes a LysM receptor lacking a cytoplasmic kinase domain. The fact that the kinase domain is missing resembles the chitin-binding LysM receptor Chitin-Elicitor Binding Protein that interacts with the LysM receptor kinase Chitin-Elicitor Receptor Kinase1 to activate plant immunity against fungal pathogens (Kaku et al., 2006; Shimizu et al., 2010). However, Medtr5g086110 is more closely related to other LYK genes, particularly LYK5 (70.9% identity at the protein level), rather than LYM1 or LYM2 (20.4% and 18.8% identity, respectively). Two related LysM-RLKs, Lysine motif domain-containing receptor-like kinase3-related3 (LRY3; Medtr5g019050) and LYK4 (Medtr5g086120), occur in G1 along with LYK3. Thus, in addition to NFP and LYK3, several members of the LysM kinase family are induced in roots within hours after inoculation in a Nod factor-dependent, ET-regulated manner.
The structural and developmental similarities between lateral roots (LRs) and nodules underlie the hypothesis that nodule development may have evolved from the LR developmental pathway (Soyano and Hayashi, 2014). Consistent with this proposal, it has been shown that Nod factor and Myc factors can both induce LR development (Oláh et al., 2005; Maillet et al., 2011). Within the G1, G2, and G3 clusters, we identified 22 genes homologous to known Arabidopsis regulators of root and LR development (Table I). In Arabidopsis, many of these genes have been described as auxin responsive and are involved in initiating cell division and primordium formation or in regulating stem cell identity and meristem structure in the root tip. Our data reveal that, in M. truncatula, rhizobium induces LR gene expression in a Nod factor-dependent manner, with massive up-regulation in the skl mutant, indicating that these LR root genes are also under negative regulation by ET.
Identification of genes related to LR formation that are up-regulated in response to rhizobium inoculation
| Time Induced | Group | M. truncatula | Closest Ortholog in Arabidopsis | BLASTP | ||
|---|---|---|---|---|---|---|
| Identifier | Annotation | Identifier | Annotation | |||
| (hpi) | ||||||
| 6 | G1 | Medtr7g020980 | ET-responsive TF | AT5G07310 | ERF115a | 2 e−28 |
| 6 | G1 | Medtr1g019240 | TF SPATULA | AT4G36930 | SPTb | 6 e−21 |
| 6 | G2 | contig_8047_1 | LOB domain protein-like protein | AT2G42430 | ASL18/LBD16c | 8 e−42 |
| 6 | G2 | Medtr5g031880 | ET-responsive TF BABY BOOM | AT5G10510 | AIL6/PLETHORA3 d | 1 e−115 |
| 6 | G2 | Medtr1g056530 | NF-YA1 | AT5G06510 | NF-YA10e | 6 e−44 |
| 6 | G2 | contig_163390_1 | RLK | AT3G55950 | CCR3f | 0 |
| 12 | G1 | contig_166109_1 | WUSCHEL-related homeobox | AT4G35550 | WOX13g | 7 e−36 |
| 12 | G2 | Medtr5g081990 | WUSCHEL-related homeobox | AT3G11260 | WOX5h | 2 e−43 |
| 12 | G2 | Medtr4g119270 | ET-responsive TF | AT5G18560 | PUCHIi | 3 e−43 |
| 12 | G2 | contig_8266_1 | LOB domain protein | AT2G45420 | LBD18j | 2 e−39 |
| 12 | G3 | Medtr1g096030 | SCARECROW | AT3G54220 | SCRh | 3 e−49 |
| 12 | G2 | contig_56175_1 | RLK | AT1G28440 | HSL1k | 0 |
| 12 | G2 | Medtr2g098910 | Ser/Thr protein kinase-like ACR4 | AT3G59420 | ACR4f | 0 |
| 12 | G3 | Medtr4g061330 | Mitogen-activated protein kinase | AT1G07880 | MPK13l | 2 e−85 |
| 24 | G2 | Medtr1g086790 | WRKY TF | AT2G47260 | WRKY23m | 2 e−46 |
| 24 | G2 | Medtr5g089750 | Short internode-related sequence | AT5G12330 | LRP1n | 3 e−65 |
| 24 | G2 | contig_73963_1 | Basic Leucine Zipper Domain (BZIP)-like TF | AT3G51290 | APSR1o | 2 e−51 |
| 24 | G3 | contig_237895_1 | RLK | AT5G45780 | LRR-RLKp | 0 |
| 36 | G1 | Medtr3g014660 | Short internodes | AT5G12330 | LRP1n | 2 e−42 |
| 36 | G2 | Medtr5g021130 | Short internode related | AT5G66350 | SHIq | 3 e−51 |
| 36 | G1 | Medtr5g040420 | NAC (for no apical meristem [NAM], Arabidopsis transcription activation factor [ATAF1-2], and cup-shaped cotyledon [CUC2]) domain protein | AT1G26870 | FEZr | 5 e−90 |
| 48 | G1 | Medtr5g014720 | RLK | AT5G65710 | HSL2k | 0 |
| Time Induced | Group | M. truncatula | Closest Ortholog in Arabidopsis | BLASTP | ||
|---|---|---|---|---|---|---|
| Identifier | Annotation | Identifier | Annotation | |||
| (hpi) | ||||||
| 6 | G1 | Medtr7g020980 | ET-responsive TF | AT5G07310 | ERF115a | 2 e−28 |
| 6 | G1 | Medtr1g019240 | TF SPATULA | AT4G36930 | SPTb | 6 e−21 |
| 6 | G2 | contig_8047_1 | LOB domain protein-like protein | AT2G42430 | ASL18/LBD16c | 8 e−42 |
| 6 | G2 | Medtr5g031880 | ET-responsive TF BABY BOOM | AT5G10510 | AIL6/PLETHORA3 d | 1 e−115 |
| 6 | G2 | Medtr1g056530 | NF-YA1 | AT5G06510 | NF-YA10e | 6 e−44 |
| 6 | G2 | contig_163390_1 | RLK | AT3G55950 | CCR3f | 0 |
| 12 | G1 | contig_166109_1 | WUSCHEL-related homeobox | AT4G35550 | WOX13g | 7 e−36 |
| 12 | G2 | Medtr5g081990 | WUSCHEL-related homeobox | AT3G11260 | WOX5h | 2 e−43 |
| 12 | G2 | Medtr4g119270 | ET-responsive TF | AT5G18560 | PUCHIi | 3 e−43 |
| 12 | G2 | contig_8266_1 | LOB domain protein | AT2G45420 | LBD18j | 2 e−39 |
| 12 | G3 | Medtr1g096030 | SCARECROW | AT3G54220 | SCRh | 3 e−49 |
| 12 | G2 | contig_56175_1 | RLK | AT1G28440 | HSL1k | 0 |
| 12 | G2 | Medtr2g098910 | Ser/Thr protein kinase-like ACR4 | AT3G59420 | ACR4f | 0 |
| 12 | G3 | Medtr4g061330 | Mitogen-activated protein kinase | AT1G07880 | MPK13l | 2 e−85 |
| 24 | G2 | Medtr1g086790 | WRKY TF | AT2G47260 | WRKY23m | 2 e−46 |
| 24 | G2 | Medtr5g089750 | Short internode-related sequence | AT5G12330 | LRP1n | 3 e−65 |
| 24 | G2 | contig_73963_1 | Basic Leucine Zipper Domain (BZIP)-like TF | AT3G51290 | APSR1o | 2 e−51 |
| 24 | G3 | contig_237895_1 | RLK | AT5G45780 | LRR-RLKp | 0 |
| 36 | G1 | Medtr3g014660 | Short internodes | AT5G12330 | LRP1n | 2 e−42 |
| 36 | G2 | Medtr5g021130 | Short internode related | AT5G66350 | SHIq | 3 e−51 |
| 36 | G1 | Medtr5g040420 | NAC (for no apical meristem [NAM], Arabidopsis transcription activation factor [ATAF1-2], and cup-shaped cotyledon [CUC2]) domain protein | AT1G26870 | FEZr | 5 e−90 |
| 48 | G1 | Medtr5g014720 | RLK | AT5G65710 | HSL2k | 0 |
| Time Induced | Group | M. truncatula | Closest Ortholog in Arabidopsis | BLASTP | ||
|---|---|---|---|---|---|---|
| Identifier | Annotation | Identifier | Annotation | |||
| (hpi) | ||||||
| 6 | G1 | Medtr7g020980 | ET-responsive TF | AT5G07310 | ERF115a | 2 e−28 |
| 6 | G1 | Medtr1g019240 | TF SPATULA | AT4G36930 | SPTb | 6 e−21 |
| 6 | G2 | contig_8047_1 | LOB domain protein-like protein | AT2G42430 | ASL18/LBD16c | 8 e−42 |
| 6 | G2 | Medtr5g031880 | ET-responsive TF BABY BOOM | AT5G10510 | AIL6/PLETHORA3 d | 1 e−115 |
| 6 | G2 | Medtr1g056530 | NF-YA1 | AT5G06510 | NF-YA10e | 6 e−44 |
| 6 | G2 | contig_163390_1 | RLK | AT3G55950 | CCR3f | 0 |
| 12 | G1 | contig_166109_1 | WUSCHEL-related homeobox | AT4G35550 | WOX13g | 7 e−36 |
| 12 | G2 | Medtr5g081990 | WUSCHEL-related homeobox | AT3G11260 | WOX5h | 2 e−43 |
| 12 | G2 | Medtr4g119270 | ET-responsive TF | AT5G18560 | PUCHIi | 3 e−43 |
| 12 | G2 | contig_8266_1 | LOB domain protein | AT2G45420 | LBD18j | 2 e−39 |
| 12 | G3 | Medtr1g096030 | SCARECROW | AT3G54220 | SCRh | 3 e−49 |
| 12 | G2 | contig_56175_1 | RLK | AT1G28440 | HSL1k | 0 |
| 12 | G2 | Medtr2g098910 | Ser/Thr protein kinase-like ACR4 | AT3G59420 | ACR4f | 0 |
| 12 | G3 | Medtr4g061330 | Mitogen-activated protein kinase | AT1G07880 | MPK13l | 2 e−85 |
| 24 | G2 | Medtr1g086790 | WRKY TF | AT2G47260 | WRKY23m | 2 e−46 |
| 24 | G2 | Medtr5g089750 | Short internode-related sequence | AT5G12330 | LRP1n | 3 e−65 |
| 24 | G2 | contig_73963_1 | Basic Leucine Zipper Domain (BZIP)-like TF | AT3G51290 | APSR1o | 2 e−51 |
| 24 | G3 | contig_237895_1 | RLK | AT5G45780 | LRR-RLKp | 0 |
| 36 | G1 | Medtr3g014660 | Short internodes | AT5G12330 | LRP1n | 2 e−42 |
| 36 | G2 | Medtr5g021130 | Short internode related | AT5G66350 | SHIq | 3 e−51 |
| 36 | G1 | Medtr5g040420 | NAC (for no apical meristem [NAM], Arabidopsis transcription activation factor [ATAF1-2], and cup-shaped cotyledon [CUC2]) domain protein | AT1G26870 | FEZr | 5 e−90 |
| 48 | G1 | Medtr5g014720 | RLK | AT5G65710 | HSL2k | 0 |
| Time Induced | Group | M. truncatula | Closest Ortholog in Arabidopsis | BLASTP | ||
|---|---|---|---|---|---|---|
| Identifier | Annotation | Identifier | Annotation | |||
| (hpi) | ||||||
| 6 | G1 | Medtr7g020980 | ET-responsive TF | AT5G07310 | ERF115a | 2 e−28 |
| 6 | G1 | Medtr1g019240 | TF SPATULA | AT4G36930 | SPTb | 6 e−21 |
| 6 | G2 | contig_8047_1 | LOB domain protein-like protein | AT2G42430 | ASL18/LBD16c | 8 e−42 |
| 6 | G2 | Medtr5g031880 | ET-responsive TF BABY BOOM | AT5G10510 | AIL6/PLETHORA3 d | 1 e−115 |
| 6 | G2 | Medtr1g056530 | NF-YA1 | AT5G06510 | NF-YA10e | 6 e−44 |
| 6 | G2 | contig_163390_1 | RLK | AT3G55950 | CCR3f | 0 |
| 12 | G1 | contig_166109_1 | WUSCHEL-related homeobox | AT4G35550 | WOX13g | 7 e−36 |
| 12 | G2 | Medtr5g081990 | WUSCHEL-related homeobox | AT3G11260 | WOX5h | 2 e−43 |
| 12 | G2 | Medtr4g119270 | ET-responsive TF | AT5G18560 | PUCHIi | 3 e−43 |
| 12 | G2 | contig_8266_1 | LOB domain protein | AT2G45420 | LBD18j | 2 e−39 |
| 12 | G3 | Medtr1g096030 | SCARECROW | AT3G54220 | SCRh | 3 e−49 |
| 12 | G2 | contig_56175_1 | RLK | AT1G28440 | HSL1k | 0 |
| 12 | G2 | Medtr2g098910 | Ser/Thr protein kinase-like ACR4 | AT3G59420 | ACR4f | 0 |
| 12 | G3 | Medtr4g061330 | Mitogen-activated protein kinase | AT1G07880 | MPK13l | 2 e−85 |
| 24 | G2 | Medtr1g086790 | WRKY TF | AT2G47260 | WRKY23m | 2 e−46 |
| 24 | G2 | Medtr5g089750 | Short internode-related sequence | AT5G12330 | LRP1n | 3 e−65 |
| 24 | G2 | contig_73963_1 | Basic Leucine Zipper Domain (BZIP)-like TF | AT3G51290 | APSR1o | 2 e−51 |
| 24 | G3 | contig_237895_1 | RLK | AT5G45780 | LRR-RLKp | 0 |
| 36 | G1 | Medtr3g014660 | Short internodes | AT5G12330 | LRP1n | 2 e−42 |
| 36 | G2 | Medtr5g021130 | Short internode related | AT5G66350 | SHIq | 3 e−51 |
| 36 | G1 | Medtr5g040420 | NAC (for no apical meristem [NAM], Arabidopsis transcription activation factor [ATAF1-2], and cup-shaped cotyledon [CUC2]) domain protein | AT1G26870 | FEZr | 5 e−90 |
| 48 | G1 | Medtr5g014720 | RLK | AT5G65710 | HSL2k | 0 |
Nod factors stimulate the formation of LRs (Oláh et al., 2005); thus, the increased expression of LR developmental genes could simply reflect additional LR initiation. Alternatively, the genes regulating LR primordium initiation may perform analogous functions required for the initiation of nodule development. To determine if the increased expression of LR-associated genes was correlated with increased nodulation or increased LR initiation, we quantified the number of LRs on nodulated wild-type cv Jemalong A17 and skl roots. Although nodulated skl roots were significantly shorter than those of cv Jemalong A17, even when corrected for root length the number of LRs in the skl mutant was significantly lower than in cv Jemalong A17 (1.26 ± 0.17 LR cm−1 for cv Jemalong A17 versus 0.76 ± 0.14 LR cm−1 for skl; P < 0.001, Student’s t test). These data suggest that the deregulated expression of LR genes in skl is associated with the initiation of nodules (Penmetsa and Cook, 1997; Penmetsa et al., 2008) and not with LRs, consistent with cross talk between the Nod factor and ET signaling pathways and the LR developmental pathway.
Early Induction of Genes Involved in Polar Root Hair Growth
With the goal of correlating transcriptional with morphological changes, we grouped genes within G1 to G3 based on the timing of initial induction relative to uninoculated wild-type samples. The outcome, shown in Figure 5, is six temporally resolved sets of genes.
