





Abstract
With the recent availability of complete genomic sequences of many organisms, high-throughput and cost-efficient systems for gene cloning and functional analysis are in great demand. Although site-specific recombination-based cloning systems, such as Gateway cloning technology, are extremely useful for efficient transfer of DNA fragments into multiple destination vectors, the two-step cloning process is time consuming and expensive. Here, we report a zero background TA cloning system that provides simple and high-efficiency direct cloning of PCR-amplified DNA fragments with almost no self-ligation. The improved T-vector system takes advantage of the restriction enzyme XcmI to generate a T-overhang after digestion and the negative selection marker gene ccdB to eliminate the self-ligation background after transformation. We demonstrate the feasibility and flexibility of the technology by developing a set of transient and stable transformation vectors for constitutive gene expression, gene silencing, protein tagging, protein subcellular localization detection, and promoter fragment activity analysis in plants. Because the system can be easily adapted for developing specialized expression vectors for other organisms, zero background TA provides a general, cost-efficient, and high-throughput platform that complements the Gateway cloning system for gene cloning and functional genomics.
Rapid advances in genome sequencing technologies in the last few years have led to the complete decoding of many complex eukaryotic genomes and have stimulated large-scale analysis of gene functions in sequenced genomes. In general, gene function can be elucidated using a variety of approaches, such as ectopic expression, gene silencing, protein subcellular localization examination, gene expression pattern analysis by promoter activity assay, structure-function analysis, and in vitro or in vivo biochemical assays (Hartley et al., 2000; Curtis and Grossniklaus, 2003; Earley et al., 2006). Typically, all these approaches require the cloning of target genes, mutated fragments, or their promoter fragments into various specialized vectors for subsequent characterization. However, the traditional approach for engineering expression constructs based on the restriction enzyme/ligase cloning method is extremely laborious and time consuming and is often hampered by lack of appropriate restriction sites; thus, the production of constructs is a significant technical obstacle for large-scale functional gene analysis in plants.
In recent years, the Gateway cloning system from Invitrogen and the Creator cloning system from CLONTECH have been developed to facilitate large-scale production of gene constructs. The recombinational cloning systems are based on a two-step process (Marsischky and LaBaer, 2004). The DNA fragment of interest is first cloned into a general donor plasmid. Subsequently, the DNA fragment flanked by two site-specific recombination sites in the donor vector can be transferred precisely into a variety of expression vectors by site-specific recombination reactions. A great advantage of the recombinational cloning technologies is that once the DNA fragment has been engineered into a donor vector, the transfer of the DNA fragment into an expression destination vector is a simple reaction that requires no traditional restriction enzyme/ligase cloning. The recombinational cloning systems, particularly the Gateway technology, have been widely used in the research community, and many Gateway-compatible open reading frame entry (donor) clone collections and expression vectors have been created for functional genomics in many organisms (Yashiroda et al., 2008), including plants (Karimi et al., 2007b). On the other hand, although extremely useful for the simple and efficient transfer of DNA fragments into multiple expression destination vectors, the usefulness of the Gateway cloning system is rather limited for many projects where only a single expression vector is required for a DNA fragment. The two-step cloning process of the Gateway technology is laborious and time consuming for the production of a single expression vector. This is particularly true when a large number of plasmids must be cloned. Although a one-step recombinational cloning method was described to eliminate the production of an entry clone (Fu et al., 2008), the approach is rather limited in scope because long primers containing the specific attachment site (att) and two-step PCR are required (Fu et al., 2008).
