





Abstract
The cell wall consists of cellulose microfibrils embedded within a matrix of hemicellulose and pectin. Cellulose microfibrils are synthesized at the plasma membrane, whereas matrix polysaccharides are synthesized in the Golgi apparatus and secreted. The trafficking of vesicles containing cell wall components is thought to depend on actin-myosin. Here, we implicate microtubules in this process through studies of the kinesin-4 family member, Fragile Fiber1 (FRA1). In an fra1-5 knockout mutant, the expansion rate of the inflorescence stem is halved compared with the wild type along with the thickness of both primary and secondary cell walls. Nevertheless, cell walls in fra1-5 have an essentially unaltered composition and ultrastructure. A functional triple green fluorescent protein-tagged FRA1 fusion protein moves processively along cortical microtubules, and its abundance and motile density correlate with growth rate. Motility of FRA1 and cellulose synthase complexes is independent, indicating that FRA1 is not directly involved in cellulose biosynthesis; however, the secretion rate of fucose-alkyne-labeled pectin is greatly decreased in fra1-5, and the mutant has Golgi bodies with fewer cisternae and enlarged vesicles. Based on our results, we propose that FRA1 contributes to cell wall production by transporting Golgi-derived vesicles along cortical microtubules for secretion.
The cell wall plays a vital role in the life of a plant. In growing cells, the tough but extensible primary wall determines the rate and direction of expansion and overall plant form. In differentiated cells, such as interfascicular fibers and xylem cells, the thick secondary cell wall provides strength to withstand gravity and large negative pressures. Other than mechanics, cell walls feature in essential processes, such as pathogen resistance, signal transduction, and cell-to-cell communication. In addition, cell wall biomass has potential as a feedstock for biofuel production. Therefore, understanding cell wall biogenesis is important fundamentally and practically.
Cell walls consist primarily of cellulose, hemicellulose, and pectin along with small amounts of protein. Typical of eukaryotic secretory products, hemicellulose and pectin are synthesized in the Golgi and then delivered to the extracellular space through the secretory system. Atypically, cellulose microfibrils are synthesized de novo at the plasma membrane by cellulose synthase (CESA) complexes, although the CESA complexes themselves are thought to be assembled in the Golgi and trafficked to the plasma membrane (McFarlane et al., 2014). How the various cell wall components are delivered to the plasma membrane and extracellular space to form a functional cell wall remains poorly understood.
Among cell wall components, cellulose has perhaps the best understood delivery process, which is influenced by cortical microtubules (Baskin, 2001; Lloyd, 2011). The cortical microtubule array organizes cellulose deposition spatially by targeting the secretion of CESA complexes (Crowell et al., 2009; Gutierrez et al., 2009) and orienting their movement (Gardiner et al., 2003; Paredez et al., 2006). What controls the delivery of other wall components is less clear. Sustained transport of organelles in plants is actin based (Sparkes, 2011), and vesicle trafficking is generally assumed to be independent of microtubules, at least during interphase. Nevertheless, in xylem tracheary cells, cortical microtubule bands have been linked to not only cellulose guidance but also, the targeted exocytosis of hemicellulose and other matrix components (Fukuda, 1997). In seed coat cells, vesicles containing pectin associate with cortical microtubules that line the mucilage secretion pockets (McFarlane et al., 2008). In addition, in maize (Zea mays) roots, vesicles bind cortical microtubules densely (Tian et al., 2004). These and other observations indicate that cortical microtubules might serve as roadways for trafficking secretory vesicles.
If microtubules are tracks, then the engines are kinesins, because seed plants lack dyneins (Zhu and Dixit, 2012). Kinesins are molecular motors that move along microtubules and transport various cargo, including organelles, vesicles, and proteins. Kinesins have proliferated in plant lineages, and many are expressed during interphase, which is surprising given that long-distance organelle motility is thought to be actin based. Recently, a particular plant kinesin of the kinesin-4 family, called Fragile Fiber1 (FRA1), was shown to move rapidly and processively (i.e. taking multiple steps) toward microtubule plus ends in vitro (Zhu and Dixit, 2011). This makes FRA1 a candidate for sustained and active vesicle transport.
An Arabidopsis (Arabidopsis thaliana) partial loss-of-function mutant, fra1-1, was reported to have altered cellulose organization in fiber cells, despite having evidently undisturbed cortical microtubule organization (Zhong et al., 2002). Those results suggested a function for FRA1 in cell wall organization rather than secretion, and FRA1 has been proposed to link motile CESA complexes in the plasma membrane to cortical microtubules (Zhong et al., 2002; Lloyd and Chan, 2004; Zhu and Dixit, 2011). Strengthening this suggestion, a null mutant of the FRA1 ortholog in rice (Oryza sativa), brittle culm12 (bc12), also was reported to have disorganized sclerenchyma cell walls (Zhang et al., 2010). However, the motility of FRA1 in vivo and its relationship to CESA complexes remain unknown.
We have reexamined FRA1 function in cell wall formation. Because the originally characterized allele, fra1-1, is predicted to give rise to a nearly full-length protein, we characterized a transfer DNA (T-DNA)-induced knockout mutant, fra1-5. Using this allele as well as imaging a functional FRA1-3GFP fusion protein, we show here that FRA1 is involved in membrane trafficking that contributes to delivery of cell wall polysaccharides, such as pectin. We propose that FRA1 drives the movement of vesicles containing cell wall cargo along cortical microtubules to facilitate their secretion.
RESULTS
The fra1-5 Knockout Mutant Has Decreased Elongation
The previously studied ethyl methanesulfonate (EMS)-induced fra1-1 allele is predicted to cause a 28-amino acid deletion (Zhong et al., 2002) and did not ablate FRA1 expression (Fig. 1A). To obtain a null allele, we identified a T-DNA insertion line (SALK_084463), in which the T-DNA resides in the second exon of the FRA1 gene. This mutant lacked an FRA1 transcript detectable by reverse transcription (RT)-PCR (Fig. 1A). In addition, the mutant did not express detectable FRA1 protein on an immunoblot probed with a polyclonal antiserum against FRA1 (Fig. 1B), confirming that it is a knockout. Because Zhong et al. (2002) mentioned three other EMS alleles, we designated the T-DNA mutant fra1-5.
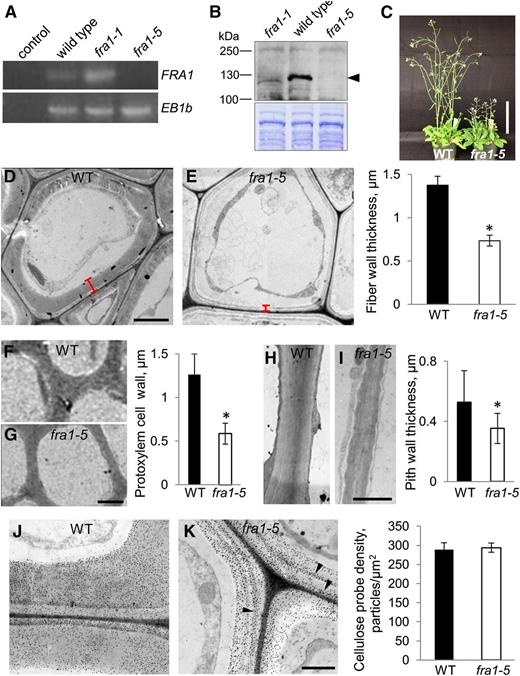
Phenotype of the fra1-5 knockout mutant. All data are for 4-week-old, continuous light-grown plants. A, RT-PCR analysis in rosette leaves. The control lane lacks RT. End Binding 1b expression is used as a loading control. B, Immunoblot of total protein extracts probed with an anti-FRA1 antibody. The arrowhead marks the expected position of FRA1. The Coomassie-stained gel is shown below. C, Whole-plant appearance. Bar = 10 cm. D and E, Transmission electron micrographs of fiber cells in basal stems. Bars plot means ± sd (n > 30 cells). Red bars indicate the width of the cell wall. Bar = 5 µm. F and G, Transmission electron micrographs of protoxylem cells in basal stems. Bars plot means ± sd (n > 40 cells). Bar = 2 µm. H and I, Transmission electron micrographs of pith cell walls in basal stems. Bars plot means ± sd (n > 40 cells). Bar = 0.5 µm. J and K, Transmission electron micrographs of fiber cell walls labeled with gold-conjugated cellobiohydrolase2. Arrowheads point to layers of high-density labeling. Bars plot means ± sd (n > 21 cells). Bar = 1 µm. WT, Wild type; asterisk, significant differences between the genotypes as determined by Student’s t test (P < 0.01).