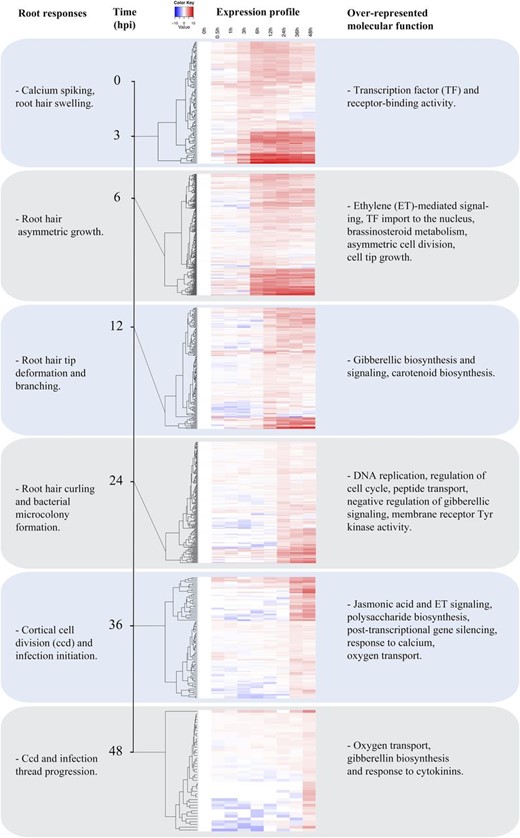
Temporal dynamics of gene expression within groups G1 to G3. To further resolve gene expression patterns within the Nod factor-responsive, ET-regulated groups (Fig. 2), we organized genes based on the kinetics of their induction (1.5-fold threshold) relative to uninoculated cv Jemalong A17. Individual heat maps represent genes induced at 3, 6, 12, 24, 36, and 48 h. In each part, the vertical axis dendrogram organizes genes according to coexpression. The horizontal axis is a time course for wild-type cv Jemalong A17. Overrepresented gene functions and biological process annotations were identified in each group using Pathway Studio 9 Desktop edition and ResNet Plant version 4.
We observed root hair deformation and branching between 3 and 6 hpi (Fig. 1, D and E). Root hair deformation results from cytoskeletal reorganization and cell wall deposition that redirects polar tip growth. Two genes up-regulated in the 3- to 6-h interval are homologs of Arabidopsis Formin-Like8 (AtFH8) and Abscisic Acid Insensitive-Like (ABIL), genes directly linked to actin nucleation and severing (Yi et al., 2005). ABIL functions in the SCAR/WAVE complex that regulates the actin cytoskeleton in tip-growing cells and during infection thread progression in legumes (Yokota et al., 2009; Miyahara et al., 2010; Hossain et al., 2012) and was recently observed to be up-regulated in root hairs undergoing infection (Breakspear et al., 2014). By contrast, AtFH8 is a formin, and a role for formins in symbiotic infection has not yet been reported. Formins regulate actin nucleation and bundling and have been associated with cell division and root hair initiation and growth (Yi et al., 2005; Xue et al., 2011). FH8 overexpression in Arabidopsis causes root hair swelling and branching similar to Nod factors in legumes. Root hair curling and infection are also associated with changes in the microtubule cytoskeleton, and β-tubulins, which are cytoskeletal proteins, have been shown to be up-regulated in root hairs (Timmers, 2008; Breakspear et al., 2014; Perrine-Walker et al., 2014). We observed that two microtubule-associated proteins, MAP65 (Medtr6g061690) and MAP70 (Medtr4g133890, contig_83417_1), were up-regulated 6 hpi in a Nod factor-dependent, ET-regulated fashion. MAP65 and MAP70 are microtubule-bundling proteins thought to cross-link and stabilize microtubules (Pesquet et al., 2010; Ho et al., 2012). MAP65 is generally thought to stabilize the central spindle during mitosis (Smertenko et al., 2008), but its role in root hair growth has not been explored yet.
Other genes up-regulated 3 to 6 hpi participate in cell wall loosening or breakdown (Fig. 5). Among these genes are several pectolytic enzymes including the M. truncatula ortholog of LjNPL, a pectate lyase that promotes root hair infection by degrading the cell wall (Xie et al., 2012). Notably, MtNPL was induced over 1,000-fold by 6 hpi, well before the onset of root hair curling (12–24 hpi) or infection thread growth (36 hpi; Fig. 1). Similarly, two expansins were up-regulated 6 hpi. Expansins are linked to root hair initiation in Arabidopsis and barley (Hordeum vulgare) and may be related to the reinitiation of polar growth during root hair branching (Cho and Cosgrove, 2002; Kwasniewski and Szarejko, 2006). Additionally, several β-glucanases were induced by 6 hpi. These enzymes are involved in the breakdown of cellulose in the cell wall and have been associated with cell wall loosening, cell growth, and lateral root development (Lewis et al., 2013). Taken together, these data suggest that cell wall degradation begins well in advance of infection and may also mediate the root hair swelling and branching observed during early time points. A second cohort of cell wall-modifying enzymes is up-regulated later, approximately 12 to 24 hpi, when root hair curling and cell division begin. Notably, these latter genes also include several cellulose biosynthetic genes, suggesting that the cell wall modifications include both anabolic and catabolic activities.
ET Regulates Multiple Hormone Biosynthetic Pathways
We observed the Nod factor-dependent, ET-regulated induction of hormone biosynthesis genes within hours of inoculation. This was accompanied by the concurrent expression of negative regulators of hormone signaling. These data suggest that, in regulating Nod factor signaling, ET plays an important role in affecting the biosynthesis and activity of other plant hormones and that the activity of these hormones must be carefully balanced to coordinate the range of hormone-dependent process.
The 2-C-methyl-d-erythritol 4-phosphate (MEP) pathway generates isopentenyl pyrophosphate (IPP) and dimethylallyl pyrophosphate (DMAPP) in plastids. These molecules function as precursors for isoprenoid compounds, including the hormones cytokinin (CK), GA, abscisic acid (ABA), and strigolactones. While all genes in the MEP pathway are expressed in the absence of rhizobium, enzymes carrying out five of the seven biochemical steps exhibited a Nod factor-dependent, ET-regulated transcriptional induction that placed them in group G1 or G2 (Fig. 6). The expression of these genes began to increase between 6 and 12 hpi, suggesting that Nod factor perception stimulates the rapid biosynthesis of DMAPP and IPP, thus providing the precursors to plant hormones and other isoprenoids.
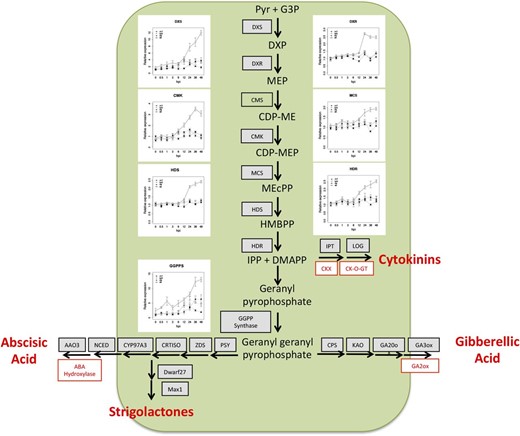
Nod factor perception initiates the biosynthesis of hormone precursors IPP and DMAPP via the MEP/1-deoxy-d-xylulose 5-phosphate biosynthetic pathway. Line graphs depicting read counts of differentially expressed MEP genes are shown. Differentially expressed genes leading to the biosynthesis of GA, ABA, and strigolactones are listed in gray, and inhibitors are depicted in red. Full depictions of the individual hormone pathways are given in Supplemental Figure S5. Error bars represent se calculated from four independent biological replicates. Abbreviations used are as follows: Pyr, pyruvate; G3P, glyceraldehyde 3-phosphate; DXS, DXP synthase; DXP, 1-deoxy-d-xylulose 5-phosphate; DXR, DXP reductase; CMS, 2-C-methyl-d-erythritol 4-phosphate cytidylyltransferase; CDP-ME, 4-diphosphocytidyl-2-C-methylerythritol; CMK, 4-diphosphocytidyl-2-C-methyl-d-erythritol kinase; CDP-MEP, 4-diphosphocytidyl-2-C-methyl-d-erythritol 2-phosphate; MCS, 2-C-methyl-d-erythritol 2,4-cyclodiphosphate synthase; MEcPP, 2-C-methyl-d-erythritol 2,4-cyclopyrophosphate; HDS, HMBPP synthase; HMBPP, (E)-4-hydroxy-3-methyl-but-2-enyl pyrophosphate; HDR, HMBPP reductase; CKX, cytokinin oxidase; CK-O-GT, cytokinin O-glucosyltransferase; PSY, phytoene synthase; ZDS, ζ-carotene desaturase; CRTISO, carotene or carotenoid isomerase; CYP, cytochrome P450, NCED, 9-cis-epoxycarotenoid dioxygenase; AAO, abscisic aldehyde oxidase; CPS, ent-copalyl diphosphate synthase; KAO, ent-kaurene acid oxidase.
CKs regulate the cell cycle in plants (Hartig and Beck, 2005). In legumes CKs function to initiate nodule organogenesis (Gonzalez-Rizzo et al., 2006; Murray et al., 2007; Tirichine et al., 2007; Op den Camp et al., 2011; Plet et al., 2011) and autoregulation of nodulation (Sasaki et al., 2014). CKs are formed when an isopetenyl moiety is conjugated to adenine nucleotides via ISOPENTENYLTRANSFERASES (IPTs). LONELY GUY (LOG) cleaves the ribose moiety from CK nucleotides to form bioactive isopentyl adenine. Hydroxylation of the nucleotide by CYP735A prior to cleavage creates the trans-zeatin form. M. truncatula homologs of IPT, CYP735A, and LOG are all up-regulated in an nfp-dependent, but lyk3-independent, manner approximately 12 hpi (Fig. 6; Supplemental Fig. S5). This suggests rapid de novo CK biosynthesis following Nod factor perception and highlights the occasional distinctiveness of NFP- and LYK3-dependent transcriptional responses. Interestingly, CK oxidase (contfig_240378_1) and CK glucosyl transferase (Medtr5g064240), CK catabolic and inactivating enzymes, respectively, were up-regulated prior to the CK biosynthetic genes. This suggests concurrent activation and attenuation of the CK pathway and may indicate that CK signaling is carefully controlled in a spatiotemporal manner. We also observed a putative equilibrative nucleoside transporter, designated ENT (contig_62928_1) expressed in an nfp- and lyk3-dependent manner beginning 3 to 6 hpi and robustly expressed in skl 12 to 48 hpi. Recent studies in Arabidopsis suggest that ENT transporters mobilize CKs from vascular tissue (Hirose et al., 2008). ENT expression following inoculation may indicate that CK mobilization complements biosynthesis to achieve the activation of CK signaling.
IPP and DMAPP derived from the MEP/1-deoxy-d-xylulose 5-phosphate pathway can be converted to geranyl geranyl pyrophosphate (GGPP) and subsequently to GA, ABA, and strigolactones. GGPP synthase is up-regulated 6 to 12 hpi, as are several members of the GA, ABA, and strigolactone biosynthetic pathways. GA suppresses NIN and NSP gene expression in L. japonicus and attenuates symbiotic infection in L. japonicus and Sesbania rostrata (Lievens et al., 2005; Maekawa et al., 2009). Our thorough sampling of early symbiotic time points reveals that the Nod factor pathway differentially activates genes involved in GA biosynthesis and catabolism and that GA biosynthetic pathways are ET regulated. GA 2-oxidase (GA2ox), a GA-inactivating enzyme, is transcriptionally up-regulated 3 hpi, peaks at 12 hpi, and slowly declines over the remainder of our time course (Fig. 6; Supplemental Fig. S5). Subsequently, the GA anabolic genes are up-regulated in a Nod factor-dependent, ET-regulated manner between 12 and 24 hpi. Thus, it appears that bioactive forms of GA are initially catabolized, which may allow Nod factor signaling to proceed, whereas GA levels increase at later time points, negatively regulating the symbiotic pathway and perhaps serving as a mechanism to regulate infection. Alternatively, these differences in transcript accumulation may reflect tissue-specific differences in GA signaling (i.e. root hair versus cortical cells).
In agreement with Breakspear et al. (2014), we observed Nod factor-induced expression of the strigolactone biosynthetic genes DWARF27 (contig_16099_1) and MAX1 (Medtr3g104560). Notably, the SL biosynthetic pathway is partially redundant with the pathway leading to ABA biosynthesis, suggesting that ABA biosynthesis is also up-regulated (Supplemental Fig. S5). Indeed, transcripts for the enzymes catalyzing six of the biosynthetic steps leading from GGPP to ABA, including 9-cis-epoxycarotenoid dioxygenase, which carries out the rate-limiting step in ABA biosynthesis, are up-regulated in a Nod factor-dependent, ET-regulated manner (Fig. 6; Supplemental Fig. S5). ABA suppresses calcium spiking, nodulin expression, and bacterial infection and nodule development (Ding et al., 2008). In addition to ABA biosynthesis, Nod factors also stimulated the transcription of ABA hydroxylase, a negative regulator of ABA signaling. This suggests that the biosynthesis and degradation of ABA must be balanced in a spatial or temporal manner to achieve the appropriate activation of the symbiotic pathway.
Auxin and jasmonic acid (JA) promote and inhibit nodulation, respectively (Sun et al., 2006; Grunewald et al., 2009). In indeterminate nodulating legumes, auxin accumulation occurs in dividing cortical cells that constitute the nascent nodule meristem (van Noorden et al., 2007). Auxin accumulation during nodulation has been associated with changes in polar auxin transport (Wasson et al., 2006), while the possibility of de novo auxin biosynthesis by the host has not been thoroughly explored. We observed Nod factor-dependent induction of an anthranilate synthase-α gene (ASA; Medtr3g070290) and several YUCCA dimethylaniline monooxygenases (contig_57674_1, Medtr7g099330, Medtr7g099160, and Medtr3g109520). ASA genes encode the α-subunit of anthranilate synthase, which catalyzes the rate-limiting step of Trp synthesis, a precursor of indole-3-acetic acid, while YUCCAs carry out the rate-limiting step in auxin biosynthesis (Zhao et al., 2001; Mashiguchi et al., 2011). Thus, in addition to altering auxin transport, Nod factor perception may stimulate auxin biosynthesis and, consequently, cell division. Two JA biosynthetic genes encoding 12-oxophytodienoate reductases (Medtr5g006730 and Medtr5g006740) were also induced 3 to 6 hpi, suggesting that JA biosynthesis is induced by Nod factors.
These results indicate that there is a symbiosis-specific transcriptional activation of multiple hormone biosynthetic and response pathways taking place within hours of inoculation with rhizobium. The facts that most of these genes are highly up-regulated in the skl mutant, and that ET biosynthetic genes are induced by 6 hpi in a Nod factor-dependent manner (see below), indicate a hierarchy of hormone perception pathways, with ET perception serving as the master negative regulator.
Rhizobium Inoculation Triggers the Transcription of ET Biosynthesis Genes, Suggesting Feedback Regulation of the Symbiotic Pathway
Although ET is a well-known negative regulator of nodulation, the ET biosynthesis and signaling pathways have not been yet characterized in detail in legumes. In order to gain insight into how ET biosynthesis is transcriptionally regulated during the legume-rhizobium symbiosis, we first identified the genes responsible for ET biosynthesis and signaling in the M. truncatula genome and analyzed their expression profiles in our RNA-seq data set.
The first committed step in ET biosynthesis is the conversion of S-adenosyl Met to 1-aminocyclopropane-1-carboxylic acid (ACC) by the enzyme ACC synthase (ACS). This is the rate-limiting step for ET production, and increased ACS protein correlates with increased ET production (Yang and Hoffman, 1984; Kende, 1993). We identified nine ACS genes in the M. truncatula genome, three of which (i.e. ACS4 [contig_12436_1], ACS7 [Medtr8g101820], and ACS8 [Medtr5g015020]) exhibit a typical symbiotic response: induced by 6 hpi in cv Jemalong A17 and hyperinduced in skl (Fig. 7A). Moreover, the hyperinduction of all three ACS genes in the skl background requires the symbiotic signaling pathway, as confirmed by qPCR analysis of gene expression in dmi1 skl double mutants (Fig. 7B).
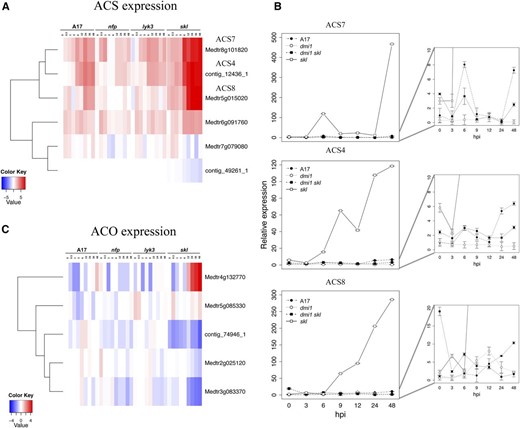
ET biosynthesis is rapidly induced by Nod factors and negatively regulated by ET in a feedback loop. A, Differential expression of several ACS genes in the RNA-seq data set. B, Specificity of symbiotic induction observed in roots for ACS4, ACS7, and ACS8 was verified in the double mutant dmi1 skl using qPCR. Detailed expression of A17, dmi1, and dmi1 skl is shown on the right. C, Expression of ACO in the RNA-seq data set. Heat maps represent relative expression values (average TMM counts normalized to cv Jemalong A17 at 0 hpi) in the four genotypes across time. In A and C, the vertical axis dendrogram organizes genes according to coexpression. The horizontal axis is a genotype-specific time course.