TA cloning is routinely used for cloning of PCR-amplified fragments. This technique exploits the terminal transferase activity of some DNA polymerases that add a 3′-A overhang to each end of the PCR product. PCR products can be easily cloned into a linearized vector with 3′-T overhangs compatible with 3′-A overhangs. Because it is difficult to generate a high-quality TA cloning vector in individual laboratories, many TA cloning kits are available in the market. Many of them use blue/white screening for recombinants, and the DNA fragments can only be cloned into the TA vector provided in the kit. To meet the need for high-throughput cloning of DNA fragments into diverse expression vectors, we have developed a significantly improved TA cloning vector system by taking advantage of the negative selection gene marker ccdB to eliminate the self-ligation background after transformation. We refer to this new method as the zero background TA cloning system (ZeBaTA). Numerous cloning tests in our laboratory have shown that ZeBaTA provides very high cloning efficiency with almost no self-ligation. Moreover, the ZeBaTA technology can be flexibly adapted for developing specialized expression vectors allowing single-step assembly of PCR-amplified genes or fragments. We demonstrate the feasibility and flexibility of the technology by developing a set of 12 transient and 12 stable transformation vectors for constitutive gene expression, gene silencing, protein tagging, protein subcellular localization, and promoter fragment activity analysis for rice (Oryza sativa) and Arabidopsis (Arabidopsis thaliana). Our results suggest that ZeBaTA technology can also be easily used to develop expression vectors for other organisms (e.g. Escherichia coli, yeast [Saccharomyces cerevisiae], insect, and mammal), thereby providing a novel and general high-throughput platform for functional genomics of target genes.
RESULTS
Construction of the ZeBaTA System
Two different strategies were used to produce T-vectors, i.e. adding a single thymidine at the 3′ blunt ends of a linearized vector (Holton and Graham, 1991; Marchuk et al., 1991) and generating single 3′-T overhangs of a linearized vector by restriction endonuclease digestion (Kovalic et al., 1991; Mead et al., 1991; Ichihara and Kurosawa, 1993; Chen et al., 2006a). Although the former has been used to produce commercial cloning kits like the pGEM-T system, we selected the restriction endonuclease digestion-mediated strategy to develop a TA cloning vector system because this approach is easy to use for individual laboratories. Previous publications have described the use of restriction enzyme XcmI (Kovalic et al., 1991; Mead et al., 1991) or AhdI/Eam1105I (Ichihara and Kurosawa, 1993; Chen et al., 2006a) to produce intermediate T-vectors. We chose XcmI as the digestion enzyme to develop the ZeBaTA cloning system because it had a better digestion efficiency than AhdI.
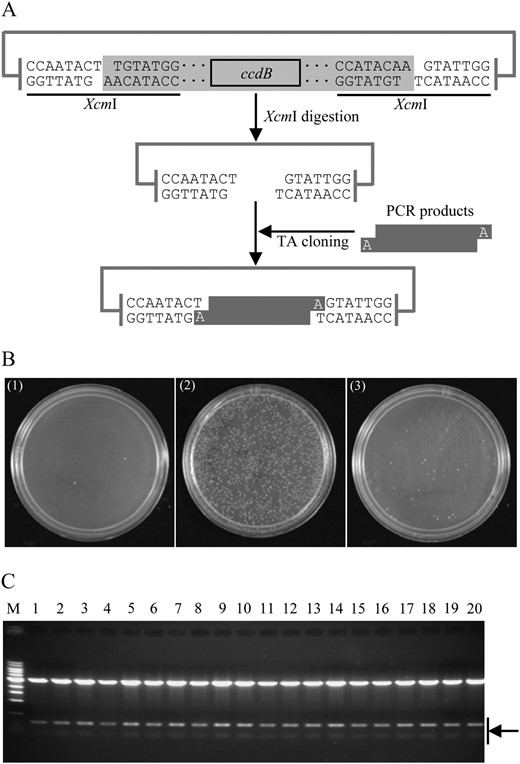
Construction of the ZeBaTA system. A, Schematic representation of direct cloning of PCR product using the ZeBaTA vector system. The linker of the vector (in gray) is removed after XcmI digestion yielding a linearized T-vector. B, TA cloning tests of the ZeBaTA system. (1) Self-ligation of XcmI-digested pGXT using T4 DNA ligase from Promega. (2) Ligation of XcmI-digested pGXT with the PCR product of the rice blast fungus M. oryzae gene MGG_07986.5 using T4 DNA ligase from Promega. (3) Ligation of XcmI-digested pGXT with the PCR product of MGG_07986.5 using T4 DNA ligase from USB Corporation. C, Samples of restriction digestion analysis of the randomly selected colonies derived from ligation of XcmI-digested pGXT with the PCR product of MGG_07986.5 using T4 DNA ligase from Promega. pGXT contains two BamHI recognition sites outside the two XcmI recognition sites (Supplemental Fig. S1), and MGG_07986.5 contains one internal BamHI site. All samples (lanes 1–20) digested by BamHI released two bands as expected. M, 1-kb DNA ladder.