The fra1-5 inflorescence stems elongated significantly more slowly than those of the wild type and ceased growth at about the same time, giving rise to a large difference in final stem length (Fig. 1C; Supplemental Fig. S1A). In addition, fra1-5 inflorescence stems were wider than wild-type controls (Supplemental Fig. S1B), indicating a decreased degree of expansion anisotropy. The alteration of organ length and width was recapitulated closely in the alterations of the lengths and widths of pith cells (Supplemental Fig. S1, C–E), indicating that the morphological phenotypes observed can be plausibly accounted for by defective expansion.
Rosette leaves and siliques of the fra1-5 mutant were also substantially shorter than those of the wild type (Supplemental Fig. S1, F and G). Interestingly, fra1-5 seedlings had only slightly shorter hypocotyls in dark-grown seedlings and slightly shorter roots in both light- and dark-grown seedlings (Supplemental Fig. S1, H and I). The reason for the relative insensitivity of the seedling organs to the loss of FRA1 function might be because of genetic redundancy with a pair of FRA1 homologs present in the genome.
The fra1-5 Mutant Has Mechanically Weaker Stems and Irregular Xylem
The inflorescence stems of fra1-5 were fragile during handling, which has been reported for fra1-1 (Zhong et al., 2002). To quantify mechanical properties of the stem, we performed three-point bending assays, because these tests are prone to fewer artifacts than breakage tests. During these experiments, fra1-5 stems always broke, whereas wild-type stems did not (Supplemental Fig. S2A). The fra1-5 stems had a reduced bending strength and bending modulus, with the latter reduced by more than a factor of two (Supplemental Fig. S2B). Mechanical defects also occurred in fra1-5 xylem cells, which had an abnormal, collapsed appearance (Supplemental Fig. S2C), similar to the appearance of the irregular xylem mutants that synthesize less secondary cell wall cellulose than the wild type (Turner and Somerville, 1997).
Cell Walls of fra1-5 Are Thin, with Minor Changes in Chemical Composition
Reduced elongation and altered mechanical properties are typically associated with defective cell wall assembly. To determine whether the fra1-5 mutant had aberrant cell walls, we examined its cell wall thickness and composition.
In transmission electron micrographs of sections from 4-week-old basal inflorescence stems, interfascicular fiber cell walls were consistently about 50% thinner in fra1-5 compared with the wild type (Fig. 1, D and E). A similar decrease in wall thickness also occurred in protoxylem cells (Fig. 1, F and G) and pith cells (Fig. 1, H and I). Evidently, FRA1 functions in the deposition of both primary and secondary cell walls. In the basal stems of 6-week-old plants, fra1-5 had one-third fewer fiber cell tiers than wild-type stems (Supplemental Fig. S3). Reduced cell wall thickness combined with fewer tiers of lignified cells likely explain the mechanical weakness of the mutant stems.
Turning to cell wall composition, we first studied the distribution of cellulose in the cell walls of interfascicular fiber cells by staining sections with gold particles coated with cellobiohydrolase 2, a probe that binds crystalline cellulose (Berg et al., 1988). In fiber cells, the mean density of the probe was not particularly different between the genotypes; however, the staining was relatively uniform in wild-type cell walls but appeared uneven or striated in mutant walls (Fig. 1, J and K).
Using 4-week-old inflorescence stems, we next analyzed cell wall composition. At 4 weeks, the apical region is elongating in both genotypes, whereas the basal region has stopped elongating (Supplemental Fig. S1A), allowing cell wall composition to be compared between growing and nongrowing tissues as well as between genotypes. Mutant walls had similar amounts of cellulose in both apical and basal stems and a modest reduction in lignin in basal stems (Table I). As for the neutral sugars present in the noncellulosic polysaccharides, mutant walls had significantly more Ara in both apical and basal stems and more Xyl in the apical but not basal stem (Table II). Although these differences could mean that specific polysaccharides were altered in the mutant, the essentially constant composition per total cell wall mass combined with the substantial decrease in cell wall thickness lead us to hypothesize that FRA1 acts to mediate overall cell wall polymer secretion.
Cellulose and lignin content of wild-type and fra1-5 plants
Alcohol insoluble residue (AIR) was prepared from apical and basal stems of wild-type and fra1-5 plants. Values are means ± sem of three biological replicates. *, Significant difference between the genotypes as determined by Student’s t test (P < 0.001).
| Tissue Source | Cellulose AIR | Lignin ABSLa |
|---|---|---|
| μg mg−1 | % | |
| Apical stem | ||
| Wild type | 182.6 ± 9.5 | —b |
| fra1-5 | 164.9 ± 1.2 | —b |
| Basal stem | ||
| Wild type | 421.7 ± 14.4 | 10.9 ± 1.0 |
| fra1-5 | 402.1 ± 2.7 | 9.1 ± 0.2* |
| Tissue Source | Cellulose AIR | Lignin ABSLa |
|---|---|---|
| μg mg−1 | % | |
| Apical stem | ||
| Wild type | 182.6 ± 9.5 | —b |
| fra1-5 | 164.9 ± 1.2 | —b |
| Basal stem | ||
| Wild type | 421.7 ± 14.4 | 10.9 ± 1.0 |
| fra1-5 | 402.1 ± 2.7 | 9.1 ± 0.2* |
Ten percent acetyl bromide soluble lignin (ABSL) is equal to 100 μg mg−1AIR.
Lignin in apical stems is close to the detection limit; therefore, it is not reported here.
Alcohol insoluble residue (AIR) was prepared from apical and basal stems of wild-type and fra1-5 plants. Values are means ± sem of three biological replicates. *, Significant difference between the genotypes as determined by Student’s t test (P < 0.001).
| Tissue Source | Cellulose AIR | Lignin ABSLa |
|---|---|---|
| μg mg−1 | % | |
| Apical stem | ||
| Wild type | 182.6 ± 9.5 | —b |
| fra1-5 | 164.9 ± 1.2 | —b |
| Basal stem | ||
| Wild type | 421.7 ± 14.4 | 10.9 ± 1.0 |
| fra1-5 | 402.1 ± 2.7 | 9.1 ± 0.2* |
| Tissue Source | Cellulose AIR | Lignin ABSLa |
|---|---|---|
| μg mg−1 | % | |
| Apical stem | ||
| Wild type | 182.6 ± 9.5 | —b |
| fra1-5 | 164.9 ± 1.2 | —b |
| Basal stem | ||
| Wild type | 421.7 ± 14.4 | 10.9 ± 1.0 |
| fra1-5 | 402.1 ± 2.7 | 9.1 ± 0.2* |
Ten percent acetyl bromide soluble lignin (ABSL) is equal to 100 μg mg−1AIR.
Lignin in apical stems is close to the detection limit; therefore, it is not reported here.