The final step in ET biosynthesis is the conversion of ACC to ET by ACC oxidase (ACO; Wang et al., 2002). Although five M. truncatula ACO genes were differentially expressed, only Medtr4g132770 exhibited a pattern consistent with strong feedback inhibition by ET (Fig. 7C). By contrast, the three ACO genes with the highest expression values in wild-type cv Jemalong A17 (Medtr2g025120, contig_74946_1, and Medtr3g083370) require an active ET signaling cascade for their induction.
Transcriptional fusions between the ACS4, ACS7, and ACS8 promoters and a GUS reporter gene revealed nearly identical expression patterns for all three promoters (Fig. 8). Both in noninoculated roots and 2 dpi, promoters were active in the epidermis, root hairs, and root primordia, particularly in young LRs (Fig. 8, A–I). We also observed ACS promoter activity in the periphery and meristem of young nodules (Fig. 8, J–L) and in the meristems of mature nodules (Fig. 8, M–O). Interestingly, ACS expression was observed in single root hair cells (Fig. 9) containing arrested rhizobial infections, in agreement with the observation that ET genetically regulates infection thread persistence and total nodule number (Penmetsa and Cook, 1997; Heidstra et al., 1997).
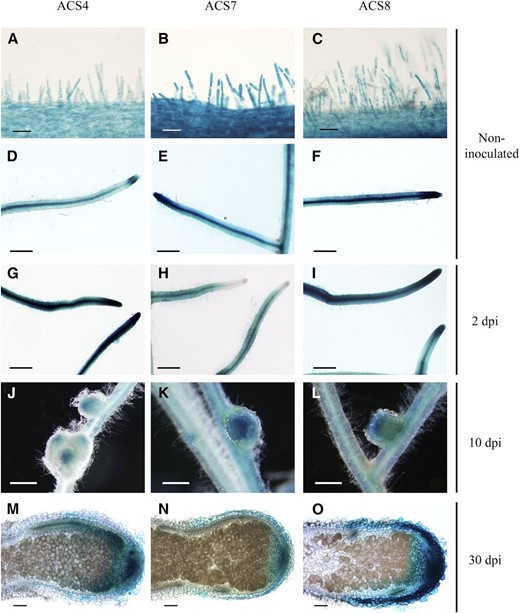
Histochemical localization of the expression of the three symbiosis-responsive ACS genes in M. truncatula roots at different stages after inoculation. The promoter of three ACS genes was cloned in fusion with the GUS reporter gene in the pLP100 binary vector. M. truncatula roots were transformed by hairy root transformation. The expression of gene ACS4 is represented in A to M, ACS7 in B to N, and ACS8 in C to O. A to F, Bright-field images of 5-bromo-4-chloro-3-indolyl-β-glucuronic acid (X-Gluc)-stained roots before inoculation. G to I, Roots at 24 hpi. J to L, Roots at 10 dpi. M to O, Roots at 30 dpi. Bars = 25 µm (A–C), 500 µm (D–L), and 100 µm (M–O).
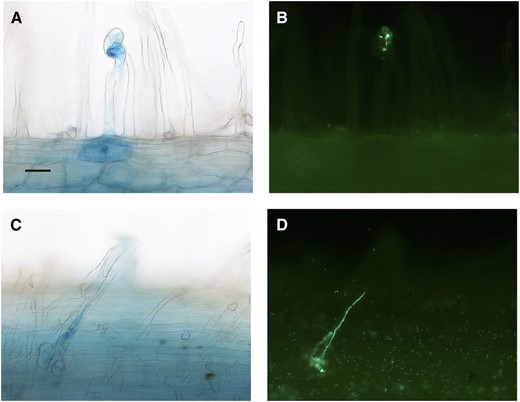
Localized ACS expression in root hairs undergoing infection arrest. Bright-field images of root hairs showing increased ProACS8:GUS expression (A and C) correlate with putative arrested infection threads of GFP-labeled S. meliloti at 10 d post inoculation (B and D). Bar = 10 µm.
Taken together, these results suggest that Nod factor perception promotes ET production in the nodulation-competent zone of roots within hours after inoculation with rhizobium and that the role of ET extends to later stages of nodule development.
Identification of ET Signaling Genes Induced by Rhizobium
ET mediates diverse plant responses by activating a cascade of TFs that effect ET responses and negatively feedback regulate the ET biosynthetic and signaling pathways. The ET signaling pathway is subject to complex negative regulation that represses signaling in the absence of ET. Briefly, the perception of ET inactivates the receptor(s) leading to pathway derepression and activation of the positive regulator EIN2 (Ju et al., 2012). Perception of ET also stabilizes key TFs (namely EIN3 and EIN3-like TFs; Chao et al., 1997; Qiao et al., 2012) by repressing two EIN3-targeting F-box proteins (Guo and Ecker, 2003; An et al., 2010), leading to an ET-induced transcriptional cascade.
Although ET signaling is well characterized in Ara-bidopsis, the pathway is sparsely studied in legumes. We queried the M. truncatula genome to identify the orthologs of ET signaling genes. We identified four ET receptors (ETRs) in IMGA 3.5 version 4, based on the presence of a conserved signal receiver domain in the predicted proteins (cl09944; NCBI conserved domains database) and the high similarity with the Arabidopsis ETR family (data not shown). Interestingly, all four M. truncatula ETRs occur in G4 and require an intact ET signaling pathway for their expression (as exemplified for contig_103103_1; Fig. 10A). ETR expression increased transiently within the first 1 hpi, independent of the Nod factor pathway, and was reinduced in an nfp- and skl-dependent manner by 6 hpi (Fig. 10A).
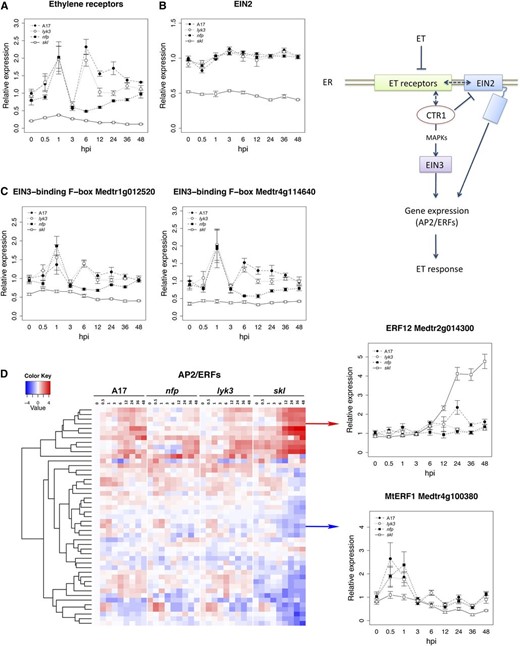
Transcriptional regulation of ET signaling pathway components during early stages of symbiotic development. A, Representative expression pattern of ETRs (contig_103103_1 is represented). B, Expression profile of the gene EIN2, whose mutation is responsible for the skl phenotype. C, Expression of two genes encoding putative EIN3-binding F-box proteins. D, Heat-map representation of differentially expressed members of the AP2/ERF family. Line graphs represent relative expression values (TMM count values relative to uninoculated cv Jemalong A17). The heat map depicts relative expression values after a log2 transformation. Error bars in the line graphs represent se calculated from four independent biological replicates.
EIN3 encodes the primary ET-responsive TF that regulates the expression of downstream AP2/ERFs in Arabidopsis (Chao et al., 1997). M. truncatula EIN3 and EIN3-like orthologs did not manifest a symbiosis-related pattern of gene expression (data not shown). Likewise, MtEIN2 expression was not impacted by Nod factor perception, and transcript was uniformly reduced in skl (Fig. 10B). skl is a loss-of-function EIN2 mutant, and the reduced MtEIN2 transcript in skl may reflect nonsense-mediated mRNA decay (Penmetsa et al., 2008). Additionally, we identified four putative EIN3-binding F-box genes with approximately 50% identity to Arabidopsis ETHYLENE INSENSITIVE3 binding F-Box protein1 (EBF1) and EBF2. Among these, Medtr1g012520 and Medtr4g114640 exhibit expression patterns similar to those of the ETRs, with Nod factor-independent induction 1 hpi, a transient decline 3 hpi, and an nfp-dependent increase 6 hpi (Fig. 10C). Similar to the ETRs, the induction of Medtr1g012520 and Medtr4g114640 required an active ET signaling pathway.
Given the rapid activation of ET biosynthesis and signaling pathways following inoculation, we examined the impact of Nod factor perception on AP2/ERF expression, TFs that are directly responsible for ET-responsive gene expression (Solano et al., 1998). We first queried the M. truncatula genome for genes containing a conserved AP2 domain and identified 216 genes. Seventy-seven of these were differentially expressed in our data set (Supplemental Table S4). Among the 47 AP2/ERFs with the largest induction, we observed two primary patterns of expression (Fig. 10D), which notably coincide with the two waves of activation of ET signaling described above: (1) genes up-regulated 0.5 to 3 hpi, dependent on ET and independent of Nod factor; and (2) genes up-regulated 6 hpi, negatively regulated by ET and variously dependent on Nod factor (top branch in the cladogram). The first set of AP2/ERF genes occurs in G4, a group of genes requiring ET for induction and enriched for plant defense genes. Members of this cluster include the ortholog (contig_70694_1) of AtERF1, a regulator of defense responses in Arabidopsis, MtERF1-1 (Medtr4g100380; Anderson et al., 2010), as well as several other AP2/ERFs that contain a conserved EDLL-like domain. EDLL is a transcriptional activation motif characteristic of pathogen-responsive AP2/ERFs in phylogenetic group IX (Nakano et al., 2006; Tiwari et al., 2012). The fact that these early (0.5–1 hpi) genes exhibit Nod factor-independent transcriptional activation that requires a functional ET signaling pathway is consistent with transient activation of host defenses by ET in response to bacterium. The second set of AP2/ERFs occurs in G1 to G3 and, based on analogy to other genes in these groups, are likely to regulate early symbiotic responses. For example, ERN1 (Medtr7g085810), an AP2/ERF that is required for nodulation (Andriankaja et al., 2007; Middleton et al., 2007; Cerri et al., 2012), is included in this group.
ERF12 is an AP2/ERF TF with expression characteristics (i.e. timing and genetic dependencies) typical of symbiosis genes. Transcriptional fusion of the ERF12 (Medtr2g014300; Fig. 10D) promoter to the GUS reporter gene was used to examine spatial patterns of expression in transformed hairy roots of wild-type cv Jemalong A17 plants. In noninoculated roots, ERF12 expression was confined to the zone of cell division close to the root cap (Fig. 11A) and to vascular tissue and inner cortical layers of the root (Fig. 11B). By 48 hpi, ERF12 expression was detected in the epidermal layer as well as in root hairs (Fig. 11, C and D). ERF12 expression was not correlated with infection thread growth, although ERF12 expression was detected at the nodule tip and in peripheral tissue throughout nodule development (Fig. 11, E and F), thus partially overlapping with the expression of the symbiotically induced ACS genes (Fig. 8, J–O).
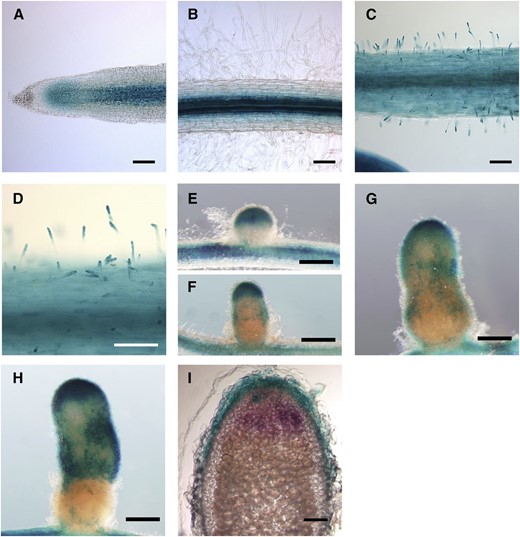
Histochemical localization of the expression of ERF12 in M. truncatula roots and nodules. A to H, M. truncatula roots transformed using ProERF12:GUS constructs and stained using X-Gluc. A and B, Uninoculated roots. C and D, Roots at 2 dpi. E to H, Nodules at different stages of development. I, Longitudinal 100-µm section of nodules at 2 to 3 weeks post inoculation. In H, S. medicae expressing a lacZ construct during early stages is stained in magenta. Bars = 100 µm (A–D and H) and 500 µm (E–G and I).
Taken together, these results depict a biphasic activation of the ET signaling pathway in response to symbiotic bacteria. An early peak at 0 to 1 hpi is independent of Nod factor perception and positively regulated by ET, and a second Nod factor-dependent peak at 6 hpi is suppressed by ET, the latter of which correlates with the induction of genes for ET biosynthesis. Both responses require MtEIN2 (skl) and involve the induction of ET receptors and EIN3-binding F-box proteins as part of a proposed feedback mechanism to regulate the pathway. Furthermore, these waves of signaling are candidates to induce the concerted expression of a suite of AP2/ERF TFs, the majority of which have not been characterized.
Modular Regulatory Networks to Predict New Regulatory and Target Genes in Response to Rhizobium and ET Signals
The transcriptome-wide clustering depicted in Figure 1 identified four major groups of genes, G1, G2, G3, and G4, that together capture the vast majority of transcriptional variation associated with Nod factor and ET signaling mutants. With the goals of identifying interactions within and among these clusters, of predicting the target genes of annotated regulatory proteins (e.g. TFs and signaling proteins), and of identifying new genes involved in symbiosis, we applied the MERLIN (for modular regulatory inference with per-gene information) regulatory network inference algorithm (Roy et al., 2013). MERLIN requires gene groupings and a set of candidate regulators as input and infers both regulator-target gene and regulator-target module relationships. A set of candidate regulators was compiled based on annotations (mainly kinases, phosphatases, and TFs) in the M. truncatula IMGA 3.5 version 4 genome. A novel aspect of MERLIN is that it predicts regulators for individual genes as well as sets of genes or modules.
We identified 4,393 regulator-target gene relationships connecting 952 regulators to 3,377 genes. Within the deduced network, a regulator connected to on average five target genes, and a target gene was on average regulated by two regulatory genes. Genes, including regulators, were grouped into modules when they had similar patterns of expression and were interconnected by a set of common inferred regulators. Thirty-three such modules included 10 or more genes, representing 85% of the total analyzed gene set (Fig. 12A). The characteristics of the nine largest modules are summarized in Supplemental Table S5. Module 227, the largest module (1,318 genes), contained most of the known symbiotic signaling genes from our data set, including NFP, LYK3, NIN, NSP2, ERN1, and NF-YA1, as well as numerous genes encoding kinases and TFs of unknown symbiotic function. The majority of genes in this module were up-regulated between 6 and 24 hpi and exhibit the characteristic Nod factor-induced, ET-repressed response that is typical of symbiotic genes. This signature expression pattern was also observed in genes in modules 230 and 207, although the genes in these groups were generally expressed at later time points. In contrast, genes in modules 184, 217, 223, 246, 229, and 137 possess expression patterns associated with ET signaling responses, with the skl mutant having an attenuated response or no response at all (Fig. 12A; Supplemental Table S5). Among these, module 246 contains the largest number of genes and is unusual because genes in this module were typically down-regulated early in response to inoculation. Interestingly, this pattern of down-regulation was accentuated in the skl background, indicating that both Nod factor and ET are positive regulators of expression. Module 223 is composed of genes for which expression is off in the skl background, including the previously described ET receptors and one of the EIN3 F-box-binding proteins (Fig. 10, A and C). To analyze whether these modules were interconnected, we focused on significant regulatory-module connections (P < 0.05) in the full network of all modules. We observed extensive cross talk and feedback within and among modules, the latter of which represent regulators for which a fraction of targets are present in one or more additional module (Fig. 12B).