Set of Expression ZeBaTA Vectors for Plants
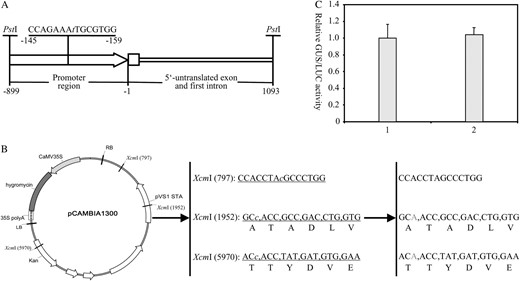
Site-specific mutagenesis of the maize ubiqutin-1 promoter (A) and the backbone of the binary vector pCAMBIA1300 (B) in which three XcmI recognition sites were deleted. The nucleotides represented in lowercase italic letters are the positions where deletions or mutations were made. Kan, Kanamycin resistance gene; LB, T-DNA left border; RB, T-DNA right border. C, Comparison of the levels of GUS expression mediated by the original and modified maize ubiquitin-1 promoter in transiently transfected rice protoplasts. GUS activities are represented as a ratio of relative GUS/LUC. The experiment was repeated three times with similar results. 1, Protoplast sample transfected with pUbiGUS; 2, protoplast sample transfected with pXUN-GUS.

ZeBaTA-based expression vectors for gene overexpression/silencing, protein tagging, protein subcellular localization, and promoter analysis in plants. A, Schematic structures of the transient expression vectors generated by XcmI digestion. B, Schematic structures of the Agrobacterium-mediated stable transformation vectors generated by XcmI digestion. LB, T-DNA left border; RB, T-DNA right border.
Because the maize ubiquitin-1 promoter used in this system was modified to block its original XcmI recognition site by deleting a single base (Fig. 2A) at the position of nucleotide −480, a gus gene (Jefferson et al., 1987) was amplified by PCR and then cloned into the XcmI-digested pXUN vector to produce an expression construct to test the expression activity of the modified maize ubiquitin-1 promoter. The derived construct pXUN-GUS and a control construct pUbi-GUS (Chen et al., 2006b), of which a gus gene is driven by the original maize ubiquitin-1 promoter, were tested transiently in the transfected rice protoplasts. Transient expression assays showed that the levels of GUS activity in rice protoplasts transfected with these two constructs were similar (Fig. 2C), indicating that the deletion of nucleotide −480 does not affect the activity of the maize ubiquitin-1 promoter.
Testing of Tagged Protein Expression
![Transient expression and protein-tagging detection of the ZeBaTA vectors in rice protoplasts. A, Fluorescence microscopy of the expression of HA-tagged GFP in rice protoplasts. B, Detection of HA-tagged GFP by western blot. Lane 1, Nontransfected control protoplast sample; lanes 2 to 4, independent protoplast samples transfected with pXUN-HA-GFP. [See online article for color version of this figure.]](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/plphys/150/3/10.1104_pp.109.137125/3/m_plphys_v150_3_1111_f4.jpeg?Expires=1750223016&Signature=v~HhqBVqfHnOCMA17xbVsV6WBFnMeiANZ14aw49j2EoS~Q6T4VixQkI20D7ZGNG4pWzgMORNkegzSyv9Ez25X8Wtfr5vGTLBn0vNb6AQPBCegDmxPmJ0p6YbPgmhnfPbD1n1d3dffCXuzpDfJ40pGdki40OYg69NYadsT~pl7AuD-JzhakZY5OWqh4~tDhNj65-ZHqO6QPBBj2BOHUFKY-nKKvIUBeZj6e0vsXDBIn2vAKqRXGeHlu4i4A~5vf9pdmG6icxvF8osRPyCGNTTJgKCnr1hNII5~QL8gb9mBJPuoPBHWrzNE0xqEPnG2B3cGANmS1kis9kVqWmfk7HOtg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Transient expression and protein-tagging detection of the ZeBaTA vectors in rice protoplasts. A, Fluorescence microscopy of the expression of HA-tagged GFP in rice protoplasts. B, Detection of HA-tagged GFP by western blot. Lane 1, Nontransfected control protoplast sample; lanes 2 to 4, independent protoplast samples transfected with pXUN-HA-GFP. [See online article for color version of this figure.]