Neutral monosaccharide composition of wild-type and fra1-5 plants
Alcohol insoluble residue (AIR) was prepared from apical and basal stems of wild-type and fra1-5 plants. Values are means ± sem of three biological replicates. *, Significant differences between the genotypes as determined by Student’s t test (P is approximately 0.001).
| Neutral Monosaccharide Composition AIR | |||||||
|---|---|---|---|---|---|---|---|
| Tissue Source | Rha | Fuc | Ara | Xyl | Man | Gal | Glc |
| μg mg−1 | |||||||
| Apical | |||||||
| Wild type | 14.3 ± 0.7 | 3.2 ± 0.1 | 51.8 ± 1.3 | 33.1 ± 0.1 | 12.8 ± 0.6 | 60.9 ± 0.1 | 21.5 ± 0.4 |
| fra1-5 | 14.5 ± 0.8 | 3.2 ± 0.1 | 71.0 ± 6.5* | 41.8 ± 4.6* | 12.7 ± 0.1 | 64.5 ± 3.4 | 22.1 ± 1.0 |
| Basal | |||||||
| Wild type | 9.2 ± 0.4 | 1.6 ± 0.1 | 14.4 ± 0.3 | 165.0 ± 5.2 | 11.6 ± 0.2 | 22.2 ± 0.6 | 17.6 ± 0.8 |
| fra1-5 | 9.4 ± 0.4 | 1.7 ± 0.1 | 16.7 ± 0.3* | 164.0 ± 4.9 | 10.4 ± 0.4 | 23.1 ± 0.4 | 16.0 ± 1.3 |
| Neutral Monosaccharide Composition AIR | |||||||
|---|---|---|---|---|---|---|---|
| Tissue Source | Rha | Fuc | Ara | Xyl | Man | Gal | Glc |
| μg mg−1 | |||||||
| Apical | |||||||
| Wild type | 14.3 ± 0.7 | 3.2 ± 0.1 | 51.8 ± 1.3 | 33.1 ± 0.1 | 12.8 ± 0.6 | 60.9 ± 0.1 | 21.5 ± 0.4 |
| fra1-5 | 14.5 ± 0.8 | 3.2 ± 0.1 | 71.0 ± 6.5* | 41.8 ± 4.6* | 12.7 ± 0.1 | 64.5 ± 3.4 | 22.1 ± 1.0 |
| Basal | |||||||
| Wild type | 9.2 ± 0.4 | 1.6 ± 0.1 | 14.4 ± 0.3 | 165.0 ± 5.2 | 11.6 ± 0.2 | 22.2 ± 0.6 | 17.6 ± 0.8 |
| fra1-5 | 9.4 ± 0.4 | 1.7 ± 0.1 | 16.7 ± 0.3* | 164.0 ± 4.9 | 10.4 ± 0.4 | 23.1 ± 0.4 | 16.0 ± 1.3 |
Alcohol insoluble residue (AIR) was prepared from apical and basal stems of wild-type and fra1-5 plants. Values are means ± sem of three biological replicates. *, Significant differences between the genotypes as determined by Student’s t test (P is approximately 0.001).
| Neutral Monosaccharide Composition AIR | |||||||
|---|---|---|---|---|---|---|---|
| Tissue Source | Rha | Fuc | Ara | Xyl | Man | Gal | Glc |
| μg mg−1 | |||||||
| Apical | |||||||
| Wild type | 14.3 ± 0.7 | 3.2 ± 0.1 | 51.8 ± 1.3 | 33.1 ± 0.1 | 12.8 ± 0.6 | 60.9 ± 0.1 | 21.5 ± 0.4 |
| fra1-5 | 14.5 ± 0.8 | 3.2 ± 0.1 | 71.0 ± 6.5* | 41.8 ± 4.6* | 12.7 ± 0.1 | 64.5 ± 3.4 | 22.1 ± 1.0 |
| Basal | |||||||
| Wild type | 9.2 ± 0.4 | 1.6 ± 0.1 | 14.4 ± 0.3 | 165.0 ± 5.2 | 11.6 ± 0.2 | 22.2 ± 0.6 | 17.6 ± 0.8 |
| fra1-5 | 9.4 ± 0.4 | 1.7 ± 0.1 | 16.7 ± 0.3* | 164.0 ± 4.9 | 10.4 ± 0.4 | 23.1 ± 0.4 | 16.0 ± 1.3 |
| Neutral Monosaccharide Composition AIR | |||||||
|---|---|---|---|---|---|---|---|
| Tissue Source | Rha | Fuc | Ara | Xyl | Man | Gal | Glc |
| μg mg−1 | |||||||
| Apical | |||||||
| Wild type | 14.3 ± 0.7 | 3.2 ± 0.1 | 51.8 ± 1.3 | 33.1 ± 0.1 | 12.8 ± 0.6 | 60.9 ± 0.1 | 21.5 ± 0.4 |
| fra1-5 | 14.5 ± 0.8 | 3.2 ± 0.1 | 71.0 ± 6.5* | 41.8 ± 4.6* | 12.7 ± 0.1 | 64.5 ± 3.4 | 22.1 ± 1.0 |
| Basal | |||||||
| Wild type | 9.2 ± 0.4 | 1.6 ± 0.1 | 14.4 ± 0.3 | 165.0 ± 5.2 | 11.6 ± 0.2 | 22.2 ± 0.6 | 17.6 ± 0.8 |
| fra1-5 | 9.4 ± 0.4 | 1.7 ± 0.1 | 16.7 ± 0.3* | 164.0 ± 4.9 | 10.4 ± 0.4 | 23.1 ± 0.4 | 16.0 ± 1.3 |
Both Cortical Microtubule and Cell Wall Organizations Are Essentially Unaltered in fra1-5
The original characterization of FRA1 reported that microtubule organization in fra1-1 plants was similar to that of wild-type plants (Zhong et al., 2002). We confirmed this for fra1-5 in both roots and hypocotyls (Supplemental Fig. S4, A and B) and in addition, for hypocotyl cells, found that the wild type and fra1-5 have indistinguishable rates of both growth and shortening at cortical microtubule plus ends (Supplemental Fig. S4C). Furthermore, roots of the mutant responded to oryzalin no differently than the wild type did (Supplemental Fig. S4D). Thus, neither microtubule organization nor dynamics seem to require FRA1 function.
To determine whether cellulose organization is disrupted in fra1-5, we used scanning electron microscopy to image the innermost cell wall layer. Because fra1-5 stems grow with decreased anisotropy, we first examined pith cells in the apical (growing) region of the inflorescence stem. Note that, although the epidermis is argued to play a starring role in controlling the rate of stem elongation, pith cells command the spotlight for anisotropy (Baskin and Jensen, 2013). The innermost cell wall layer seemed highly organized in both genotypes (Fig. 2, A and B). To determine whether the genotypes differ quantitatively in wall organization, we analyzed orientation as a function of feature size in the images by using a Fourier transform-based method (Marga et al., 2005). Although the wild type had slightly more uniformly oriented cell walls than the fra1-5 mutant for features around 20 nm, the difference was invisible by eye. Considering the 50% difference in elongation rate, cell wall organization in the two genotypes was strikingly similar (Fig. 2C). Additionally, the net orientation of the cell wall fibrils, recovered in the same analysis, for each genotype averaged to 90° (i.e. transverse to the long axis of the stem), although the distribution of angles was modestly wider for fra1-5 (Fig. 2D). The difference in distribution was small but statistically significant; it indicates a difficulty among cells in fra1-5 stems to achieve a strictly transverse orientation of microfibrils. Similar patterns occur in roots growing with reduced anisotropy and have been hypothesized to weaken transverse mechanical reinforcement and hence, increase radial growth (Baskin, 2005).
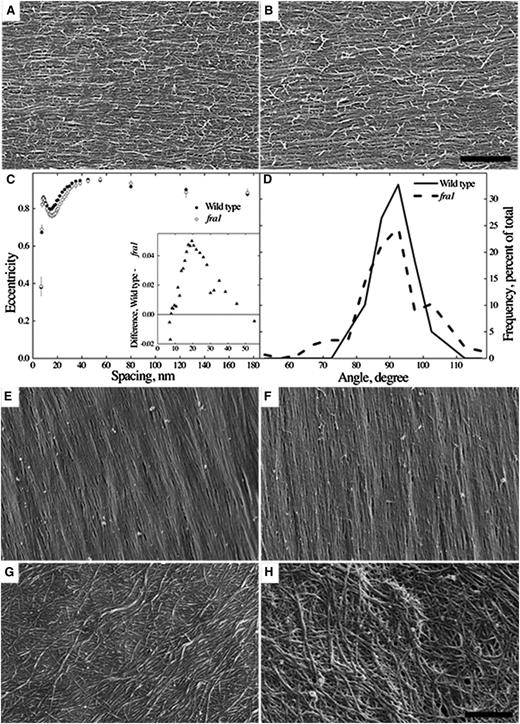
Cell wall organization. A and B, Representative images of the newly deposited cell wall layer in pith cells from the apical region of 4-week-old wild-type (A) and fra1-5 (B) inflorescence stems. Bar = 500 nm. C, Eccentricity of the Fourier transform as a function of spatial frequency. A value of 0 indicates complete radial symmetry, and a value of 1 indicates perfect alignment. Symbols plot means ± sem of four stems per genotype, with about 40 images per stem (approximately 150 images total per genotype). C, Inset shows the difference for the curves in C over a selected range of frequency. D, Distribution of the net orientation for the images used in C. Orientation per image was averaged for features between 30 and 60 nm. Homogeneity of variance is rejected with P < 0.001 using Levene’s test (Supplemental Methods S1). E to H, Representative images of cell wall organization in interfascicular fiber cells from the basal region of 4-week-old wild-type (E and G) and fra1-5 (F and H) inflorescence stems. Bar = 500 nm.