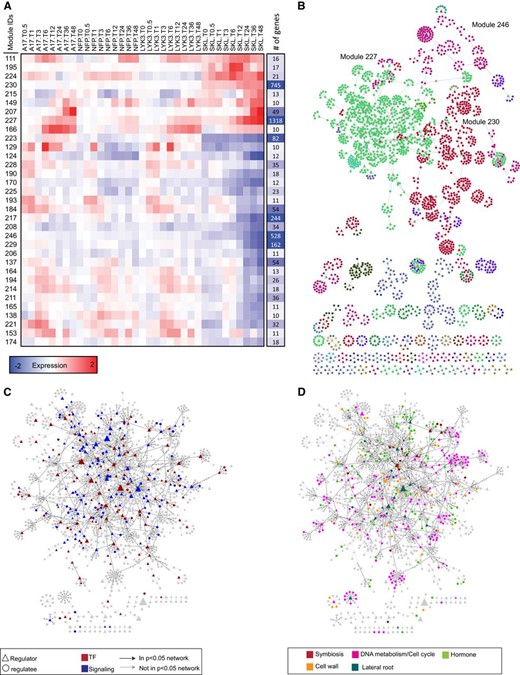
MERLIN network analysis on genes in groups G1 to G4. A, Average expression patterns of the main modules identified by MERLIN. Columns on the right represent the size of the modules. B, Diagram of the regulatory network predicted by MERLIN. Each module is assigned a different color. Regulators are represented as triangles and regulated genes as circles. When a gene is predicted to regulate others, a directed arrow is shown. C, Structure of the regulatory network of TF and signaling genes associated with module 227, the most symbiosis-related module. D, Interconnection between functional categories in the regulatory network predicted for cluster 227. Black arrows indicate significant regulatory relationships (P < 0.05).
To further characterize the properties of these modules, we examined them for enrichment of terms in the GO biological processes schema. We also analyzed the promoters of genes within modules for known transcriptional cis-elements using curated position-weight matrices for sequence-specific TFs. Significant enrichment for GO process terms and promoter cis-elements was identified in the three largest modules (Table II). Module 227, which contains the majority of symbiosis-related genes, is enriched with GO terms associated with assembly and organization of cellular components, protein complexes, and DNA-related processes, while gene promoters within this module are enriched with motifs related to cell cycle regulation and membrane transport. Module 230 is enriched for GO terms associated with biosynthesis and metabolism of nitrogen compounds and RNA-related processes, including coding and noncoding RNA transcription and translation. Interestingly, gene promoters in module 230 are enriched in motifs related to regulation of root primordia and meristem function. Module 246, which possesses a clear ET-related pattern of expression, is enriched for terms associated with cell wall organization and response to oxidative stress, with enrichment in cis-elements implicated in the regulation of plant cell development and seed maturation.
GO enrichment and cis-element analysis in genes present in G1 to G4
| Cluster No. | GO Term Enrichment | cis-Element Enrichment | Biological Function Associated with the cis-Element |
|---|---|---|---|
| 227 | Assembly, biogenesis, and organization of cellular components and macromolecular and protein complexes; DNA conformation change and DNA metabolic process | E2FA and DEL3-binding motif | Regulation of the expression of genes involved in the cell cyclea |
| Sclareol box1 | Regulation of the expression of ATP-binding cassette transportersb | ||
| 230 | Biosynthesis and metabolism of nitrogen compounds (amino acids, nucleic acids, etc.), RNA and noncoding RNA metabolism, transcription, translation, and protein metabolism | TEF box | Required for gene expression in root primordiac |
| Up2 motif | Regulation of gene expression during axillary bud outgrowthd | ||
| 246 | Plant cell wall structure organization and biogenesis, response to oxidative stress | Athb-1 and Athb-2 binding motif | Homeodomain related to plant cell development and elongatione |
| Pyrimidine and purine repeat motif | Element identified in the promoter of genes related to seed maturation and developmentf |
| Cluster No. | GO Term Enrichment | cis-Element Enrichment | Biological Function Associated with the cis-Element |
|---|---|---|---|
| 227 | Assembly, biogenesis, and organization of cellular components and macromolecular and protein complexes; DNA conformation change and DNA metabolic process | E2FA and DEL3-binding motif | Regulation of the expression of genes involved in the cell cyclea |
| Sclareol box1 | Regulation of the expression of ATP-binding cassette transportersb | ||
| 230 | Biosynthesis and metabolism of nitrogen compounds (amino acids, nucleic acids, etc.), RNA and noncoding RNA metabolism, transcription, translation, and protein metabolism | TEF box | Required for gene expression in root primordiac |
| Up2 motif | Regulation of gene expression during axillary bud outgrowthd | ||
| 246 | Plant cell wall structure organization and biogenesis, response to oxidative stress | Athb-1 and Athb-2 binding motif | Homeodomain related to plant cell development and elongatione |
| Pyrimidine and purine repeat motif | Element identified in the promoter of genes related to seed maturation and developmentf |
| Cluster No. | GO Term Enrichment | cis-Element Enrichment | Biological Function Associated with the cis-Element |
|---|---|---|---|
| 227 | Assembly, biogenesis, and organization of cellular components and macromolecular and protein complexes; DNA conformation change and DNA metabolic process | E2FA and DEL3-binding motif | Regulation of the expression of genes involved in the cell cyclea |
| Sclareol box1 | Regulation of the expression of ATP-binding cassette transportersb | ||
| 230 | Biosynthesis and metabolism of nitrogen compounds (amino acids, nucleic acids, etc.), RNA and noncoding RNA metabolism, transcription, translation, and protein metabolism | TEF box | Required for gene expression in root primordiac |
| Up2 motif | Regulation of gene expression during axillary bud outgrowthd | ||
| 246 | Plant cell wall structure organization and biogenesis, response to oxidative stress | Athb-1 and Athb-2 binding motif | Homeodomain related to plant cell development and elongatione |
| Pyrimidine and purine repeat motif | Element identified in the promoter of genes related to seed maturation and developmentf |
| Cluster No. | GO Term Enrichment | cis-Element Enrichment | Biological Function Associated with the cis-Element |
|---|---|---|---|
| 227 | Assembly, biogenesis, and organization of cellular components and macromolecular and protein complexes; DNA conformation change and DNA metabolic process | E2FA and DEL3-binding motif | Regulation of the expression of genes involved in the cell cyclea |
| Sclareol box1 | Regulation of the expression of ATP-binding cassette transportersb | ||
| 230 | Biosynthesis and metabolism of nitrogen compounds (amino acids, nucleic acids, etc.), RNA and noncoding RNA metabolism, transcription, translation, and protein metabolism | TEF box | Required for gene expression in root primordiac |
| Up2 motif | Regulation of gene expression during axillary bud outgrowthd | ||
| 246 | Plant cell wall structure organization and biogenesis, response to oxidative stress | Athb-1 and Athb-2 binding motif | Homeodomain related to plant cell development and elongatione |
| Pyrimidine and purine repeat motif | Element identified in the promoter of genes related to seed maturation and developmentf |
We used the inferred network to predict new regulators involved in symbiotic and/or ET responses based on the number of incoming (regulated gene) and outgoing (regulator) connections as well as graph-based ranking approaches to prioritize genes based on their connectivity to genes known to be associated with symbiosis (Supplemental Table S6). We focused on regulators with the largest number of connections, as this has been used as a measure of importance in network-based analyses (Barabási and Oltvai, 2004). On this basis, we defined regulatory hubs as those regulators connected to significantly more genes than an average regulator (with 17 or more connections) in the high-confidence network. We identified a total of 47 such regulatory hubs (Table III), the majority of which were in modules 230 (19) and 227 (16), the module closely related to symbiotic responses. These network hubs encode diverse classes of regulatory proteins, including MYB, WRKY, and AP2/ERF TFs, kinases, GTPase-related proteins, and chromatin-modifying enzymes. Although the majority of these genes are uncharacterized in M. truncatula, several hub homologs have been characterized in Arabidopsis, including those that regulate meristem identity and may have comparable roles in nodule development, such as Nucleostemin-Like1 (contig_83847_1), Breast Cancer1-Associated RING Domain1 (Medtr7g098170), and PLETHORA3 (Medtr5g031880).
Regulatory hubs with the greatest number of connections inferred by MERLIN
| Gene | No. of Connections | Description | Original Cluster | MERLIN Module |
|---|---|---|---|---|
| Medtr1g105350 | 49 | DNA damage repair | G4 | 246 |
| Medtr5g010650 | 39 | Myb TF | G2 | 230 |
| Medtr5g080780 | 39 | Histone acetyltransferase type B | G3 | 230 |
| contig_59380_1 | 38 | Unknown | G2 | 154 |
| Medtr1g086790 | 37 | WRKY TF | G2 | 227 |
| Medtr1g072890 | 34 | Ribosomal RNA-Processing Protein12-Like | G3 | 230 |
| contig_101096_1 | 33 | Receptor-like kinase | G2 | 227 |
| Medtr5g089220 | 33 | Unknown | G2 | 227 |
| contig_54218_1 | 32 | Chromodomain helicase DNA binding | G3 | 230 |
| contig_49136_1 | 31 | Chromodomain helicase DNA binding | G3 | 230 |
| Medtr5g025960 | 30 | RopGEF14 | G2 | 227 |
| contig_9054_1 | 28 | AP2 ERF TF | G1 | 230 |
| Medtr1g087290 | 26 | BRCA1 C Terminus domain containing protein domain | G3 | 227 |
| contig_63535_1 | 26 | DNA repair | G3 | 230 |
| Medtr1g072970 | 26 | Ribosomal RNA-Processing Protein12-Like | G3 | 230 |
| Medtr7g080780 | 25 | TF NAI1 | G4 | 137 |
| Medtr1g050750 | 25 | Pentatricopeptide repeat containing | G3 | 230 |
| Medtr7g098610 | 25 | Receptor-like kinase | G4 | 246 |
| contig_61021_1 | 24 | BZIP TF bZIP50 | G4 | 246 |
| Medtr7g025130 | 24 | Unknown | G4 | 246 |
| Medtr7g017530 | 23 | Pentatricopeptide repeat | G2 | 227 |
| contig_83847_1 | 23 | Nucleolar GTP binding/Nucleostemin-Like1 | G3 | 230 |
| Medtr5g010260 | 22 | Scaffold attachment region DNA binding1 | G3 | 230 |
| Medtr4g116300 | 21 | Zinc binding | G2 | 161 |
| Medtr7g098170 | 21 | Plant homeodomain zinc finger protein11-like/BRCA1-Associated RING Domain1 | G3 | 227 |
| contig_239504_1 | 21 | Calmodulin-binding | G4 | 246 |
| Medtr7g091990 | 20 | Unknown | G4 | 137 |
| AC229724_1053 | 20 | Unknown | G2 | 227 |
| Medtr7g050990 | 20 | (+)-Ɗ-Cadinene synthase | G4 | 229 |
| Medtr7g089770 | 20 | Pentatricopeptide repeat containing | G3 | 230 |
| Medtr1g085980 | 19 | Palmitoyltransferase ERF2 | G3 | 227 |
| Medtr5g025500 | 19 | Upstream activation factor subunit 30 | G3 | 227 |
| Medtr5g031880 | 19 | AP2 ERF/PLETHORA3 | G2 | 227 |
| Medtr4g033790 | 18 | Unknown | G3 | 227 |
| Medtr5g045910 | 18 | Receptor-like kinase | G2 | 227 |
| Medtr3g099850 | 18 | Neuroguidin | G3 | 230 |
| Medtr1g085040 | 18 | R2R3-MYB TF | G4 | 246 |
| Medtr5g053920 | 17 | AP2 domain TF | G4 | 184 |
| AC233109_38 | 17 | DNA (cytosine-5)-methyltransferase | G3 | 227 |
| Medtr3g031270 | 17 | Wound induced | G2 | 227 |
| Medtr8g094070 | 17 | DNA excision repair cross-complementation group 6 like | G2 | 227 |
| contig_114217_1 | 17 | WD repeat | G3 | 230 |
| contig_73997_1 | 17 | Proliferating cell nuclear antigen | G3 | 230 |
| Medtr3g116200 | 17 | ARID/BRIGHT DNA-binding domain | G3 | 230 |
| Medtr4g128240 | 17 | 3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase | G3 | 230 |
| Medtr7g113360 | 17 | DNA polymerase | G3 | 230 |
| Medtr8g018150 | 17 | Unknown | G3 | 230 |
| Gene | No. of Connections | Description | Original Cluster | MERLIN Module |
|---|---|---|---|---|
| Medtr1g105350 | 49 | DNA damage repair | G4 | 246 |
| Medtr5g010650 | 39 | Myb TF | G2 | 230 |
| Medtr5g080780 | 39 | Histone acetyltransferase type B | G3 | 230 |
| contig_59380_1 | 38 | Unknown | G2 | 154 |
| Medtr1g086790 | 37 | WRKY TF | G2 | 227 |
| Medtr1g072890 | 34 | Ribosomal RNA-Processing Protein12-Like | G3 | 230 |
| contig_101096_1 | 33 | Receptor-like kinase | G2 | 227 |
| Medtr5g089220 | 33 | Unknown | G2 | 227 |
| contig_54218_1 | 32 | Chromodomain helicase DNA binding | G3 | 230 |
| contig_49136_1 | 31 | Chromodomain helicase DNA binding | G3 | 230 |
| Medtr5g025960 | 30 | RopGEF14 | G2 | 227 |
| contig_9054_1 | 28 | AP2 ERF TF | G1 | 230 |
| Medtr1g087290 | 26 | BRCA1 C Terminus domain containing protein domain | G3 | 227 |
| contig_63535_1 | 26 | DNA repair | G3 | 230 |
| Medtr1g072970 | 26 | Ribosomal RNA-Processing Protein12-Like | G3 | 230 |
| Medtr7g080780 | 25 | TF NAI1 | G4 | 137 |
| Medtr1g050750 | 25 | Pentatricopeptide repeat containing | G3 | 230 |
| Medtr7g098610 | 25 | Receptor-like kinase | G4 | 246 |
| contig_61021_1 | 24 | BZIP TF bZIP50 | G4 | 246 |
| Medtr7g025130 | 24 | Unknown | G4 | 246 |
| Medtr7g017530 | 23 | Pentatricopeptide repeat | G2 | 227 |
| contig_83847_1 | 23 | Nucleolar GTP binding/Nucleostemin-Like1 | G3 | 230 |
| Medtr5g010260 | 22 | Scaffold attachment region DNA binding1 | G3 | 230 |
| Medtr4g116300 | 21 | Zinc binding | G2 | 161 |
| Medtr7g098170 | 21 | Plant homeodomain zinc finger protein11-like/BRCA1-Associated RING Domain1 | G3 | 227 |
| contig_239504_1 | 21 | Calmodulin-binding | G4 | 246 |
| Medtr7g091990 | 20 | Unknown | G4 | 137 |
| AC229724_1053 | 20 | Unknown | G2 | 227 |
| Medtr7g050990 | 20 | (+)-Ɗ-Cadinene synthase | G4 | 229 |
| Medtr7g089770 | 20 | Pentatricopeptide repeat containing | G3 | 230 |
| Medtr1g085980 | 19 | Palmitoyltransferase ERF2 | G3 | 227 |
| Medtr5g025500 | 19 | Upstream activation factor subunit 30 | G3 | 227 |
| Medtr5g031880 | 19 | AP2 ERF/PLETHORA3 | G2 | 227 |
| Medtr4g033790 | 18 | Unknown | G3 | 227 |
| Medtr5g045910 | 18 | Receptor-like kinase | G2 | 227 |
| Medtr3g099850 | 18 | Neuroguidin | G3 | 230 |
| Medtr1g085040 | 18 | R2R3-MYB TF | G4 | 246 |
| Medtr5g053920 | 17 | AP2 domain TF | G4 | 184 |
| AC233109_38 | 17 | DNA (cytosine-5)-methyltransferase | G3 | 227 |
| Medtr3g031270 | 17 | Wound induced | G2 | 227 |
| Medtr8g094070 | 17 | DNA excision repair cross-complementation group 6 like | G2 | 227 |
| contig_114217_1 | 17 | WD repeat | G3 | 230 |
| contig_73997_1 | 17 | Proliferating cell nuclear antigen | G3 | 230 |
| Medtr3g116200 | 17 | ARID/BRIGHT DNA-binding domain | G3 | 230 |
| Medtr4g128240 | 17 | 3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase | G3 | 230 |
| Medtr7g113360 | 17 | DNA polymerase | G3 | 230 |
| Medtr8g018150 | 17 | Unknown | G3 | 230 |
| Gene | No. of Connections | Description | Original Cluster | MERLIN Module |
|---|---|---|---|---|
| Medtr1g105350 | 49 | DNA damage repair | G4 | 246 |
| Medtr5g010650 | 39 | Myb TF | G2 | 230 |
| Medtr5g080780 | 39 | Histone acetyltransferase type B | G3 | 230 |
| contig_59380_1 | 38 | Unknown | G2 | 154 |
| Medtr1g086790 | 37 | WRKY TF | G2 | 227 |
| Medtr1g072890 | 34 | Ribosomal RNA-Processing Protein12-Like | G3 | 230 |
| contig_101096_1 | 33 | Receptor-like kinase | G2 | 227 |
| Medtr5g089220 | 33 | Unknown | G2 | 227 |
| contig_54218_1 | 32 | Chromodomain helicase DNA binding | G3 | 230 |
| contig_49136_1 | 31 | Chromodomain helicase DNA binding | G3 | 230 |
| Medtr5g025960 | 30 | RopGEF14 | G2 | 227 |
| contig_9054_1 | 28 | AP2 ERF TF | G1 | 230 |
| Medtr1g087290 | 26 | BRCA1 C Terminus domain containing protein domain | G3 | 227 |
| contig_63535_1 | 26 | DNA repair | G3 | 230 |
| Medtr1g072970 | 26 | Ribosomal RNA-Processing Protein12-Like | G3 | 230 |
| Medtr7g080780 | 25 | TF NAI1 | G4 | 137 |
| Medtr1g050750 | 25 | Pentatricopeptide repeat containing | G3 | 230 |