Gene Silencing by Hairpin RNAi or Artificial MicroRNA
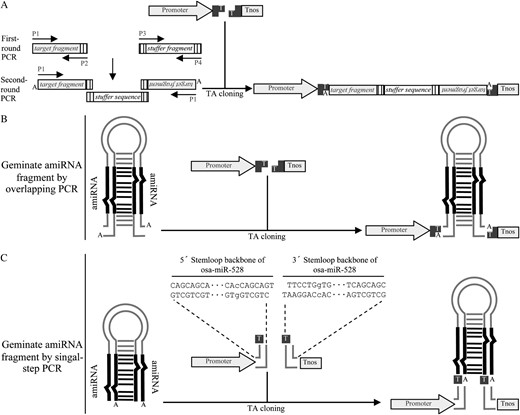
Schematic illustration of the construction of hpRNAi or amiRNA constructs by single-step cloning. A, Generation of hpRNAi constructs by overlapping PCR approach. The target gene fragment and the stuffer sequence fragment are amplified in the first-round PCR. Primers P2, P3, and P4 introduce complementary adapters (indicated by vertically lined boxes) to the amplified fragments. The two amplified fragments are fused together as an inverted-repeat cassette in the second-round PCR by using single P1 primer. The resulting fragment is then directly cloned into the plant expression T-vector. B, Generation of amiRNA constructs by overlapping PCR approach. C, Generation of amiRNA constructs for rice genes by single-step PCR. The expression vectors pXUN-osaMIR528 and pCXUN-osaMIR528 were preassembled with 5′ and 3′ stemloop backbone sequences of a rice miRNA precursor Osa-MIR-528 (Warthmann et al., 2008). Thus, making amiRNA constructs for rice target genes only requires an amiRNA-amiRNA* fragment generated from single-step PCR. The nucleotides represented in lowercase letters are the positions where mutations were made to introduce two XcmI recognition sites.

Silencing of the PDS gene in Arabidopsis and rice by the ZeBaTA-based hpRNAi or amiRNA approaches. A, Arabidopsis plants transformed with the hpRNAi construct pCXSN-atPDS-RNAi showing the PDS silencing albino phenotype. (1) Control plant; (2 and 3) two examples of transgenic Arabidopsis plants. B, Rice plants transformed with the amiRNA vectors showing the albino phenotype. (1) Control plant; (2) example of pCXUN-amiPDS-transformed plants; and (3) example of pCXUN528-PDS-transformed plants. C, RT-PCR analysis of PDS suppression transgenic rice plants. Five independent primary plants (1, 2, 3, 4, and 5) transformed with pCXUN-amiPDS and five independent primary plants (6, 7, 8, 9, and 10) transformed with pCXUN528-PDS were selected for the analysis. CK, Wild-type Nipponbare plant used as the control.