We next imaged interfascicular fiber cells in basal regions of the stem to study secondary cell walls. Cell wall organization among these fiber cells was surprisingly variable: some were highly organized (Fig. 2, E and F), but others were disorganized, resembling nongrowing parenchyma (Fig. 2, G and H). The variability precluded quantitative analysis, but cell wall organization could not be distinguished between the two genotypes qualitatively. The presence of numerous fra1-5 fiber cells with highly organized walls contradicts the previous claim that FRA1 is essential for fiber cell wall organization (Zhong et al., 2002). Taken together, the scanning electron microscopy analyses indicate that cell wall organization is unlikely to account for the mechanical weakness of fra1-5 stems.
FRA1 Moves Processively along Cortical Microtubules and Its Motility Correlates with Expansion
To understand the function of FRA1, we studied its localization and dynamics by expressing full-length FRA1 fused to a triple GFP tag under the control of the native FRA1 promoter. The FRA1-3GFP construct essentially complemented fra1-5 transcriptionally and morphologically (Fig. 3, A–C), indicating that the resulting fusion protein is functional. To determine whether FRA1 is motile in vivo, we imaged living cells with variable-angle epifluorescence microscopy (Konopka and Bednarek, 2008). In cotyledon and hypocotyl epidermis, FRA1-3GFP localized to puncta at the cortex (Figs. 3E and 4A). The cortical FRA1-3GFP particles were dynamic, undergoing diffusive and directional movement (Fig. 3F; Supplemental Movies S1–S4). Directional movement of FRA1-3GFP occurred at an average velocity of 0.24 μm s−1 and a characteristic run length of 2.4 μm (Fig. 3, H and I). Single-molecule photobleaching analysis showed that the linearly moving FRA1-3GFP particles consist primarily of one or two FRA1 dimers (Fig. 3J), consistent with kinesin-driven motility in other systems (Hendricks et al., 2010).
![Motility of FRA1-3GFP in vivo. A, Overall appearance of 4-week-old plants grown under continuous light. Bar = 10 cm. B, Transcript level in rosette leaves measured by qRT-PCR and normalized to the FRA1 expression level in the wild type. Values are means ± sem from three biological replicates. C, Stem height of 4-week-old plants. Values are means ± sd (n = 20 plants). D, Representative image of FRA1-3GFP (green) in a vascular cell of the root of a 4-d-old seedling. Fei Mao4-64 (FM4-64) (red) was used to label the plasma membrane. Bar = 2 µm. E, Image of FRA1-3GFP in a cotyledon pavement cell of a 4-d-old seedling (same in E–J). Bar = 10 µm. F, Representative kymograph showing the movement of FRA1-3GFP. Diagonal lines represent motile events. Vertical bar = 20 s; horizontal bar = 1 μm. G, Dual-channel image of FRA1-3GFP (green) and RFP-TUB6 (red). The arrowheads label FRA1-3GFP puncta along a cortical microtubule. Bar = 10 µm. H, Distribution of the velocity of processive FRA1-3GFP puncta. The average velocity is 0.24 ± 0.09 μm s−1 (means ± sd, n = 233). I, Distribution of the run length of FRA1-3GFP puncta. The characteristic run length (L 1/2) is 2.4 μm (n = 228). J, Distribution of the number of GFP fluorophores in motile FRA1-3GFP puncta (n = 45). The puncta that contain 6 and 12 GFP fluorophores indicate one and two FRA1-3GFP dimers, respectively. The FRA1-3GFP puncta that contain nine GFP fluorophores probably represent two FRA1-3GFP dimers that got photobleached before image acquisition. K, Time projections of 200 images of FRA1-3GFP from pavement cells of seedlings treated as indicated. Linear tracks in these images indicate motility of FRA1-3GFP. Insets show the cortical microtubules after the stated treatments. Bar = 10 µm. L, Motile parameters of FRA1-3GFP from the experiments shown in K. Values are means ± sd (n = 233, n = 107, and n = 87 molecules for control, taxol, and latrunculin-B [Lat-B], respectively). NA, Not applicable; WT, wild type; asterisk, significant difference from control as determined by Student’s t test (P < 0.01).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/plphys/167/3/10.1104_pp.114.251462/2/m_plphys_v167_3_780_f3.jpeg?Expires=1750195246&Signature=VS77C4yE6sCzSueegrVmL18vyDOZyHwPlIBxIsqaeiSwu-uBrHnCuMEnU6UYGEuHG~EUOY4aYj40IRI8uIJbF0PZE~iA03MLYyn40ABpIChIfKj0B4ETx8pWA9OsnZsh6FyAfEBKNsdrhFvNU2zy7ByTRn6lAgznlQwwcqTR40WC9povQg3Q4YQaKoWDp6C3pMJNWRICJdQFu6Ypi2EHGGVuk9-3lWOiGLKhj5GvzPT4u4vtF~gHQGvgV6LEiu1S~4fubVAKR9Cp8VgxlTII6ZOUO3Z3BXMuK8~Dyw376YhTUUP5CIhaq9CUl~7IcxMAtC-p0-AsfIkHHkyjidVO4g__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Motility of FRA1-3GFP in vivo. A, Overall appearance of 4-week-old plants grown under continuous light. Bar = 10 cm. B, Transcript level in rosette leaves measured by qRT-PCR and normalized to the FRA1 expression level in the wild type. Values are means ± sem from three biological replicates. C, Stem height of 4-week-old plants. Values are means ± sd (n = 20 plants). D, Representative image of FRA1-3GFP (green) in a vascular cell of the root of a 4-d-old seedling. Fei Mao4-64 (FM4-64) (red) was used to label the plasma membrane. Bar = 2 µm. E, Image of FRA1-3GFP in a cotyledon pavement cell of a 4-d-old seedling (same in E–J). Bar = 10 µm. F, Representative kymograph showing the movement of FRA1-3GFP. Diagonal lines represent motile events. Vertical bar = 20 s; horizontal bar = 1 μm. G, Dual-channel image of FRA1-3GFP (green) and RFP-TUB6 (red). The arrowheads label FRA1-3GFP puncta along a cortical microtubule. Bar = 10 µm. H, Distribution of the velocity of processive FRA1-3GFP puncta. The average velocity is 0.24 ± 0.09 μm s−1 (means ± sd, n = 233). I, Distribution of the run length of FRA1-3GFP puncta. The characteristic run length (L 1/2) is 2.4 μm (n = 228). J, Distribution of the number of GFP fluorophores in motile FRA1-3GFP puncta (n = 45). The puncta that contain 6 and 12 GFP fluorophores indicate one and two FRA1-3GFP dimers, respectively. The FRA1-3GFP puncta that contain nine GFP fluorophores probably represent two FRA1-3GFP dimers that got photobleached before image acquisition. K, Time projections of 200 images of FRA1-3GFP from pavement cells of seedlings treated as indicated. Linear tracks in these images indicate motility of FRA1-3GFP. Insets show the cortical microtubules after the stated treatments. Bar = 10 µm. L, Motile parameters of FRA1-3GFP from the experiments shown in K. Values are means ± sd (n = 233, n = 107, and n = 87 molecules for control, taxol, and latrunculin-B [Lat-B], respectively). NA, Not applicable; WT, wild type; asterisk, significant difference from control as determined by Student’s t test (P < 0.01).