| Medtr7g098610 | 25 | Receptor-like kinase | G4 | 246 |
| contig_61021_1 | 24 | BZIP TF bZIP50 | G4 | 246 |
| Medtr7g025130 | 24 | Unknown | G4 | 246 |
| Medtr7g017530 | 23 | Pentatricopeptide repeat | G2 | 227 |
| contig_83847_1 | 23 | Nucleolar GTP binding/Nucleostemin-Like1 | G3 | 230 |
| Medtr5g010260 | 22 | Scaffold attachment region DNA binding1 | G3 | 230 |
| Medtr4g116300 | 21 | Zinc binding | G2 | 161 |
| Medtr7g098170 | 21 | Plant homeodomain zinc finger protein11-like/BRCA1-Associated RING Domain1 | G3 | 227 |
| contig_239504_1 | 21 | Calmodulin-binding | G4 | 246 |
| Medtr7g091990 | 20 | Unknown | G4 | 137 |
| AC229724_1053 | 20 | Unknown | G2 | 227 |
| Medtr7g050990 | 20 | (+)-Ɗ-Cadinene synthase | G4 | 229 |
| Medtr7g089770 | 20 | Pentatricopeptide repeat containing | G3 | 230 |
| Medtr1g085980 | 19 | Palmitoyltransferase ERF2 | G3 | 227 |
| Medtr5g025500 | 19 | Upstream activation factor subunit 30 | G3 | 227 |
| Medtr5g031880 | 19 | AP2 ERF/PLETHORA3 | G2 | 227 |
| Medtr4g033790 | 18 | Unknown | G3 | 227 |
| Medtr5g045910 | 18 | Receptor-like kinase | G2 | 227 |
| Medtr3g099850 | 18 | Neuroguidin | G3 | 230 |
| Medtr1g085040 | 18 | R2R3-MYB TF | G4 | 246 |
| Medtr5g053920 | 17 | AP2 domain TF | G4 | 184 |
| AC233109_38 | 17 | DNA (cytosine-5)-methyltransferase | G3 | 227 |
| Medtr3g031270 | 17 | Wound induced | G2 | 227 |
| Medtr8g094070 | 17 | DNA excision repair cross-complementation group 6 like | G2 | 227 |
| contig_114217_1 | 17 | WD repeat | G3 | 230 |
| contig_73997_1 | 17 | Proliferating cell nuclear antigen | G3 | 230 |
| Medtr3g116200 | 17 | ARID/BRIGHT DNA-binding domain | G3 | 230 |
| Medtr4g128240 | 17 | 3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase | G3 | 230 |
| Medtr7g113360 | 17 | DNA polymerase | G3 | 230 |
| Medtr8g018150 | 17 | Unknown | G3 | 230 |
| Gene | No. of Connections | Description | Original Cluster | MERLIN Module |
|---|---|---|---|---|
| Medtr1g105350 | 49 | DNA damage repair | G4 | 246 |
| Medtr5g010650 | 39 | Myb TF | G2 | 230 |
| Medtr5g080780 | 39 | Histone acetyltransferase type B | G3 | 230 |
| contig_59380_1 | 38 | Unknown | G2 | 154 |
| Medtr1g086790 | 37 | WRKY TF | G2 | 227 |
| Medtr1g072890 | 34 | Ribosomal RNA-Processing Protein12-Like | G3 | 230 |
| contig_101096_1 | 33 | Receptor-like kinase | G2 | 227 |
| Medtr5g089220 | 33 | Unknown | G2 | 227 |
| contig_54218_1 | 32 | Chromodomain helicase DNA binding | G3 | 230 |
| contig_49136_1 | 31 | Chromodomain helicase DNA binding | G3 | 230 |
| Medtr5g025960 | 30 | RopGEF14 | G2 | 227 |
| contig_9054_1 | 28 | AP2 ERF TF | G1 | 230 |
| Medtr1g087290 | 26 | BRCA1 C Terminus domain containing protein domain | G3 | 227 |
| contig_63535_1 | 26 | DNA repair | G3 | 230 |
| Medtr1g072970 | 26 | Ribosomal RNA-Processing Protein12-Like | G3 | 230 |
| Medtr7g080780 | 25 | TF NAI1 | G4 | 137 |
| Medtr1g050750 | 25 | Pentatricopeptide repeat containing | G3 | 230 |
| Medtr7g098610 | 25 | Receptor-like kinase | G4 | 246 |
| contig_61021_1 | 24 | BZIP TF bZIP50 | G4 | 246 |
| Medtr7g025130 | 24 | Unknown | G4 | 246 |
| Medtr7g017530 | 23 | Pentatricopeptide repeat | G2 | 227 |
| contig_83847_1 | 23 | Nucleolar GTP binding/Nucleostemin-Like1 | G3 | 230 |
| Medtr5g010260 | 22 | Scaffold attachment region DNA binding1 | G3 | 230 |
| Medtr4g116300 | 21 | Zinc binding | G2 | 161 |
| Medtr7g098170 | 21 | Plant homeodomain zinc finger protein11-like/BRCA1-Associated RING Domain1 | G3 | 227 |
| contig_239504_1 | 21 | Calmodulin-binding | G4 | 246 |
| Medtr7g091990 | 20 | Unknown | G4 | 137 |
| AC229724_1053 | 20 | Unknown | G2 | 227 |
| Medtr7g050990 | 20 | (+)-Ɗ-Cadinene synthase | G4 | 229 |
| Medtr7g089770 | 20 | Pentatricopeptide repeat containing | G3 | 230 |
| Medtr1g085980 | 19 | Palmitoyltransferase ERF2 | G3 | 227 |
| Medtr5g025500 | 19 | Upstream activation factor subunit 30 | G3 | 227 |
| Medtr5g031880 | 19 | AP2 ERF/PLETHORA3 | G2 | 227 |
| Medtr4g033790 | 18 | Unknown | G3 | 227 |
| Medtr5g045910 | 18 | Receptor-like kinase | G2 | 227 |
| Medtr3g099850 | 18 | Neuroguidin | G3 | 230 |
| Medtr1g085040 | 18 | R2R3-MYB TF | G4 | 246 |
| Medtr5g053920 | 17 | AP2 domain TF | G4 | 184 |
| AC233109_38 | 17 | DNA (cytosine-5)-methyltransferase | G3 | 227 |
| Medtr3g031270 | 17 | Wound induced | G2 | 227 |
| Medtr8g094070 | 17 | DNA excision repair cross-complementation group 6 like | G2 | 227 |
| contig_114217_1 | 17 | WD repeat | G3 | 230 |
| contig_73997_1 | 17 | Proliferating cell nuclear antigen | G3 | 230 |
| Medtr3g116200 | 17 | ARID/BRIGHT DNA-binding domain | G3 | 230 |
| Medtr4g128240 | 17 | 3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase | G3 | 230 |
| Medtr7g113360 | 17 | DNA polymerase | G3 | 230 |
| Medtr8g018150 | 17 | Unknown | G3 | 230 |
Module 227 was analyzed in greater detail because it includes both the largest number of genes and the largest number of characterized symbiosis genes. We found a strong interconnection between TFs and genes involved in signaling processes, which primarily include kinases, GTPases, and calcium signaling proteins, which is graphically represented in Figure 12C. Major regulatory hubs in this module include the well-known symbiotic genes NIN, ERN1, NSP2, NF-YA1, LYK3, LYK4, and LYK5 as well as numerous uncharacterized genes. Similarly, significant interconnection is found in the network between genes involved in symbiosis, DNA metabolism, cell cycle, cell wall, LR development, and hormone responses (Fig. 12D). For instance, NF-YA1 is connected to known LR regulators such as PUCHI, PLETHORA3, and WUSCHEL-RELATED HOMEOBOX5 (WOX5; Galinha et al., 2007; Hirota et al., 2007; Tian et al., 2014) as well as the symbiotic flotillin FLOT2, which is involved in both nodulation and root development (Haney and Long, 2010). A complete list of genes and their predicted GO functions is given in Supplemental Table S7.
MERLIN has been shown to infer modules of coexpressed genes enriched in similar biological functions (Roy et al., 2013). Thus, it is expected that genes regulating common biological processes will display greater connectivity in the predicted signaling network than do genes with more disparate functions. Given that several of the annotated gene sets described above exhibited high interconnectivity, we asked whether the network associated with module 227 could be further subdivided into subnetworks representing potentially novel functional pathways. To address this, we applied a graph-clustering algorithm to the network associated with module 227. We identified 10 subnetworks that were more densely connected within each subnetwork than they were among the subnetworks. Six of the 10 subnetworks are enriched for genes involved in DNA metabolism, cell cycle, hormone, and symbiosis (Supplemental Table S8). Among the remaining networks, subnetwork 2 contained seven putative LR-associated genes, including homologs of well-known regulators of the LR pathway in Arabidopsis PLETHORA3, WOX5 and WOX13, LATERAL ORGAN BOUNDARY DOMAIN18 (LBD18), and PUCHI.
We next asked if we could predict new regulators of symbiosis based on their connectivity to known symbiotic regulatory proteins within module 227, or to genes with a known symbiotic mutant phenotype, by using a network diffusion algorithm (Supplemental Protocol S1). We identified novel genes that were highly ranked, with network diffusion scores at least 2 sd above the average (Table IV). NSP2, NF-YA1, LYK3, LYK4, NIN, and ERN1 were among the symbiosis regulators used to seed the identification of novel candidate regulatory genes and were themselves found to be highly connected. This approach yielded 56 additional regulators that were highly connected to the known symbiosis genes (Table IV). These novel regulators included several genes involved in LR development (LBD16 and ERF115), CK signaling (Arabidopsis Response Regulator [ARR]), and polar growth (RopGEF12). In addition, numerous uncharacterized TFs, kinases, and other signaling molecules were also implicated in early Nod factor signal transduction.
Identification of candidate genes based on their connection with known symbiosis genes in the 227 subnetwork
| Gene | Gene Description | Connectivity Scorea |
|---|---|---|
| Medtr3g072710 | Nodulation-signaling pathway2 protein (NSP2) | 0.153 |
| Medtr5g086110 | LysM domain-containing receptor-like kinase | 0.122 |
| Medtr1g056530 | Nuclear transcription factor Y subunit A-10 (NF-YA1) | 0.083 |
| Medtr5g086130 | LysM receptor kinase (LYK3) | 0.076 |
| Medtr7g082260 | Zinc finger protein | 0.075 |
| contig_119095_1 | SUPERMAN-like zinc finger protein | 0.072 |
| Medtr3g031270 | Wound-induced protein | 0.071 |
| Medtr5g099060 | Nodule inception protein (NIN) | 0.070 |
| Medtr5g086120 | Receptor-like protein kinase (LYK4) | 0.069 |
| Medtr7g085810 | ET-responsive TF (ERN1) | 0.069 |
| contig_56789_1 | Calmodulin-binding protein | 0.067 |
| Medtr4g079760 | TF basic helix-loop-helix30 | 0.064 |
| Medtr3g015490 | Two-component response regulator ARR9 | 0.064 |
| Medtr2g101830 | Rop guanine nucleotide-exchange factor (RopGEF12) | 0.064 |
| contig_103831_1 | Myb-like transcription factor Myb | 0.062 |
| contig_60935_1 | Protein phosphatase2C | 0.056 |
| Medtr7g011900 | Isoliquiritigenin 2′-O-methyltransferase | 0.056 |
| Medtr5g025840 | Kinase-like protein | 0.053 |
| contig_8047_1 | LOB domain protein-like protein (LBD16) | 0.052 |
| Medtr4g127140 | MADS box transcription factor | 0.050 |
| Medtr5g025850 | Receptor-like protein kinase | 0.048 |
| contig_49939_2 | Receptor-like kinase | 0.044 |
| AC235664_10 | 4-Hydroxy-3-methyl-but-2-enyl diphosphate reductase (HDR) | 0.044 |
| contig_167979_1 | Chaperone protein DnaJ-like protein | 0.044 |
| contig_245351_1 | Receptor protein kinase | 0.044 |
| contig_76132_1 | HDR | 0.044 |
| Medtr4g104060 | Unknown protein | 0.044 |
| Medtr5g010010 | 2-C-Methyl-d-erythritol 2 4-cyclodiphosphate synthase (MCS) | 0.044 |
| Medtr6g082180 | Unknown protein | 0.044 |
| Medtr7g104250 | Unknown protein | 0.044 |
| Medtr5g019070 | LRR receptor-like Ser/Thr-protein kinase FEI | 0.043 |
| Medtr3g086320 | Pectate lyase | 0.043 |
| Medtr2g068960 | ACC oxidase homologb | 0.040 |
| Medtr2g069020 | ACC oxidase homologb | 0.040 |
| Medtr8g097190 | Unknown protein | 0.040 |
| contig_30837_1 | Receptor-like kinase | 0.040 |
| Medtr2g068650 | Kinase-like protein | 0.039 |
| Medtr4g051330 | Two-component response regulator ARR9 | 0.039 |
| CX549419 | Weakly similar to UniRef100 A7P585 (Vitis vinifera) | 0.039 |
| contig_60385_1 | Basic helix-loop-helix TF-like protein | 0.039 |
| Medtr1g031540 | Receptor Ser/Thr kinase | 0.039 |
| contig_68802_2 | Cytochrome P450 | 0.038 |
| Medtr4g107970 | WRKY transcription factor | 0.038 |
| Medtr5g033030 | Multidrug resistance protein ATP-binding cassette transporter family | 0.038 |
| Medtr2g069250 | Nodule-specific Gly-rich protein3C | 0.038 |
| Medtr2g019750 | ET-responsive transcription factor3b | 0.038 |
| Medtr8g026680 | Rac GTPase-activating protein | 0.038 |
| contig_55524_1 | Zinc finger protein | 0.037 |
| AC233082_23 | Receptor-like kinase | 0.037 |
| Medtr5g026760 | Kinase-like protein | 0.036 |
| Medtr8g074530 | Unknown protein | 0.033 |
| Medtr7g020980 | ET-responsive TF (ERF115) | 0.033 |
| contig_62786_1 | Inositol-tetrakisphosphate 1-kinase | 0.032 |
| Medtr3g008760 | 21-kD protein | 0.032 |
| Medtr6g052300 | Metal transporter cyclin and CBS domain divalent metal cation transport mediator2 | 0.032 |
| contig_94824_1 | LRR-RLK protein | 0.032 |
| Gene | Gene Description | Connectivity Scorea |
|---|---|---|
| Medtr3g072710 | Nodulation-signaling pathway2 protein (NSP2) | 0.153 |
| Medtr5g086110 | LysM domain-containing receptor-like kinase | 0.122 |
| Medtr1g056530 | Nuclear transcription factor Y subunit A-10 (NF-YA1) | 0.083 |
| Medtr5g086130 | LysM receptor kinase (LYK3) | 0.076 |
| Medtr7g082260 | Zinc finger protein | 0.075 |
| contig_119095_1 | SUPERMAN-like zinc finger protein | 0.072 |
| Medtr3g031270 | Wound-induced protein | 0.071 |
| Medtr5g099060 | Nodule inception protein (NIN) | 0.070 |
| Medtr5g086120 | Receptor-like protein kinase (LYK4) | 0.069 |
| Medtr7g085810 | ET-responsive TF (ERN1) | 0.069 |
| contig_56789_1 | Calmodulin-binding protein | 0.067 |
| Medtr4g079760 | TF basic helix-loop-helix30 | 0.064 |
| Medtr3g015490 | Two-component response regulator ARR9 | 0.064 |
| Medtr2g101830 | Rop guanine nucleotide-exchange factor (RopGEF12) | 0.064 |
| contig_103831_1 | Myb-like transcription factor Myb | 0.062 |
| contig_60935_1 | Protein phosphatase2C | 0.056 |
| Medtr7g011900 | Isoliquiritigenin 2′-O-methyltransferase | 0.056 |
| Medtr5g025840 | Kinase-like protein | 0.053 |
| contig_8047_1 | LOB domain protein-like protein (LBD16) | 0.052 |
| Medtr4g127140 | MADS box transcription factor | 0.050 |
| Medtr5g025850 | Receptor-like protein kinase | 0.048 |
| contig_49939_2 | Receptor-like kinase | 0.044 |
| AC235664_10 | 4-Hydroxy-3-methyl-but-2-enyl diphosphate reductase (HDR) | 0.044 |
| contig_167979_1 | Chaperone protein DnaJ-like protein | 0.044 |
| contig_245351_1 | Receptor protein kinase | 0.044 |
| contig_76132_1 | HDR | 0.044 |
| Medtr4g104060 | Unknown protein | 0.044 |
| Medtr5g010010 | 2-C-Methyl-d-erythritol 2 4-cyclodiphosphate synthase (MCS) | 0.044 |
| Medtr6g082180 | Unknown protein | 0.044 |
| Medtr7g104250 | Unknown protein | 0.044 |
| Medtr5g019070 | LRR receptor-like Ser/Thr-protein kinase FEI | 0.043 |
| Medtr3g086320 | Pectate lyase | 0.043 |
| Medtr2g068960 | ACC oxidase homologb | 0.040 |
| Medtr2g069020 | ACC oxidase homologb | 0.040 |
| Medtr8g097190 | Unknown protein | 0.040 |
| contig_30837_1 | Receptor-like kinase | 0.040 |
| Medtr2g068650 | Kinase-like protein | 0.039 |
| Medtr4g051330 | Two-component response regulator ARR9 | 0.039 |
| CX549419 | Weakly similar to UniRef100 A7P585 (Vitis vinifera) | 0.039 |
| contig_60385_1 | Basic helix-loop-helix TF-like protein | 0.039 |
| Medtr1g031540 | Receptor Ser/Thr kinase | 0.039 |
| contig_68802_2 | Cytochrome P450 | 0.038 |
| Medtr4g107970 | WRKY transcription factor | 0.038 |
| Medtr5g033030 | Multidrug resistance protein ATP-binding cassette transporter family | 0.038 |
| Medtr2g069250 | Nodule-specific Gly-rich protein3C | 0.038 |
| Medtr2g019750 | ET-responsive transcription factor3b | 0.038 |
| Medtr8g026680 | Rac GTPase-activating protein | 0.038 |
| contig_55524_1 | Zinc finger protein | 0.037 |
| AC233082_23 | Receptor-like kinase | 0.037 |
| Medtr5g026760 | Kinase-like protein | 0.036 |
| Medtr8g074530 | Unknown protein | 0.033 |
| Medtr7g020980 | ET-responsive TF (ERF115) | 0.033 |
| contig_62786_1 | Inositol-tetrakisphosphate 1-kinase | 0.032 |
| Medtr3g008760 | 21-kD protein | 0.032 |
| Medtr6g052300 | Metal transporter cyclin and CBS domain divalent metal cation transport mediator2 | 0.032 |
| contig_94824_1 | LRR-RLK protein | 0.032 |
The connectivity score, obtained by applying a regularized Laplacian kernel method to the inferred regulatory network, measures connectivity to the known symbiosis genes. The top 56 genes shown were selected such that their score was 2 sd above the mean. Input symbiosis genes are shown in boldface.