Recently, the artificial microRNA (amiRNA) approach has been introduced for highly specific gene silencing in both dicot and monocot plants (Niu et al., 2006; Schwab et al., 2006; Ossowski et al., 2008; Warthmann et al., 2008). Typically, the amiRNA is generated by site-directed mutagenesis on precursors of endogenous miRNAs to exchange the natural miRNA sequences with those of amiRNAs using overlapping PCR (Ossowski et al., 2008). The same ZeBaTA-based vector system developed for ectopic gene expression and hpRNAi can also be used for making amiRNA expression constructs by simple TA cloning (Fig. 5B), thus bypassing the time-consuming two-step procedure for the regular restriction enzyme digestion-mediated cloning or the Gateway cloning (Ossowski et al., 2008). We further developed a ZeBaTA-amiRNA system to simplify the generation of rice amiRNA constructs because our lab is focusing on rice functional genomics. The new ZeBaTA-amiRNA vector was designed based on the stemloop backbone derived from Osa-MIR528, an endogenous rice miRNA precursor that has been used to efficiently express amiRNAs for highly specific silencing of targeted genes in rice (Warthmann et al., 2008). By site-directed mutagenesis of a single base on the 5′ and 3′ stemloop backbones of Osa-MIR528, respectively, a cassette of 5′ Osa-MIR528 stemloop backbone-XcmI-ccdB-XcmI-3′ Osa-MIR528 was assembled and cloned into the expression vectors where the expression of amiRNA is under the control of the maize ubiquitin-1 promoter. Figure 5C illustrates the structural maps of the Osa-MIR528-based vectors pXUN-osaMIR528 and pCXUN-osaMIR528. The vectors allow for high-throughput generation of rice amiRNA constructs by cloning the amiRNA-amiRNA* fragment generated from a single-step PCR into the ZeBaTA vector with the preassembled Osa-MIR528 stemloop backbone (Fig. 5C; Supplemental Fig. S3), thus avoiding the time-consuming overlapping PCR. The modified vector was evaluated by expression of the amiRNA for silencing of the OsPDS gene. The two constructs pCXUN-amiPDS and pCXUN528-PDS, which contain original or modified Osa-MIR528 stemloop backbone with amiRNA sequence targeting OsPDS, respectively, were introduced into rice cv Nipponbare by Agrobacterium-mediated transformation. Consistent with a previous study (Warthmann et al., 2008), 70.1% of the primary transgenic lines transformed with pCXUN-amiPDS had a bleaching PDS silencing phenotype (Fig. 6B; Table I
PDS silencing frequency of transgenic rice mediated by the ZeBaTA-amiRNA system
amiRNA Vector | Total Independent Transformants | Albino Phenotype | Efficiency |
|---|---|---|---|
| % | |||
| pCXUN-amiPDS | 55 | 39 | 70.1 |
| pCXUN528-PDS | 35 | 27 | 77.1 |
amiRNA Vector | Total Independent Transformants | Albino Phenotype | Efficiency |
|---|---|---|---|
| % | |||
| pCXUN-amiPDS | 55 | 39 | 70.1 |
| pCXUN528-PDS | 35 | 27 | 77.1 |
PDS silencing frequency of transgenic rice mediated by the ZeBaTA-amiRNA system
amiRNA Vector | Total Independent Transformants | Albino Phenotype | Efficiency |
|---|---|---|---|
| % | |||
| pCXUN-amiPDS | 55 | 39 | 70.1 |
| pCXUN528-PDS | 35 | 27 | 77.1 |
amiRNA Vector | Total Independent Transformants | Albino Phenotype | Efficiency |
|---|---|---|---|
| % | |||
| pCXUN-amiPDS | 55 | 39 | 70.1 |
| pCXUN528-PDS | 35 | 27 | 77.1 |
Protein Subcellular Localization/Colocalization and Promoter Activity Assay
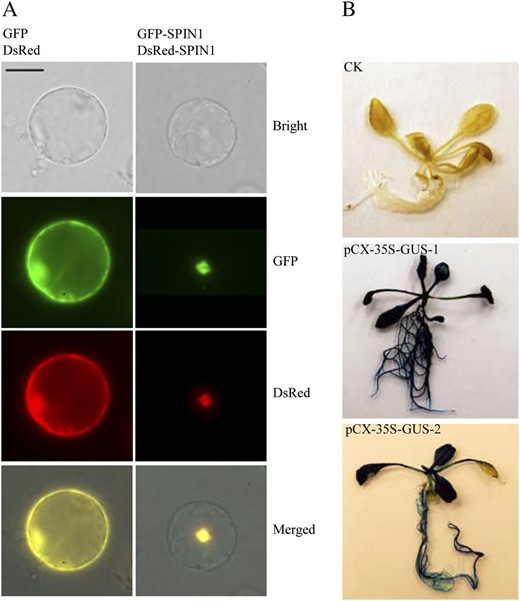
Protein subcellular localization and promoter activity analysis using the ZeBaTA vectors. A, Fluorescence microscopy of the coexpression of GFP and DsRed, or GFP-SPIN1 and DsRed-SPIN1 fusions in rice protoplasts. Scale bar = 20 μm. The RNA binding nuclear protein SPIN1 was used as a tester (Vega-Sánchez et al., 2008). B, GUS staining of Arabidopsis transformed with pCX-35S-GUS, where the 35S promoter was cloned into the vector pCXGUS-P to test the system. CK, Plant transformed with control vector pCAMBIA1300 (www.cambia.org); pCX-35S-GUS-1 and pCX-35S-GUS-2, two independent primary transgenic plants.