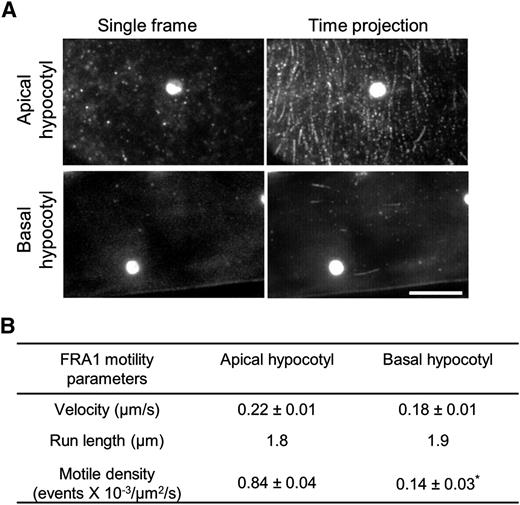
FRA1 motility correlates with growth. A, FRA1-3GFP motility assessed from image sequences. In the time projection of 200 images, motile FRA1-3GFP puncta appear as linear tracks. The bright round structure in these images is a chloroplast imaged because of chlorophyll autofluorescence. Bar = 10 μm. B, Motile parameters of processive FRA1-3GFP puncta. Values are means ± sd (n = 174 and n = 55 molecules in apical and basal hypocotyls, respectively, from at least three seedlings). Asterisk, Significant difference between apical and basal hypocotyl as determined by Student’s t test (P < 0.01).
To characterize FRA1-3GFP motility with respect to cortical microtubules, we crossed a red fluorescent protein-β-tubulin6 (RFP-TUB6) marker (Ambrose et al., 2011) into these plants, allowing for dual-channel imaging. FRA1-3GFP moved processively and unidirectionally along cortical microtubules (Fig. 3G; Supplemental Movie S2). FRA1 motility depended on microtubules but not actin filaments based on treatments with the microtubule-depolymerizing drug oryzalin or the actin depolymerizing drug latrunculin-B (Fig. 3, K and L). FRA1 seemed to prefer moving on stable or bundled microtubules insofar as treatment with taxol, which stabilizes and bundles microtubules, increased the number of motile FRA1-3GFP particles (Fig. 3, K and L). Imaging in guard cells, in which the cortical array has a defined polarity (Marc et al., 1989), showed that FRA1-3GFP moved toward microtubule plus ends (Supplemental Movie S3), consistent with previous in vitro experiments (Zhu and Dixit, 2011).
In view of the high demand for cell wall precursors during expansion, we compared FRA1 activity in growing and nongrowing tissues. In hypocotyls of 4-d-old seedlings, cortical FRA1 puncta were abundant in the growing apical region, and those undergoing directed, persistent motility were relatively easy to find. In contrast, FRA1 puncta were scarce and rarely motile in the nongrowing basal region (Fig. 4; Supplemental Movies S4 and S5), although cortical microtubules are present in this region (Supplemental Fig. S5A). Likewise, FRA1 was less abundant and moved less often in nongrowing 10-d-old cotyledon pavement cells compared with growing cells in 4-d-old cotyledons (Supplemental Fig. S5B). These results are consistent with a role for FRA1 in sustaining high rates of cell wall secretion.
FRA1 Has Partially Distinct Functions in Arabidopsis and Rice
The putative rice FRA1 ortholog (Brittle Culm12 [BC12]; also known as Gibberellin-Deficient Dwarf1 [GDD1]) surprisingly acts as a transcription factor, spending time in the nucleus and regulating expression of the gibberellin synthesis gene Ent-Kaurene Oxidase (KO2; Li et al., 2011). To determine whether phenotypes in fra1-5 are caused by anomalous transcription, we used quantitative real-time PCR to assay the expression of representative genes. In fra1-5, the KO2 ortholog (Yamaguchi, 2008) was transcribed at essentially wild-type levels (Supplemental Fig. S6A), and in contrast to the rice gdd1 mutant, the dwarf phenotype of fra1-5 was unaffected by applying gibberellin (Supplemental Fig. S6B). In addition, a suite of genes involved in the synthesis and modification of cellulose, hemicellulose, pectin, and lignin (Hall and Ellis, 2013) was expressed marginally lower in fra1-5 compared with the wild type (Supplemental Fig. S6C), but none of the changes were of the magnitude expected from the loss of a transcription factor. Finally, FRA1-3GFP resides in the cytoplasm (Fig. 3D), unlike the rice ortholog, which localizes to both cytoplasm and nucleus (Zhang et al., 2010). This is consistent with a canonical nuclear localization signal being present in BC12/GDD1 but absent in FRA1 (Zhang et al., 2010). Together, these data indicate that FRA1 has diverged between the crucifer and the grass family.
FRA1 Does Not Contribute to CESA Motility within the Plasma Membrane or Trafficking of Complexes to the Membrane
The velocity of FRA1 is about 50 times faster than that of the plasma membrane-embedded CESA complexes (Paredez et al., 2006), a difference that makes it unlikely that FRA1 directly guides the movement of CESA complexes along microtubules. Nevertheless, to investigate the relationship between FRA1 and CESA motility, we used two cellulose synthesis inhibitors: isoxaben and 2,6-dichlorobenzonitrile (DCB). Isoxaben depletes CESA complexes from the plasma membrane (Paredez et al., 2006), whereas DCB immobilizes CESA complexes at the membrane (DeBolt et al., 2007). As imaged in hypocotyls, neither drug affected FRA1 motility (Fig. 5A), indicating that its motility is insensitive to CESA status. Reciprocally, the sensitivity of fra1-5 seedlings to isoxaben or DCB was unaltered (Supplemental Fig. S5C), and the velocity distribution of yellow fluorescent protein (YFP)-CESA6 complexes in the plasma membrane of fra1-5 seedlings largely matched that of the wild type (Fig. 5B).
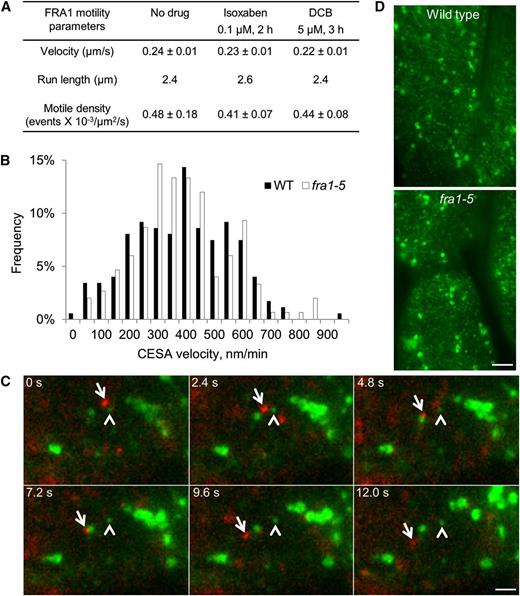
Relationship between FRA1 and CESA complexes. A, Motile parameters of processive FRA1-3GFP puncta in apical hypocotyl cells treated as indicated. Values are means ± sd (n ≥ 95 molecules from at least three seedlings). B, Distribution of the velocity of YFP-CESA6 at the plasma membrane. The average velocity of YFP-CESA6 is 409 ± 173 nm min−1 (n = 174 molecules) in the wild type and 430 ± 192 nm min−1 (n = 150 molecules) in fra1-5. C, Movement of FRA1-tdTomato (arrows) and YFP-CESA6-labeled vesicles (arrowheads) in hypocotyl epidermal cells. Bar = 2 μm. D, Images of plasma membrane-localized YFP-CESA6 particles in hypocotyl epidermal cells. The average particle density ± sd in the wild type (0.42 ± 0.05 μm−2, n = 10 cells) and fra1-5 (0.39 ± 0.06 μm−2, n = 11 cells) is not significantly different as determined by Student’s t test. Bar = 5 μm. WT, Wild type.
The delivery of CESA complexes from the Golgi apparatus to the plasma membrane is thought to occur through specialized vesicular compartments called either small CESA compartments (Gutierrez et al., 2009) or microtubule-associated CESA compartments (Crowell et al., 2009). To determine whether FRA1 transports these compartments, we imaged hypocotyls in plants coexpressing FRA1-tandem-dimeric Tomato (tdTomato) and YFP-CESA6 in the fra1-5 background. The FRA1-tdTomato construct rescued the fra1-5 phenotype, indicating that it is functional (Supplemental Fig. S7). Among 56 motile FRA1-tdTomato particles, none were labeled with YFP (Fig. 5C; Supplemental Movie S6). Likewise, among 30 YFP-CESA6-labeled vesicles that showed directional motility, none were labeled with tdTomato (Fig. 5C; Supplemental Movie S6). In addition, the YFP-CESA6 particle density in the plasma membrane of wild-type (0.42 ± 0.05 μm−2) and fra1-5 (0.39 ± 0.06 μm−2) hypocotyl cells is statistically indistinguishable (Fig. 5D). Together, these findings indicate that FRA1 does not mediate the transport and insertion of CESA complexes.