Annotated as 2-oxoglutarate-Fe(II) oxygenase family oxidoreductase in IMGA 4.0 version 1.
| Gene | Gene Description | Connectivity Scorea |
|---|---|---|
| Medtr3g072710 | Nodulation-signaling pathway2 protein (NSP2) | 0.153 |
| Medtr5g086110 | LysM domain-containing receptor-like kinase | 0.122 |
| Medtr1g056530 | Nuclear transcription factor Y subunit A-10 (NF-YA1) | 0.083 |
| Medtr5g086130 | LysM receptor kinase (LYK3) | 0.076 |
| Medtr7g082260 | Zinc finger protein | 0.075 |
| contig_119095_1 | SUPERMAN-like zinc finger protein | 0.072 |
| Medtr3g031270 | Wound-induced protein | 0.071 |
| Medtr5g099060 | Nodule inception protein (NIN) | 0.070 |
| Medtr5g086120 | Receptor-like protein kinase (LYK4) | 0.069 |
| Medtr7g085810 | ET-responsive TF (ERN1) | 0.069 |
| contig_56789_1 | Calmodulin-binding protein | 0.067 |
| Medtr4g079760 | TF basic helix-loop-helix30 | 0.064 |
| Medtr3g015490 | Two-component response regulator ARR9 | 0.064 |
| Medtr2g101830 | Rop guanine nucleotide-exchange factor (RopGEF12) | 0.064 |
| contig_103831_1 | Myb-like transcription factor Myb | 0.062 |
| contig_60935_1 | Protein phosphatase2C | 0.056 |
| Medtr7g011900 | Isoliquiritigenin 2′-O-methyltransferase | 0.056 |
| Medtr5g025840 | Kinase-like protein | 0.053 |
| contig_8047_1 | LOB domain protein-like protein (LBD16) | 0.052 |
| Medtr4g127140 | MADS box transcription factor | 0.050 |
| Medtr5g025850 | Receptor-like protein kinase | 0.048 |
| contig_49939_2 | Receptor-like kinase | 0.044 |
| AC235664_10 | 4-Hydroxy-3-methyl-but-2-enyl diphosphate reductase (HDR) | 0.044 |
| contig_167979_1 | Chaperone protein DnaJ-like protein | 0.044 |
| contig_245351_1 | Receptor protein kinase | 0.044 |
| contig_76132_1 | HDR | 0.044 |
| Medtr4g104060 | Unknown protein | 0.044 |
| Medtr5g010010 | 2-C-Methyl-d-erythritol 2 4-cyclodiphosphate synthase (MCS) | 0.044 |
| Medtr6g082180 | Unknown protein | 0.044 |
| Medtr7g104250 | Unknown protein | 0.044 |
| Medtr5g019070 | LRR receptor-like Ser/Thr-protein kinase FEI | 0.043 |
| Medtr3g086320 | Pectate lyase | 0.043 |
| Medtr2g068960 | ACC oxidase homologb | 0.040 |
| Medtr2g069020 | ACC oxidase homologb | 0.040 |
| Medtr8g097190 | Unknown protein | 0.040 |
| contig_30837_1 | Receptor-like kinase | 0.040 |
| Medtr2g068650 | Kinase-like protein | 0.039 |
| Medtr4g051330 | Two-component response regulator ARR9 | 0.039 |
| CX549419 | Weakly similar to UniRef100 A7P585 (Vitis vinifera) | 0.039 |
| contig_60385_1 | Basic helix-loop-helix TF-like protein | 0.039 |
| Medtr1g031540 | Receptor Ser/Thr kinase | 0.039 |
| contig_68802_2 | Cytochrome P450 | 0.038 |
| Medtr4g107970 | WRKY transcription factor | 0.038 |
| Medtr5g033030 | Multidrug resistance protein ATP-binding cassette transporter family | 0.038 |
| Medtr2g069250 | Nodule-specific Gly-rich protein3C | 0.038 |
| Medtr2g019750 | ET-responsive transcription factor3b | 0.038 |
| Medtr8g026680 | Rac GTPase-activating protein | 0.038 |
| contig_55524_1 | Zinc finger protein | 0.037 |
| AC233082_23 | Receptor-like kinase | 0.037 |
| Medtr5g026760 | Kinase-like protein | 0.036 |
| Medtr8g074530 | Unknown protein | 0.033 |
| Medtr7g020980 | ET-responsive TF (ERF115) | 0.033 |
| contig_62786_1 | Inositol-tetrakisphosphate 1-kinase | 0.032 |
| Medtr3g008760 | 21-kD protein | 0.032 |
| Medtr6g052300 | Metal transporter cyclin and CBS domain divalent metal cation transport mediator2 | 0.032 |
| contig_94824_1 | LRR-RLK protein | 0.032 |
| Gene | Gene Description | Connectivity Scorea |
|---|---|---|
| Medtr3g072710 | Nodulation-signaling pathway2 protein (NSP2) | 0.153 |
| Medtr5g086110 | LysM domain-containing receptor-like kinase | 0.122 |
| Medtr1g056530 | Nuclear transcription factor Y subunit A-10 (NF-YA1) | 0.083 |
| Medtr5g086130 | LysM receptor kinase (LYK3) | 0.076 |
| Medtr7g082260 | Zinc finger protein | 0.075 |
| contig_119095_1 | SUPERMAN-like zinc finger protein | 0.072 |
| Medtr3g031270 | Wound-induced protein | 0.071 |
| Medtr5g099060 | Nodule inception protein (NIN) | 0.070 |
| Medtr5g086120 | Receptor-like protein kinase (LYK4) | 0.069 |
| Medtr7g085810 | ET-responsive TF (ERN1) | 0.069 |
| contig_56789_1 | Calmodulin-binding protein | 0.067 |
| Medtr4g079760 | TF basic helix-loop-helix30 | 0.064 |
| Medtr3g015490 | Two-component response regulator ARR9 | 0.064 |
| Medtr2g101830 | Rop guanine nucleotide-exchange factor (RopGEF12) | 0.064 |
| contig_103831_1 | Myb-like transcription factor Myb | 0.062 |
| contig_60935_1 | Protein phosphatase2C | 0.056 |
| Medtr7g011900 | Isoliquiritigenin 2′-O-methyltransferase | 0.056 |
| Medtr5g025840 | Kinase-like protein | 0.053 |
| contig_8047_1 | LOB domain protein-like protein (LBD16) | 0.052 |
| Medtr4g127140 | MADS box transcription factor | 0.050 |
| Medtr5g025850 | Receptor-like protein kinase | 0.048 |
| contig_49939_2 | Receptor-like kinase | 0.044 |
| AC235664_10 | 4-Hydroxy-3-methyl-but-2-enyl diphosphate reductase (HDR) | 0.044 |
| contig_167979_1 | Chaperone protein DnaJ-like protein | 0.044 |
| contig_245351_1 | Receptor protein kinase | 0.044 |
| contig_76132_1 | HDR | 0.044 |
| Medtr4g104060 | Unknown protein | 0.044 |
| Medtr5g010010 | 2-C-Methyl-d-erythritol 2 4-cyclodiphosphate synthase (MCS) | 0.044 |
| Medtr6g082180 | Unknown protein | 0.044 |
| Medtr7g104250 | Unknown protein | 0.044 |
| Medtr5g019070 | LRR receptor-like Ser/Thr-protein kinase FEI | 0.043 |
| Medtr3g086320 | Pectate lyase | 0.043 |
| Medtr2g068960 | ACC oxidase homologb | 0.040 |
| Medtr2g069020 | ACC oxidase homologb | 0.040 |
| Medtr8g097190 | Unknown protein | 0.040 |
| contig_30837_1 | Receptor-like kinase | 0.040 |
| Medtr2g068650 | Kinase-like protein | 0.039 |
| Medtr4g051330 | Two-component response regulator ARR9 | 0.039 |
| CX549419 | Weakly similar to UniRef100 A7P585 (Vitis vinifera) | 0.039 |
| contig_60385_1 | Basic helix-loop-helix TF-like protein | 0.039 |
| Medtr1g031540 | Receptor Ser/Thr kinase | 0.039 |
| contig_68802_2 | Cytochrome P450 | 0.038 |
| Medtr4g107970 | WRKY transcription factor | 0.038 |
| Medtr5g033030 | Multidrug resistance protein ATP-binding cassette transporter family | 0.038 |
| Medtr2g069250 | Nodule-specific Gly-rich protein3C | 0.038 |
| Medtr2g019750 | ET-responsive transcription factor3b | 0.038 |
| Medtr8g026680 | Rac GTPase-activating protein | 0.038 |
| contig_55524_1 | Zinc finger protein | 0.037 |
| AC233082_23 | Receptor-like kinase | 0.037 |
| Medtr5g026760 | Kinase-like protein | 0.036 |
| Medtr8g074530 | Unknown protein | 0.033 |
| Medtr7g020980 | ET-responsive TF (ERF115) | 0.033 |
| contig_62786_1 | Inositol-tetrakisphosphate 1-kinase | 0.032 |
| Medtr3g008760 | 21-kD protein | 0.032 |
| Medtr6g052300 | Metal transporter cyclin and CBS domain divalent metal cation transport mediator2 | 0.032 |
| contig_94824_1 | LRR-RLK protein | 0.032 |
The connectivity score, obtained by applying a regularized Laplacian kernel method to the inferred regulatory network, measures connectivity to the known symbiosis genes. The top 56 genes shown were selected such that their score was 2 sd above the mean. Input symbiosis genes are shown in boldface.
Annotated as 2-oxoglutarate-Fe(II) oxygenase family oxidoreductase in IMGA 4.0 version 1.
DISCUSSION
Comprehensive Analysis of Very Early Transcriptional Responses to Nod Factors
In this work, we implemented a sequencing-based, whole-transcriptome approach to investigate the relationship between the Nod factor and ET signaling pathways during the earliest stages of the M. truncatula-S. medicae symbiosis. We analyzed patterns of gene expression between 0 and 48 hpi, encompassing morphological changes to root hairs and their infection by bacteria, as well as the initiation of cortical cell divisions. Throughout this early time course, changes to the host are driven largely by the perception of bacterially delivered Nod factor and host-derived ET signals, with attendant cellular and biochemical changes. In order to identify symbiosis- and ET-related changes in gene expression, we utilized M. truncatula mutants characterized by aberrant Nod factor (nfp and lyk3) and ET responses (skl) and compared their transcriptional profiles with those of wild-type cv Jemalong A17 plants. Information gained by including the skl mutant was particularly valuable because skl is hypersensitive to Nod factor (Oldroyd et al., 2001) and Nod factor signaling responses are amplified. Importantly, we verified that hyperinduction of symbiosis transcripts in the skl mutant was dependent on Nod factor signaling by testing gene induction in the dmi1 skl double mutant. Thus, weak and potentially cell-specific (e.g. root hair-specific) responses that may have been masked in the wild type were easily identified in skl mutants.
Our analysis identified 10,985 differentially expressed genes (FDR = 0.001), which clustered into 11 groups based on common expression patterns across genotypes and time points. Of particular interest are expression groups G1, G2, and G3, which contain genes exhibiting Nod factor-dependent induction that are negatively regulated by ET; this is the predominant expression pattern of genes involved in early symbiosis. Indeed, included within these groups are previously characterized nodulin and Nod factor signaling genes (Fig. 4). Thus, we have uncovered a set of 2,356 novel genes coexpressed with classical symbiotic markers that are likely to function at or downstream of Nod factor recognition during early symbiotic development.
The lyk3 mutant was previously observed to be partially responsive to rhizobium (Catoira et al., 2001; El Yahyaoui et al., 2004; Mitra et al., 2004), and our data are consistent with and extend this observation. While many of the G1, G2, and G3 genes are not expressed in lyk3, many others are expressed with similar kinetics as cv Jemalong A17 but at a reduced level. In contrast, the nfp mutant, which is a loss-of-function mutation, is typically regarded as Nod factor insensitive. Nevertheless, we observed consistent elevations in read counts for several symbiosis-related genes in the nfp mutant, although these responses were significantly attenuated and delayed relative to the wild type. For example, G2 genes such as the rhizobium-induced peroxidase Rhizobium-induced peroxidase1 were induced between 3 and 6 hpi in cv Jemalong A17 and skl, while their expression did not increase until approximately 12 hpi in nfp, and their peak transcript abundance in nfp was markedly lower than in cv Jemalong A17 (Fig. 3; Supplemental Table S1). Rose et al. (2012) came to a similar conclusion in their study of phosphorylation and transcriptional responses 1 h after Nod factor treatment. There are several possibilities to explain these observations, including that there may be additional Nod factor receptors present and functional; alternatively, these attenuated responses may reflect the perception of microbe-associated molecular patterns distinct from Nod factors.
Interestingly, expression group G10 reveals a set of genes with constitutively elevated expression in the nfp mutant but that are unchanged in the wild type or other mutant backgrounds. Such an expression pattern could arise as an artifact of normalization, but the fact that G10 is enriched for genes with particular functions (i.e. defense and signal transduction) strongly suggests that G10 represents a specific class of genes that are constitutively expressed in the root and suppressed by nfp function, where NFP represses basal transcription.