FRA1 Supports Secretion of Cell Wall Components
To test whether the fra1-5 mutation results in defects in the secretion of matrix polysaccharides, we took advantage of a unique method for assaying the secretion of the pectic polymer rhamnogalacturonan-I based on the uptake and incorporation into pectin of a Fuc analog, Fuc-alkyne (Anderson et al., 2012). In this method, wall-localized Fuc-alkyne is labeled by a specific reaction with the membrane-impermeable compound Alexa 488-azide. After a 2-h incubation with the alkyne, wild-type cell walls within the growth zone were brightly and uniformly labeled, indicating abundant secretion of this pectic moiety; in contrast, fra1-5 cell walls were dimly and unevenly labeled (Fig. 6, A–C), consistent with a reduced rate of pectin secretion.
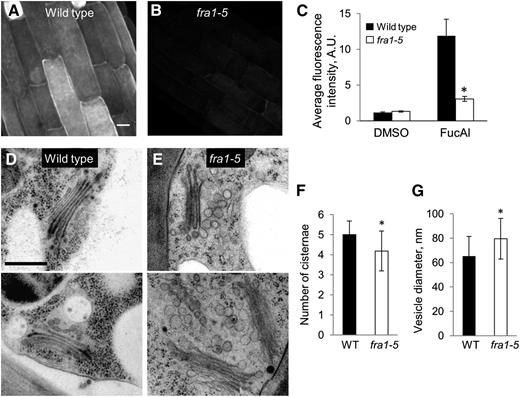
Secretion of pectin and endomembrane morphology. A and B, Representative fluorescence images in root elongation zone epidermal cells of 4-d-old seedlings incubated in Fuc-alkyne (FucAl) for 2 h and reacted with Alexa 488-azide. Bar = 10 µm. C, Quantification of the fluorescence for the experiment shown in A and B. Values are means ± sem (n ≥ 6 cells per seedling with nine seedlings per treatment from two experiments). Dimethyl sulfoxide (DMSO) is used as a solvent control. D and E, Representative images showing Golgi and vesicle morphology in protoxylem cells of the inflorescence prepared by high-pressure cryofixation. Bar = 500 nm. F and G, Quantification of the number of cisternae per Golgi body (n > 30 Golgi from three plants) and the diameter of Golgi-associated vesicles (n > 200 vesicles from three plants) in the wild type and fra1-5. Values are means ± sd. WT, Wild type; asterisk, significant differences between the genotypes as determined by Student’s t test (P < 0.001).
Membrane trafficking from the Golgi to the plasma membrane is responsible for secretion of matrix polysaccharides. To examine the Golgi apparatus, we used high-pressure freezing and freeze substitution followed by transmission electron microscopy. We examined protoxylem cells in the apical (growing) region of the inflorescence stem, because their walls are strikingly thinner in fra1-5 compared with the wild type (Fig. 1, F and G). Golgi bodies had fewer cisternae in fra1-5 compared with the wild type and accumulated larger vesicles in their vicinity (Fig. 6, D–G). These abnormalities are consistent with inhibited secretion in fra1-5.
DISCUSSION
FRA1 Functions in Both Primary and Secondary Cell Wall Production
Our characterization of the fra1-5 knockout mutant revealed defects in the cell walls of growing pith, protoxylem, and mature interfascicular fibers, indicating that FRA1 is important for the production of both primary and secondary cell walls. The most conspicuous cell wall defect was the loss of mass: fra1-5 cell walls were 40% to 50% the thickness of wild-type counterparts. This finding is consistent with the expression of FRA1 throughout the plant (Zhong et al., 2002; Zhou et al., 2007). In the report on the EMS-induced fra1-1 allele, cell wall thickness in fiber cells was not quantified but seemed to be unaffected in the micrograph shown (Zhong et al., 2002). Whether this reflects the true status of fra1-1 cell walls requires additional study.
Cellulose microfibrils were reported to be disorganized in fra1-1 fiber cells (Zhong et al., 2002) and rice bc12 sclerenchyma (Zhang et al., 2010). We saw disorganized cell wall textures in fra1-5 fiber cells but also observed them in the wild-type cells. We examined fiber cells in about 20 plants per genotype prepared in a dozen independent experiments and consistently saw cells with highly organized and poorly organized textures in both genotypes. Neither of the previous studies indicated how many cells and plants were imaged, and they might, therefore, have sampled insufficiently. Nevertheless, for the wild type, the presence of disorganized wall textures among fiber cells is disquieting. Perhaps, the disorganized texture reflects cell wall delamination during sectioning, thus exposing old, intrinsically disordered regions of the wall rather than the fresh innermost layer. Regardless, many fra1-5 fiber cells had highly organized cell wall textures, and we found only minor cell wall disorganization for growing inflorescence stem pith. Because cell wall patterning is not consistently altered in fra1-5, we conclude that cell wall organization is not a primary function of the FRA1 kinesin.
Expansion Is Impaired in fra1-5
The major morphological phenotype of fra1-5 is reduced axial expansion. All organs of the mature fra1-5 plant were shorter than the wild type, and our analysis of the inflorescence showed that this was caused by lower expansion rate rather than altered timing of growth.
The process of expansion depends on the extensibility of the cell wall. Reduced delivery of agents that weaken the cell wall, such as expansins, might account for the decreased expansion seen in fra1-5. Although it has been less explored, the provision of new cell wall polymers themselves also alters cell wall extensibility (Hoson and Masuda, 1991; Boyer, 2009). This is because a polymer within the cell wall that is bearing load might have only a limited capacity for additional deformation, whereas at the moment of its incorporation, a polymer would have considerable slack. Therefore, taking the reduced expansion and cell wall thickness together, we hypothesize that FRA1 acts to ensure efficient secretion of vesicles, which is needed to support both rapid expansion in the primary cell wall and the massive secondary thickening in fiber and xylem cells.
FRA1 Is a Motile Kinesin in Living Plant Cells
To the best of our knowledge, this work is the first example of single-molecule imaging of kinesins in living plants. Similar to other kinesins (Cai et al., 2007; Verhey et al., 2011), the majority of FRA1-3GFP molecules shows diffusive motility in the cytoplasm. These molecules are probably in an autoinhibited state to prevent futile ATP consumption (Ganguly and Dixit, 2013). A subpopulation of the FRA1-3GFP molecules shows directional movement along cortical microtubules. The mechanism that activates FRA1 motility is unknown but could involve binding of the C-terminal tail domain of FRA1 to cargo or phosphorylation of FRA1 (Ganguly and Dixit, 2013). The motile FRA1-3GFP molecules move in a plus end-directed manner, with an average velocity of 0.24 µm s−1 and a characteristic run length of 2.4 µm, consistent with our in vitro motility data (Zhu and Dixit, 2011). The small differences between in vivo and in vitro FRA1 velocity and run length might be caused by different ionic conditions and regulatory mechanisms present in cells. The velocity and run length of FRA1 are comparable with other motile kinesins in eukaryotic systems (Cai et al., 2007; Verhey et al., 2011). Importantly, the ability to move long distances makes FRA1 suited for efficient transport of cargo along cortical microtubule tracks.
FRA1 motility in hypocotyl and cotyledon epidermal cells was positively correlated with growth status. FRA1 abundance and motile density were high in expanding cells and about 6-fold lower in cells that had stopped expanding. Interestingly, CESA complexes show a similar pattern of expression and activity in the hypocotyl (Crowell et al., 2009). Together, these correlations support the hypothesis that FRA1 activity is deployed specifically during stages of active wall deposition.
FRA1 Does Not Contribute to the Movement of CESA Complexes
The plasma membrane-embedded CESA complexes track cortical microtubules (Paredez et al., 2006), and these elements have been proposed to be linked by FRA1 (Zhong et al., 2002; Lloyd and Chan, 2004; Zhu and Dixit, 2011). However, our data indicate that this is unlikely. FRA1 moved about 50 times faster than plasma membrane-embedded CESA complexes, and the motility of FRA1 and CESA complexes was independent of one another. Furthermore, mutation of another protein, cellulose synthase interacting1, that is widely accepted as somehow linking CESA complexes to cortical microtubules gives rise to plants with decreases in cellulose content and increases in radial expansion that are larger than those caused by mutations in FRA1 (Bringmann et al., 2012; Li et al., 2012).
Vesicular compartments carrying CESA complexes have also been observed to interact with cortical microtubules (Crowell et al., 2009; Gutierrez et al., 2009), and kinesins, such as FRA1, have been hypothesized to mediate this interaction (Crowell et al., 2010). However, we found that FRA1 does not colocalize with motile, vesicular CESA compartments, indicating that FRA1 is unlikely to mediate their association with cortical microtubules.
FRA1 Contributes to the Export of Matrix Polysaccharide
The hypothesis that the FRA1 kinesin is required to maintain high rates of secretion receives support from our observations of pectin delivery to cell walls. In fra1-5, alkynylated Fuc was incorporated into the cell walls of rapidly elongating root epidermal cells to a lower extent and in an uneven pattern. Both characteristics resemble the lower incorporation seen for the mature zone of wild-type roots, which presumably delivers matrix polysaccharides to the epidermal apoplast at a reduced rate in the absence of either expansion or secondary cell wall thickening (Anderson et al., 2012). Fuc-alkyne predominantly labels rhamnogalacturonan-I, meaning that our data are consistent with FRA1 being important for the delivery of that pectic component to the cell wall. However, given our chemical composition analysis and analysis from another recent study (Kong et al., 2015) showing that major polysaccharides of the cell wall are present at similar proportions in both genotypes, we think that FRA1 does not handle pectin specifically but that it functions in maintaining high rates of secretion for multiple matrix polysaccharides.
We observed abnormal Golgi morphology and accumulation of enlarged vesicles in the fra1-5 mutant, which are characteristic symptoms of defective post-Golgi trafficking and vesicle secretion (Synek et al., 2006; Feraru et al., 2012; Boutté et al., 2013; Li et al., 2013). Whether the accumulated vesicles in fra1-5 are destined to the plasma membrane and represent FRA1 cargo remains to be determined.
Microtubules and Oriented Cell Wall Assembly
The cell wall phenotype of fra1-5 along with the processive movement of FRA1 and its abundance in regions of rapid growth implicate this kinesin in the process of cell wall secretion. This is surprising. Except for specialized cell types, microtubules have long been considered inconsequential for secretory vesicle delivery (Hepler and Palevitz, 1974; Steinborn et al., 2002). Certainly, phenotypes associated with the removal of microtubules do not involve notable reduction of either expansion or the amount of secreted cell wall (Shibaoka, 1972; Shibaoka and Hogetsu, 1977).
Nevertheless, when no longer governed by microtubules, cell wall assembly becomes far less patterned, and this might simplify requirements for cell wall delivery and secretion. Cortical microtubules are dense, cross linked to the plasma membrane, and bound by many enzymes; consequently, the removal of microtubules is apt to alter conditions in the cell’s cortex pervasively, including changing limits on when and where vesicles may fuse with the membrane. We hypothesize that FRA1-driven transport of secretory vesicles is needed to support rapid rates of secretion where cell walls are undergoing highly patterned assembly, such as in the anisotropically expanding inflorescence pith or secondarily thickening fibers. The motor-driven transport might efficiently target vesicles to exocytotic sites positioned along cortical microtubules or otherwise, allow microtubules to guide patterned assembly around oriented cellulose microfibrils.
MATERIALS AND METHODS
Plant Material and Growth
The Arabidopsis (Arabidopsis thaliana) Columbia-0 accession was used throughout. The fra1-5 mutant was isolated from a T-DNA insertion line (SALK_084463) obtained from Arabidopsis Biological Resource Center (http://abrc.osu.edu/). Homozygous mutants were identified by using primers listed in Supplemental Table S1. Seeds of fra1-1 were obtained from Zheng-Hua Ye.
For growth on plates, seeds were sterilized with 5% (v/v) bleach for 10 min, rinsed four times with water, and planted on 1× Murashige and Skoog medium (Caisson Laboratories). Seeds were stratified at 4°C for 2 d and then grown at 20°C under 16 h of light.
For growth in soil, seeds were grown under continuous light, 70% humidity, and 21°C after stratification at 4°C for 2 d. Continuous light was chosen, because it produced more severe developmental defects in adult fra1-5 plants than growth under the 16-h photoperiod.
RT-PCR and Quantitative Reverse Transcription-PCR
Total RNA was extracted from basal internodes or rosette leaves of 4-week-old plants, DNAse treated, and used for complementary DNA (cDNA) synthesis using qScript cDNA Supermix (Quanta BioSciences). Primers for RT-PCR and quantitative reverse transcription (qRT)-PCR are listed in Supplemental Table S1. The qRT-PCR was conducted using the SYBR Green method. Three biological replicates, each with three technical replicates, were used to estimate fold change in gene expression relative to the wild type. Actin2 was used as an internal control.
Histology
To image pith cells, 5-mm segments from apical and middle regions of 4-week-old inflorescence stems were cut longitudinally by a Vibratome into 100-µm sections. Middle plane sections were stained with 5 µg mL−1 propidium iodide for 5 min and imaged using confocal microscopy. Cell lengths and widths were measured using ImageJ (http://imagej.nih.gov/ij/). To observe the lignification pattern, transverse sections of stems were stained with 1% (w/v) phloroglucinol in 6 n HCl for 5 min and imaged under a dissecting light microscope.
Generation of the FRA1-3GFP and FRA1-tdTomato Constructs and Transgenic Plants
The pFRA1::FRA1-3GFP construct was generated using a 1.3-kb sequence upstream of the FRA1 start codon and the full-length FRA1 cDNA followed by three tandem copies of enhanced green fluorescent protein cDNA. The pFRA1::FRA1-tdTomato construct was generated using a 3-kb sequence upstream of the FRA1 start codon and the full-length FRA1 cDNA followed by tdTomato cDNA. Primers for making these constructs are listed in Supplemental Table S1. These constructs were ligated into the pCAMBIA 1300 vector and introduced into the fra1-5 mutant through Agrobacterium spp.-mediated floral dip transformation. Transgenic plants were selected using 20 µg mL−1 hygromycin, and homozygous lines expressing a single copy of the transgene were used for phenotypic analysis and imaging. To determine the relationship between FRA1 motility and microtubules, the pFRA1::FRA1-3GFP line was crossed to plants expressing ubiquitin promoter-driven RFP-TUB6 (Ambrose et al., 2011), and progeny expressing both markers were selected for imaging.
Live Cell Imaging and Image Analysis
FRA1-3GFP was imaged using variable-angle epifluorescence microscopy at 22°C to 23°C. Seedlings were gently mounted in water between two layers of double-sided adhesive tape. For drug treatments, seedlings were incubated with 0.1 µm isoxaben for 2 h, 5 µm DCB for 3 h, 20 µm taxol (Cytoskeleton, Inc.) for 1.5 h, 20 µm oryzalin for 3 h, or 2 µm latrunculin B (Enzo Life Science) for 2 h before mounting and imaging. For controls, seedlings were treated with either water or 0.1% (v/v) dimethyl sulfoxide as appropriate. Epidermal cells in the apical or subapical region of the hypocotyl were imaged unless otherwise indicated. GFP and RFP were excited using 2-mW, 488-nm and 2-mW, 561-nm diode-pumped solid-state lasers (Melles Griot), and images were collected using a 100× (numerical aperture 1.45) objective and a back-illuminated electron-multiplying CCD camera (ImageEM; Hamamatsu) at 1-s intervals in the GFP channel and 4-s intervals in the RFP channel for 3 min.
Velocity and run length for individual motile events were measured using kymograph analysis in SlideBook 5.0 (Intelligent Imaging Innovations). Motile density was calculated as the number of motile FRA1-3GFP particles per micrometer−2 per second−1. Single-molecule photobleaching analysis was conducted as described earlier (Ross and Dixit, 2010). Briefly, we treated seedlings with 1 mm sodium azide for 2 h to deplete cellular ATP, which caused FRA1-3GFP particles to become immobilized on cortical microtubules. The fluorescence intensity of individual FRA1-3GFP particles was then recorded over time, and clearly detectable single bleach steps were used to estimate the fluorescence intensity of a single GFP molecule. This information was used to calculate the number of GFP molecules in motile FRA1-3GFP particles as (initial fluorescence intensity of a motile particle − background fluorescence intensity) fluorescence intensity of a single GFP molecule−1. Data were collected from at least three cells from three independent seedlings.