LYK3 and NFP are members of the LysM gene family (Limpens et al., 2003; Arrighi et al., 2006). We identified several other related genes, notably LYR3, LYK4, LYK10, and Medtr5g086110, whose expression patterns following inoculation are strikingly similar to LYK3 and NFP (Fig. 4). MERLIN network analysis predicted that these genes are part of the same subnetwork and are coregulated (Fig. 12C; Supplemental Table S7). M. truncatula LYR3 was recently identified as a high-affinity lipochitooligosaccharide-binding protein able to recognize both fungal Myc and rhizobial Nod factors (Fliegmann et al., 2013). A role in symbiosis has also been to assigned LYK4, since expression of a gene-specific RNA interference construct impaired infection thread development (Limpens et al., 2003). Symbiotic function has not been assigned to LYK10 or to Medtr5g086110. Their weak and delayed transcriptional induction in lyk3 and nfp mutants suggests that one or more of these LysM genes may be partially compensating for lyk3 and nfp mutations, perhaps with altered affinity for Nod factors or downstream signaling proteins. The inferred LysM gene subnetwork is consistent with a multiprotein complex composed of several members of the LysM family, or of partially redundant parallel LysM receptors, involved in Nod factor perception and symbiotic signaling (Moling et al., 2014). These additional LysM homologs are good candidates for novel symbiosis-related receptor proteins and deserve priority in reverse genetic and biochemical studies to test their role in the host’s symbiotic responses.
Nod Factors Affect the Transcriptional Regulation of Hormone Biosynthetic and Signaling Pathways
Our analysis depicts the complex activation of multiple hormone biosynthetic pathways by Nod factors within the first 48 hpi. The MEP pathway is rapidly up-regulated to produce IPP and DMAPP, molecules that serve as precursors for many plant hormones, including CK, GA, ABA, and strigolactones. Multiple hormone biosynthetic and signaling pathways are subsequently activated in a Nod factor-dependent, ET-regulated manner. These pathways are likely to interact both synergistically and antagonistically to affect downstream symbiotic responses. Previous evidence has shown that hormone perception directly impacts the expression of the symbiotic signaling genes, suggesting feedback regulation of the symbiotic pathway. Understanding when and where these regulators are transcribed relative to hormone biosynthesis and the developing symbiosis will help elucidate what processes the hormone perception pathways are affecting.
CK exhibits both stimulatory and inhibitory activity during nodulation, as it initiates nodule development and is also translocated from shoot to root to autoregulate nodule number (Gonzalez-Rizzo et al., 2006; Sasaki et al., 2014). Type A ARR expression is one hallmark of the activation of CK pathways, and the up-regulation of several type A ARRs has been observed following treatments with CK, rhizobium, and purified Nod factors (Op den Camp et al., 2011; Plet et al., 2011; Ariel et al., 2012). We observed the induction of several of these ARRs following inoculation with rhizobium, with most genes induced by 12 to 24 hpi, consistent with the up-regulation of IPT (and presumably de novo CK biosynthesis) at 12 hpi (Supplemental Fig. S5). It is noteworthy that IPT expression was reduced in the skl mutant, indicating that ET perception is required for the symbiotic induction of IPT. However, in contrast to IPT, the symbiosis-related ARRs were hyperinduced in skl, suggesting that additional sources of CK or alternative signals may be involved in their symbiotic induction. Sasaki et al. (2014) recently demonstrated that rhizobial infection induces Hypernodulation aberrant root formation protein1-dependent CK biosynthesis in the shoot and that CKs can be transported from shoot to root. The authors proposed a model whereby CK is translocated to the root system as part of the autoregulatory feedback inhibition of nodulation. Interestingly, we observed the early up-regulation of an equilibrative nucleoside transporter that has been proposed to function as a CK transporter (Hirose et al., 2008). This could serve as the mechanism to mobilize CK from the vascular system to responding cortical cells, where it may impact nodule organogenesis.
In addition to directly impacting both nodule development and the symbiotic pathway, CK may influence the biosynthesis and signaling of other plant hormones. Ariel et al. (2012) noted that treating M. truncatula roots with CKs induces the expression of GA biosynthetic genes, suggesting that the Nod factor-induced biosynthesis of CK may indirectly stimulate GA pathways. Our sampling of multiple early time points reveals nuanced aspects of GA signaling during the legume-rhizobium interaction. Both GA anabolic and catabolic enzymes are up-regulated in a Nod factor-dependent manner, providing a potential means to fine-tune GA biosynthesis and thereby regulate Nod factor signal output in a spatiotemporal or tissue-specific manner. GA2ox expression occurs concomitant with NIN and NSP2 induction approximately 3 hpi, consistent with a down-regulation of GA signaling that may facilitate Nod factor signaling. Conversely, in the 12- to 24-hpi window, we observed an up-regulation of enzymes of the GA biosynthetic pathway. This may serve as a mechanism to restrict infection by inhibiting the Nod factor signaling pathway. Alternatively, GA may positively affect nodule organogenesis. GA is classically known as a regulator of cell expansion via the DELLA proteins but was recently shown to also modulate cell cycle activity in Arabidopsis roots (Achard et al., 2009). Thus, GA may be required for cell cycle activation, leading to cell division and persistence of the nodule meristem. In this regard, pea (Pisum sativum) mutants affected in GA biosynthesis exhibit reduced nodulation and aberrant nodule meristem formation (Ferguson et al., 2011).
Cross Talk between Nod Factors, Hormone Biosynthesis, and the Lateral Root Developmental Network
This analysis identified putative regulators of LR development that were transcribed in a Nod factor-dependent, ET-regulated manner (Table I), and our network analysis suggests that many of these genes are associated with or regulated by essential symbiosis genes (Fig. 12D). This relationship may reflect the parallel activation of the LR developmental pathway by Nod factors, as observed by Oláh et al. (2005), and potentially the recruitment of LR genes to initiate nodule development (Markmann and Parniske, 2009; Desbrosses and Stougaard, 2011; Soyano and Hayashi, 2014). The skl mutant exhibits an approximately 10 times increase in the number of infections and a large increase in cortical cell divisions but manifests a reduced number of LRs when nodulated (Penmetsa and Cook., 1997; this study). Thus, the massive, Nod factor-induced up-regulation of LR genes in skl reflects the initiation of nodule primordia and not additional LR growth. Indeed, many of the Nod factor-induced LR genes are homologous to Arabidopsis genes that regulate cell fate and meristem patterning in root and LR meristems. LBD16, LBD18, and PUCHI are transcriptionally regulated by the auxin response factors ARF7 and ARF19 following auxin accumulation and are essential for LR initiation and patterning (Hirota et al., 2007; Kang et al., 2013). WOX5 is induced by auxin (Gonzali et al., 2005) and is essential for cells in the root meristem to maintain stem cell identity (Sarkar et al., 2007). WOX5 expression was previously observed in the root cortex following inoculation with rhizobium and may participate in the dedifferentiation of cortical cells to initiate nodule primordium formation (Osipova et al., 2012). In our experiment, WOX5 and WOX13 are both induced within 12 hpi, just before the onset of cortical cell division. Thus, our data are consistent with LR regulatory genes functioning in nodule organogenesis.
A common feature of many of the LR regulatory genes is that their expression is induced by auxin. While auxin biosynthesis can take place in the shoot and be translocated to the root via the phloem, it can also be synthesized locally. In root nodules, the activation of auxin-responsive promoters is correlated with initial cell divisions in pericycle and cortical cells in both determinate and indeterminate nodules (Grunewald et al., 2009). We observed the up-regulation of several genes encoding enzymes involved in auxin biosynthesis, including one ASA gene and several members of the YUCCA gene family. Their coordinated expression in our experiment suggests that auxin biosynthesis in roots is rapidly induced by rhizobium. Interestingly, our data here reveal hyperinduction of anthranilate synthase in skl, suggesting that ET is a negative regulator of auxin biosynthesis during symbiosis. This contrasts with results in Arabidopsis, where ET stimulates the expression of anthranilate synthase genes (ASA1 and ASB1; Stepanova et al., 2005). Taken together, these observations suggest a distinct relationship between ET and auxin in M. truncatula during nodulation, perhaps providing the signal to initiate the LR/nodule organogenesis program.
Nod Factors Stimulate ET Biosynthesis in an ET-Regulated Manner
Although ET is a well-established negative regulator of nodulation, gene families associated with ET biosynthesis have not been characterized in M. truncatula, nor has the impact of Nod factor perception on the ET biosynthesis pathway been described. Phylogenetic and conserved motif analysis identified nine ACS and six ACO genes in IMGA 3.5 version 4 of the M. truncatula genome. Several of these genes were expressed in roots, but only three ACSs (ACS4, ACS7, and ACS8) and a single ACO gene were induced upon inoculation in a Nod factor-dependent, ET-regulated manner. Because ACS catalyzes the rate-limiting step in ET biosynthesis, this strongly suggests that Nod factors stimulate ET biosynthesis and that ET-mediated negative feedback regulates this process. Moreover, ACS promoter:GUS constructs indicate that ET biosynthesis occurs in distinct symbiotic tissues and stages of development, implicating ET as a subtle regulator of early symbiotic processes. Basal expression of ACS in the epidermal layer of roots, particularly in the infection-competent zone, correlates well with the involvement of ET in epidermal cell differentiation and in the induction and growth of root hairs (Tanimoto et al., 1995; Růzicka et al., 2007; Swarup et al., 2007). Furthermore, the localized expression of ACS observed in root hairs undergoing arrested infections suggests that cell autonomous increases in ET may control infection thread progression and the establishment of a successful symbiotic interaction. This hypothesis fits with the observed hyperinfection phenotype in MtEIN2 (skl) mutants during nodulation (Penmetsa and Cook, 1997; Penmetsa et al., 2003, 2008). ACS promoters are also active in nodule primordia and in the meristematic zone of mature nodules. At these stages ET may act as a regulator of nodule elongation and growth, most likely through an interplay with other hormones. Such a dual role for ET at different symbiotic stages would extend the model of ET as a regulator of infection thread initiation and growth (Penmetsa and Cook, 1997) and is in agreement with a model in which nodulation becomes insensitive to exogenous ACC application after the appearance of macroscopic nodule primordia (Penmetsa and Cook, 1997; Oldroyd et al., 2001).
Nod Factor Perception Activates the ET Signaling Pathway in Roots
ET signaling is poorly characterized in legumes. Here, we identified homologs of ET signaling genes in M. truncatula and analyzed the impact of Nod factor and ET perception on their expression. EIN2 is a highly conserved ET signaling gene for which ortholog genes have been characterized in diverse plant species, including M. truncatula (Penmetsa et al., 2008). In Arabidopsis, ET perception induces EIN2 phosphorylation and cleavage at Ser-645, which allows the translocation of the EIN2 C terminus to the nucleus and the subsequent induction of ET-related gene transcription (Ju et al., 2012; Qiao et al., 2012). By analogy, a similar processing of EIN2 can be expected in M. truncatula. Although inoculation with rhizobium did not affect EIN2 transcription, differential phosphorylation of Ser-648 has been observed upon the application of isolated Nod factors in wild-type M. truncatula roots but, interestingly, not in an nfp mutant background (Rose et al., 2012; http://more.biotech.wisc.edu). This suggests that ET signaling is modulated through the Nod factor-triggered phosphorylation of EIN2, integrating both Nod factor and ET signaling in the same regulatory circuit.
Following inoculation, we observed highly amplified symbiotic responses in the ET-insensitive skl mutant (MtEIN2), likely due to both increased Nod factor sensitivity during early phases of the interaction (Oldroyd et al., 2001) and to the approximately 10 times larger number of root hairs undergoing sustained infection at later time points (i.e. after 24 h in our analysis; Penmetsa and Cook, 1997). In addition to the hyperinduction of symbiotic genes from G1, G2, and G3, a number of genes in G4 were transiently activated in the three ET-sensing genotypes but not in skl. Within this group, we identified homologs of the ET signaling cascade of M. truncatula, including four ET receptors, two EIN3-binding F-box proteins, and several members of the AP2/ERF family of TFs. Phylogenetic analysis of these G4 AP2/ERF genes, which were all down-regulated in skl, classifies them in group IX (Nakano et al., 2006), a clade known to contain members that bind to conserved GCC boxes at the promoters of plant defense and pathogen resistance genes (Zhou et al., 1997; Berrocal-Lobo et al., 2002). Notably, G4 is significantly enriched for defense genes, including the putative M. truncatula ortholog to AtERF1, a well-known transcriptional regulator of plant defense responses (Solano et al., 1998; Lorenzo et al., 2003).
Numerous studies describe a rapid defense response following inoculation with compatible rhizobium, including the production of reactive oxygen species, ion fluxes, accumulation of phenolic compounds, and induction of defense gene expression (Gamas et al., 1998; Ramu et al., 2002; Mitra et al., 2004). The transient induction of MtERF1 and other AP2/ERFs in M. truncatula ‘Jemalong A17’ but not in skl fits a model wherein defense-related gene induction serves to regulate the number of successful infections that develop into nodules. This model is consistent with the higher susceptibility of skl to pathogen attack and rhizobium infection (Penmetsa and Cook, 1997; Penmetsa et al., 2008).
Among the TFs up-regulated in a symbiosis-specific manner is the AP2/ERF ERN1, which activates the nodule organogenesis pathway downstream of CCaMK (Andriankaja et al., 2007; Middleton et al., 2007). Homologs with symbiotic function and lower expression levels than ERN1, namely ERN2 (Medtr6g029180) and ERN3 (Medtr8g085960), were also identified in our analysis but not found to be differentially expressed. Vernié et al. (2008) characterized a fourth AP2/ERF that is required for nodule differentiation at later nodule stages, EFD (contig_56456_1). In agreement with previous studies, our data show that EFD was expressed in M. truncatula roots but that EFD expression levels do not change following inoculation. Instead, we identified six new members of the AP2/ERF family induced in a symbiosis-dependent manner in M. truncatula, with kinetics similar to that of ERN1. We constructed promoter:GUS fusions for one of these genes, ERF12, and demonstrated its increased expression in the epidermis and root hairs 48 hpi and also in cells of the nodule periphery throughout nodule development. Other AP2/ERFs, namely Medtr4g119270 and Medtr7g020980, are close homologs of Arabidopsis AtPUCHI and AtERF115, respectively (Table I). AtPUCHI is an auxin-induced AP2/ERF that functions in LR initiation (Hirota et al., 2007), whereas AtERF115 functions in the maintenance of root meristems by regulating the division of quiescent cells (Heyman et al., 2013). Thus, these M. truncatula AP2/ERFs may connect the LR and nodule primordium developmental pathways. With the exception of ERN1, none of these Nod factor-induced AP2/ERF genes have been identified in forward genetic screens for nodulation mutants. The large size of the AP2/ERF gene family and the potential functional redundancy among members may explain their lack of discovery via forward genetic screens. Determining if and how these genes function during nodulation will be an important future challenge.
Identification of cis- and trans-Acting Factors That Control Transcriptional Regulatory Networks
We implemented MERLIN network analysis to identify regulatory relationships among the genes in groups G1, G2, G3, and G4. In recent years, bioinformatic and molecular analyses have identified DNA cis-elements targeted by several symbiotic TFs. NSP1 has been shown to bind to NODULATION-RESPONSIVE ELEMENT cis-elements in the promoters of ENOD11, ERN1, and NIN, while NSP2 likely binds to this region indirectly by way of its interaction with NSP1 (Hirsch et al., 2009). Likewise, NIN binds to the promoters of NF-YA1, CLE-RS1, CLE-RS2, and NPL in L. japonicus, while NF-YA1 binds to the ERN1 promoter in M. truncatula and is believed to coregulate its expression together with NSP1 and NSP2 (Xie et al., 2012; Soyano et al., 2013, 2014; Laloum et al., 2014). MERLIN predicts 4,393 regulator-target gene relationships, including several of these known associations. For instance, NIN is predicted to regulate ERN1 and vice versa (experimental work shows that this is done indirectly through NF-YA1), while NF-YA1 is predicted to regulate ENOD11 (via ERN1). The inferred network was used to prioritize regulators based on their connectivity to the full network as well as their connectivity specifically to genes already associated with symbiosis based on functional studies. Symbiotic genes clustered in module 227, with significant connectivity observed among known NIN, ERN1, NSP2, NF-YA1, LYK3, LYK4, and LYK5 and 56 new highly connected putative regulators of symbiosis.