Plants expressing YFP-CESA6 in the procuste1-1 background (Paredez et al., 2006) were crossed to fra1-5, and F3 progeny homozygous for YFP-CESA6 and fra1-5 were used for imaging. The motility of YFP-CESA6 at the cell cortex was imaged as above for FRA1-3GFP. Time-lapse images were captured at 10-s intervals using a 2-mW, 488-nm laser for 10 min. For plasma membrane YFP-CESA6 particle density measurements, single images of hypocotyl epidermal cells were captured and then cropped to select regions of the plasma membrane of cells of interest. CESA particles within the region of interest were identified as puncta with a diameter of 0.5 µm using the Imaris (Bitplane) software’s Spots tool, setting the minimum quality threshold to that automatically assigned by running the algorithm on the corresponding uncropped image. A maximum quality threshold was used to exclude spurious puncta arising from edge effects. Particle density was calculated by dividing the total remaining puncta by the plasma membrane surface area.
To determine the relationship between FRA1 and vesicular CESA compartments, a pFRA1::FRA1-tdTomato construct was transformed into plants homozygous for YFP-CESA6 and fra1-5. T2 progeny expressing both markers were selected and imaged at 1-s intervals using 1-mW, 488-nm and 2-mW, 561-nm lasers for 2 min.
To quantify microtubule organization and dynamics in fra1-5, plants expressing ubiquitin promoter-driven RFP-TUB6 (Ambrose et al., 2011) were crossed to fra1-5, and F3 progeny were used for imaging. Time-lapse images were captured at 3-s intervals using a 1-mW, 561-nm laser for 5 min. The growth and shortening rates of cortical microtubule plus ends were measured using kymograph analysis in ImageJ. We noticed that the RFP-TUB6 signal is lower in fra1-5, which made it difficult to reliably score catastrophe and rescue events; therefore, these data were not quantified.
Transmission Electron Microscopy
To image cell walls, freshly excised 2-mm basal stem segments were fixed for 90 min in 2% (w/v) glutaraldehyde buffered with 0.1 m pipes buffer, pH 6.8. The tissue was then postfixed for 90 min in buffered 2% (w/v) osmium tetroxide, dehydrated, and embedded in Spurr’s resin. Thin sections were stained with uranyl and lead salts and imaged in an LEO 912 AB Energy-Filtered Transmission Electron Microscope operated at 120 kV. Cell wall thickness was measured using ImageJ for the outermost two layers of interfascicular fiber cells, pith cells in the center of the stem, and cells with thick walls in the protoxylem. Cellulose was labeled with colloidal gold using enzyme-gold affinity cytochemistry as described earlier (Berg et al., 1988) and detailed in Supplemental Methods S1.
To image Golgi and vesicles, transverse sections of 4-week-old apical and basal stems were loaded in specimen carriers that contained packing buffer (100 mm PIPES, pH 6.8 and 150 mm Suc) and ultrarapidly frozen using a BAL-TEC HPM 010 High-Pressure Freezer. Samples were freeze substituted over 5 d at −85°C in acetone containing 2% (w/v) osmium tetroxide and 0.1% (w/v) uranyl acetate, slowly thawed to room temperature, rinsed in acetone, and embedded in Spurr’s resin. Thin sections were stained and imaged as described above. The diameter of vesicles within approximately 1 µm of Golgi stacks was measured in ImageJ. The number of cisternae per Golgi was counted manually.
Scanning Electron Microscopy
Freshly excised segments (approximately 5 mm long) from 4-week-old plants were sectioned in phosphate-buffered saline at a nominal thickness of 100 µm on a Vibratome. The sections were rinsed with 1% (v/v) Triton X-100 and dehydrated using a graded ethanol series. Dehydrated samples were critically point dried, mounted on stubs, coated with platinum (approximately 2 nm), and imaged in a scanning electron microscope (FEI Magellan) equipped with a field emission gun. Samples were imaged at 1 kV and 25 pA. For analysis of organization in the apical material, four sections from four independent stems per genotype were imaged (approximately 150 images per genotype). For each section, approximately 10 cells were imaged along a transverse transect. For quantification, the fit-ellipse routine was used as described by Marga et al. (2005) and detailed in Supplemental Methods S1.
Metabolic Labeling of Pectin Using Fuc-Alkyne
Fuc-alkyne-based labeling of pectin was conducted essentially as described previously (Anderson et al., 2012). The root elongation zones of labeled seedlings were imaged with a Zeiss Cell Observer SD Spinning Disc Confocal Microscope (488-nm laser excitation and 525/25 emission filter) using a 63×, 1.40 numerical aperture oil immersion objective. Z stacks of epidermal cells were collected, and cell wall-associated fluorescence intensity in maximum projections was measured using ImageJ by calculating mean pixel intensity values for individual cells.
Immunoblotting
A polyclonal antiserum was generated against a 24-amino acid peptide (P931–P954) from the FRA1 tail domain and purified using affinity chromatography against the same peptide (Epitomics). For FRA1 immunoblotting, 4-week-old light-grown shoots were first ground in liquid N2 and then homogenized in protein isolation buffer (50 mm Tris-acetate, pH 7.5, and 2 mm EDTA); a protease inhibitor tablet from Roche and total protein (approximately 100 µg each) were separated by SDS-PAGE and transferred to a 0.45-µm polyvinylidene difluoride membrane (Thermo Scientific). Proteins were probed with FRA1 primary antibody (1:2,000) and anti-rabbit IgG horseradish peroxidase secondary antibody (1:5,000; Jackson Immuno Research). Detection was conducted using SuperSignal West Dura chemiluminescence substrate (Thermo Scientific).
Sequence data from this article can be found in the GenBank/EMBL data libraries under accession number At5g47820 for FRA1.
Supplemental Data
The following supplemental materials are available.
Supplemental Figure S1. Reduced anisotropic growth of fra1-5.
Supplemental Figure S2. Cell wall mechanical properties.
Supplemental Figure S3. Stem lignification.
Supplemental Figure S4. Cortical microtubule organization in fra1-5.
Supplemental Figure S5. Motility of FRA1-3GFP in pavement cells.
Supplemental Figure S6. Transcriptional and GA response.
Supplemental Figure S7. Phenotype of fra1-5 plants expressing FRA1-tdTomato.
Supplemental Table S1. Primers used in this work.
Supplemental Movie S1. Motility of FRA1-3GFP in a pavement cell.
Supplemental Movie S2. FRA1-3GFP moves along cortical microtubules.
Supplemental Movie S3. FRA1-3GFP moves toward microtubule plus-ends.
Supplemental Movie S4. Motility of FRA1-3GFP in the apical region of a hypocotyl.
Supplemental Movie S5. Motility of FRA1-3GFP in the basal region of a hypocotyl.
Supplemental Movie S6. Two-color imaging of FRA1-tdTomato and CESA6-YFP.
ACKNOWLEDGMENTS
We thank Geoffrey Wasteneys (University of British Columbia) for the gift of the RFP-TUB6 tubulin line and David Ehrhardt (Carnegie Institution, Stanford, CA) for the gift of the YFP-CESA6 line.
Glossary
- cDNA
complementary DNA
- DCB
2,6-dichlorobenzonitrile
- EMS
ethyl methanesulfonate
- qRT
quantitative reverse transcription
- RT
reverse transcription
LITERATURE CITED
Author notes
This work was supported by Washington University (Monsanto/Norman Borlaug Corporate Fellowship to C.Z.); the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences of the U.S. Department of Energy (grant no. DE–FG–03ER15421 to the laboratory of T.I.B for scanning electron microscopy experiments of the cell wall); the Center for Lignocellulose Structure and Formation, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences (award no. DE–SC0001090 to the laboratory of C.T.A. for Fuc-labeling experiments); and the National Science Foundation (grant no. MCB–1121287 to R.D.).
Address correspondence to [email protected].
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (www.plantphysiol.org) is: Ram Dixit ([email protected]).
Articles can be viewed without a subscription.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}