MERLIN also identified numerous connections between the regulators of symbiosis and M. truncatula orthologs of Arabidopsis genes that control LR development (Fig. 12; Table I). Thus, NF-YA1 occurs in a network neighborhood that includes key LR regulators PUCHI, PLETHORA3, and WOX5 as well as the symbiotic flotillin FLOT2. PUCHI, PLETHORA3, and WOX5 are all required for proper LR development in Arabidopsis, whereas silencing M. truncatula FLOT2 affects both nodulation and the development of primary roots and LRs (Haney and Long, 2010). Interestingly, the symbiotic TFs NIN and ERN1 were closely associated with LBD16 in module 227. LBD16 activates the cell cycle leading to LR primordium initiation, and it may perform a similar function in mediating Nod factor-induced cell division leading to nodule primordia. In Arabidopsis, auxin mediates the derepression of the LR pathway by inducing the transcription of these key LR regulatory genes via the TFs ARF7 and ARF19. The Nod factor-induced expression of YUCCA and ASA genes suggests that the symbiotic activation of an LR/nodule pathway may also be auxin regulated. It is thus noteworthy that two ARF TFs (contig_130161_1 and TC202592) are also included in the NF-YA1/PUCHI/PLETHORA3/WOX5 subnetwork.
Further insights gained from this analysis include the significant enrichment of cis-elements in promoters of genes coexpressed with symbiosis markers, including the E2FA- and DEL3-binding motif in module 227. We hypothesize that these motifs function to regulate GO processes that are also enriched in module 227, namely cortical cell division and specific membrane transport mechanisms, for which transcripts are induced between 6 and 48 hpi (Fig. 12; Table II). Similarly, module 246 contains genes whose normal expression depends on ET signaling and is enriched for promoter motifs associated with processes in which ET is known to play a major role, including cellular development and elongation and seed maturation (Wang et al., 2002). We underscore the fact that cis-element resources for plants remain highly underdeveloped, often with meager validation. The fact that we find putative cis-element enrichment is an indication of the potential power of this approach, as information about validated cis-elements increases.
The magnitude and complexity of genome-scale expression data can complicate the identification of suitable candidate genes for functional characterization, a challenge that we have addressed using network analysis and related statistical tools. In defining regions within the symbiotic network that are regulatory hot spots or in close proximity to known symbiotic genes, MERLIN identified high-value targets for reverse genetic analysis. Forty-seven hubs in the main network manifest significantly higher connectivity within the network (at least 17 connections, which is greater than 3-fold that of the average gene), suggesting that these genes have a major effect on downstream symbiotic gene expression (Table III). Likewise, network-based gene prioritization using the MERLIN inferred network identified a subset of 56 uncharacterized regulatory genes adjacent to known symbiosis genes in module 227 that constitute additional candidates for reverse genetic experimentation (Table IV). The clustering of the essential and functionally related symbiosis genes NIN, NSP2, ERN1, and NF-YA1 within cluster 227 supports the robustness of our network model. We note, however, that NFP, a known key regulator of symbiosis, was not identified among the highly connected genes. Upstream regulators of a transcriptional cascade need not themselves be regulated along with the transcriptional response that they control, as may be the case for NFP. However, NFP transcription did respond to nodulation in the wild type, albeit at low levels, and its expression was markedly enhanced in skl, placing it in module 227. Importantly, the proximity of candidate regulatory genes to subnetworks enriched with genes of known function (Fig. 12D) may predict which biological processes are being regulated, thus facilitating the development of testable hypotheses.
CONCLUSION
This work describes the early dynamic changes occurring in the M. truncatula root transcriptome within minutes to hours after inoculation with symbiotic bacteria. The use of two Nod factor receptor mutants and an ET-insensitive hypernodulating mutant facilitated the exploration of both Nod factor- and ET-mediated responses. Moreover, double mutant analysis underscores the fact that many ET-mediated responses during nodulation are the consequence of Nod factor signaling, likely through Nod factor-induced provision of new ET biosynthetic, perception, and transcription systems. We conclude both that Nod factor-induced ET is the major negative regulatory circuit for nodulation and also that ET has other more subtle roles, sometimes serving to positively regulate transcription in a Nod factor-dependent manner, sometimes acting independent of Nod factor to up-regulate host defense response and also to modulate the basal transcription of certain Nod factor signaling components prior to Nod factor perception. The dynamic nature of ET during nodulation is suggested by the spatial distribution of ET biosynthetic and signaling gene transcription at sites of both successful and arrested rhizobial infections.
Furthermore, we provide evidence for the critical role of ET in the activation of other hormone pathways, including CK, GA, auxin, and strigolactone, as well as the involvement of genes related to LR growth. Such pathways were typically positively regulated by Nod factor and negatively regulated by ET. Network analysis of most ET- and symbiosis-responsive genes has identified numerous candidate regulatory genes, providing the basis for the selection and functional characterization of novel genes with a likely role in Nod factor signaling and early symbiotic processes. Information gathered in this work contributes toward a more complete understanding of the Nod factor and ET signaling cascades in M. truncatula and lays the foundation for future research efforts to functionally characterize candidate regulators during the early symbiotic interaction.
MATERIALS AND METHODS
Biological Material Growth, RNA Isolation, and Illumina Library Preparation
Medicago truncatula seeds were scarified, germinated and grown in aeroponic tanks (caissons), and inoculated as described in Supplemental Protocol S1. Five-day-old plantlets were inoculated with Sinorhizobium medicae ABS7M (pXLGD4), which harbors the β-galactosidase (lacZ) gene as a constitutive marker to a final optical density at 600 nm of 0.001. Four M. truncatula genotypes were used: wild-type cv Jemalong A17, nfp (C31; Amor et al., 2003), lyk3 (hcl-1, B56; Catoira et al., 2001), and skl (skl1-1; Penmetsa and Cook, 1997). Root samples were harvested at 0.5, 1, 3, 6, 12, 24, 36, and 48 hpi. Four independent biological replicates per time point and genotype were collected and immediately frozen in liquid nitrogen for RNA-seq analysis. A parallel experiment was run to verify the developmental stages of the roots. Roots were harvested, fixed, and stained using 5-bromo-4-chloro-3-indolyl-β-d-galacto-pyranoside as described previously (Lohar et al., 2006) using PIPES instead of HEPES buffer.
Total RNA was isolated from root tissues using the GeneAll HybridR+ kit (GeneAll), and mRNA was purified using the TruSeq RNA sample preparation kit (Illumina) according to the manufacturer’s instructions. Purified mRNA was chemically fragmented to 200-bp fragments, and the first and second strand complementary DNAs were synthesized, followed by end repair and index adapter ligation. The quality and quantity of the resulting libraries were evaluated using a Bioanalyzer 2100 (Agilent Technologies). Bar-coded libraries were pooled with up to 12 samples per flow cell lane for cluster generation on the Illumina flow cell using cBot isothermal amplification.
RNA Sequencing and Bioinformatic Analysis
Library clusters were sequenced on the Illumina HiSeq 2000 sequencer using the TruSeq SBS kit v3-HS to generate single-end sequences (100 bp). Real-time analysis and base calling were performed using the CASAVA 1.8.2 software package (Illumina). The number of reads collected from each library ranged from 11 to 32 million, after imposing a minimum Illumina quality score of 31 (Supplemental Fig. S1). Prior to full analysis, RNA-seq reads were preprocessed to remove low-quality reads and to trim adaptor sequences based on quality scores. Reads in which no adaptor sequences were detected were also discarded. The resulting quality-filtered single-end sequence reads were aligned to several reference genomic data sets, including (1) the M. truncatula genome IMGA 3.5 version 4 and (2) the M. truncatula Gene Index release 11.0, both frozen as of February 2012, (3) the Sinorhizobium meliloti 1021 genome (http://iant.toulouse.inra.fr/bacteria/annotation/cgi/rhime.cgi), and (4) the NCBI Plant Sequences Database. Alignment was conducted using CLC Assembly Cell 3.2 (CLC Bio) with a minimum similarity fraction of 0.9, a minimum length fraction of 0.9, and a maximum of two mismatches allowed. After the removal of sequences that mapped to multiple regions and correcting for redundancy among databases, 80.2% of the total reads were mapped, including 77.8% that had unique assignment to 69,237 genes in the IMGA 3.5 version 4 release of the M. truncatula genome. Mapped reads were normalized using the TMM (Robinson and Oshlack, 2010) method for each gene to report the expression of genes. The TMM process estimates scale factors from the raw data that can be used in downstream statistical analysis procedures such as the detection of differential expression. Data from biological replicates were used to evaluate variance and calculate expression statistics using edgeR, a software package implemented in the R/Bioconductor package (www.bioconductor.org). For comparison, a parallel analysis of expression was conducted using DESeq (Anders and Huber, 2010), yielding similar results (data not shown).
qPCR Analysis
To evaluate the Nod factor dependence of the ET response, qPCR was used to quantify expression in a fully independent transcriptional time course, comparing transcript levels among M. truncatula ‘Jemalong A17’, dmi1-1 (C71; Ané et al., 2004), skl, and the double mutant dmi1-1 skl. Analyses were conducted following the Minimum Information for Publication of Quantitative Real-Time PCR Experiments guidelines (Bustin et al., 2009) as described previously (Riely et al., 2013). For relative quantification, the comparative cycle threshold (Ct) method, also known as the 2–ƊƊCt method, was employed using the 26S proteasome regulatory subunit S5A (Medicago Gene Index TC108192) and a ubiquitin carrier protein (Medicago Gene Index TC176441) as reference genes. Similar results were obtained for both reference genes, so the data presented here represent relative expression values calculated using TC108192. A list of primers used in this work is provided in Supplemental Table S9.
Promoter Cloning, Hairy Root Transformation, and Histochemical Staining for GUS Activity
DNA fragments containing 1.2 to 2 kb upstream of the predicted coding regions of the three ACS genes (ACS4, contig_12436_1; ACS7, Medtr8g101820; and ACS8, Medtr5g015020) and ERF12 (Medtr2g014300) were amplified using the primer pairs listed in Supplemental Table S9 and cloned into entry vectors. Promoter regions were then cloned either into the HindIII and KpnI sites of the binary plasmid pLP100 (Boisson-Dernier et al., 2001) for the ACS constructs or into pBGWFS7 (Karimi et al., 2002) using the Gateway system for ERF12 to make transcriptional fusions with the uidA (GUS) gene. Plasmids were electroporated into the Agrobacterium rhizogenes strain ARqua1, and transformed hairy roots were generated in M. truncatula ‘Jemalong A17’ as described previously (Boisson-Dernier et al., 2001). Four-week-old transformed plantlets were transferred to pots containing a 1:1 perlite:vermiculite mixture and inoculated with S. meliloti 2011 constitutively expressing GFP (Limpens et al., 2003). Prior to staining for GUS activity, roots were prefixed under vacuum in 0.3% (w/v) paraformaldehyde in 100 mm sodium phosphate buffer (pH 7) for 30 min. After washing in phosphate buffer, roots were immersed in GUS staining solution (1 mm X-Gluc, 0.5 mm potassium ferricyanide, 0.5 mm potassium ferrocyanide, 5 mm EDTA, and 100 mm sodium phosphate buffer, pH 7) and incubated at 37°C in the dark from 2 h to overnight. Uninoculated roots were washed in phosphate buffer, cut, and mounted on glass slides in 50% glycerol for microscopic observation. Nodules were embedded in 4% (w/v) agarose, and 100-µm sections were made using a vibratome. Roots and nodule sections were imaged on either an Olympus SZX16 stereomicroscope or a Leica DM5000B compound fluorescence microscope (JH Technologies), and images were collected using SPOT Advanced software version 4.1.
MERLIN Analysis of Regulatory Networks
The MERLIN network inference algorithm (Roy et al., 2013) was applied on genes in groups G1 to G4, specifying as input 1,734 signaling proteins or TFs as putative regulators of the network. The algorithm was run 30 times with different initializations to evaluate the consistency of the inferred regulatory connections. A high-confidence network was derived by selecting regulatory connections observed in 60% or more of the random initializations. Consensus modules based on genes that cooccurred in the same module with a frequency of 50% or higher were obtained using a consensus clustering approach (Knaack et al., 2014). After defining modules, regulators were assigned based on a hypergeometric statistical test using the Benjamini-Hochberg procedure to control for FDR < 0.05. The same statistical analysis was applied to examine modules for the enrichment of GO processes and binding sites of TFs annotated in publicly available motif databases (Athamap, Agris, PLACE, PPDB, and JASPAR). The prediction of new regulators and subnetwork identification were carried out as described in Supplemental Protocol S1.
The RNA-seq data used in this article have been deposited in NCBI’s Bioproject collection under the Bioproject identifier PRJNA269201 (SRR1726469–SRR1726612).
Supplemental Data
The following supplemental materials are available.
Supplemental Figure S1. Number of Illumina reads per sample.
Supplemental Figure S2. Curve for the determination of the FDR value.
Supplemental Figure S3. Venn diagram representing the set of differentially expressed genes across genotypes.
Supplemental Figure S4. qPCR data on ENOD11, NIN, and ERN1 expression at early symbiotic stages (0–48 hpi).
Supplemental Figure S5. Schematic representation of the CK, ABA, GA, and strigolactone biosynthetic pathways and their expression in the RNA-seq experiment.
Supplemental Table S1. Normalized TMM count values for the 10,985 genes identified as differentially expressed in the RNA-seq experiment.
Supplemental Table S2. Studio Pathway GO functional overrepresentation analysis.
Supplemental Table S3. Expression of symbiosis-related genes.
Supplemental Table S4. List of members of the AP2/ERF family found differentially expressed in the RNA-seq experiment.
Supplemental Table S5. Description of the main clusters identified by MERLIN.
Supplemental Table S6. MERLIN regulatory network using genes in groups G1 to G4.
Supplemental Table S7. Gene composition in cluster 227.
Supplemental Table S8. Subnetworks identified in cluster 227.
Supplemental Table S9. List of primers used.
Supplemental Protocol S1. Biological material growth, prediction of new regulators based on network proximity to known symbiosis genes, and novel subnetwork identification.
Glossary
- hpi
hours post inoculation
- TF
transcription factor
- ET
ethylene
- RNA-seq
transcriptome sequencing
- FDR
false discovery rate
- GO
Gene Ontology
- qPCR
quantitative real-time PCR
- NCBI
National Center for Biotechnology Information
- LR
lateral root
- MEP
2-C-methyl-d-erythritol 4-phosphate
- IPP
isopentenyl pyrophosphate
- DMAPP
dimethylallyl pyrophosphate
- CK
cytokinin
- ABA
abscisic acid
- GGPP
geranyl geranyl pyrophosphate
- JA
jasmonic acid
- ACC
1-aminocyclopropane-1-carboxylic acid
- TMM
Trimmed Mean of M component
- X-Gluc
5-bromo-4-chloro-3-indolyl-β-glucuronic acid
LITERATURE CITED
Fraley C, Raftery AE (2006) MCLUST version 3: an R package for normal mixture modeling and model-based clustering. Technical report no. 504. Department of Statistics, University of Washington, Seattle, WA
Author notes
This work was supported by the Woo Jang Choon Project of the Rural Development Administration of Korea (grant no. PJ906910 to J.-H.M. and D.R.C.), by the Basic Science Research Program through the National Research Foundation of Korea, funded by the Ministry of Education (grant no. 2013R1A1A2060244 to J.-H.M.), by the U.S. National Science Foundation (grant no. IOS–1122261 to B.K.R. and D.R.C. and grant no. IOS–1237936 to S.R.), and by a Marie Curie International Outgoing fellowship (SymBioSignal; to E.L.).
These authors contributed equally to the article.
Present address: Environmental Sciences Department, Public University of Navarra, Campus Arrosadia s/n, 31006 Pamplona, Spain.
Present address: Samsung Genome Institute, Yangjae Road, Seoul 135–710, Republic of Korea.
Address correspondence to [email protected] and [email protected].
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (www.plantphysiol.org) is: Douglas R. Cook ([email protected]).
E.L., B.K.R., J.-H.M., and D.R.C. conceived the study; E.L., B.K.R., S.C.K., N.C.-G., H.-J.H., M.O., G.B.K., A.K.S., and R.V.P. performed the experiments; E.L., B.K.R., S.C.K., H.-J.Y., D.C., A.F.S., G.-S.L., N.K., S.R., J.-H.M., and D.R.C. analyzed the data; E.L., B.K.R., S.R., J.-H.M., and D.R.C. wrote and revised the article.
Articles can be viewed without a subscription.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}