For promoter activity assays, two reporters, gus and gfp, were used for constructing pXGUS-P/pCXGUS-P and pXGFP-P/pCXGFP-P, respectively. The linear T-vectors of pXGUS-P/pCXGUS-P or pXGFP-P/pCXGFP-P (Fig. 3, A and B) allow direct cloning of PCR-amplified promoter fragments located in front of the reporter genes. As proof of concept, the 35S promoter was cloned into pCXGUS-P to drive expression of the reporter gene gus. Arabidopsis plants stably transformed with the construct pCX-35S-GUS showed constitutive GUS expression in the whole plants (Fig. 7B), confirming the feasibility of the system for assaying promoter activity.
DISCUSSION
With the rapid development of the next-generation sequencing technology, more plant genomes will be sequenced in the near future. How to rapidly determine the function of the identified genes on a large scale is a daunting challenge. The ability to efficiently make constructs to transiently and stably express specific genes in cells, tissues, or whole plants is a fundamental aspect and bottle neck of plant functional genomics research. Traditionally, the cloning vectors for plant research carry a multicloning site (MCS) within their target gene expression cassettes. The restriction sites in the MCS are rather limited, making cloning of most target genes difficult. Although TA cloning vectors have been widely used for cloning of PCR-amplified fragments, the system has not yet been incorporated in the cloning vectors for transient and stable expression of target genes because of the technical challenge of generating low-background TA cloning vectors. The Gateway system has been a popular choice for generating various constructs because it allows the gene of interest to be easily cloned into specifically designed plasmids without DNA restriction digestions. The two-step cloning and expensive reagents, however, make the Gateway system impractical for large-scale cloning in most individual laboratories when the entry clone collections are not available. The ZeBaTA system described here overcomes the limitations of both the TA and Gateway cloning systems. After two XcmI recognition sites have been introduced into the MCS, any PCR fragments with a T-overhang can be easily cloned into a ZeBaTA vector. With the introduction of the negative selection marker gene ccdB between the two XcmI sites, any self-ligation transformants are eliminated. Using this technology, we constructed a set of 12 transient and 12 stable transformation vectors for plant gene expression studies and tested the vectors in rice or Arabidopsis in our laboratories. These vectors can be used in a wide range of functional genomics projects in plants and will be distributed to the research community upon request.
Under certain conditions, cloning with T-vectors generated by digestion with AhdI or XcmI gave low efficiency and the T residue of the insert-vector junction in the recombinant clones is often missing (Mead et al., 1991; Chen et al., 2006a). Chen et al. (2006a) speculated that this may be due to the presence of unknown factors that, during digestion and preparation of the T-vectors, influence the stability of 3′-T overhangs. In this study, we found that the main factor affecting successful cloning is the use of an appropriate T4 DNA ligase. We tested the Promega T4 DNA ligase, which is included in the pGEM-T easy vector system, and the T4 DNA ligase from USB Corporation. The ligations using Promega T4 DNA ligase consistently gave a very high cloning efficiency; most of the ligations using USB Corporation T4 DNA ligases yielded low efficiency. Many of the recombinant plasmids from the latter ligations missed a T residue in the insert-vector junction, consistent with observations by Mead et al. (1991) and Chen et al. (2006a). The T residue is missing mainly because regular commercial T4 DNA ligases contain exonuclease activities that can remove the 3′-T tails from the vector, as reported in the technical manual of the pGEM-T and pGEM-T easy vector systems (http://www.promega.com/tbs/tm042/tm042.pdf); removal of the 3′-T tails from the vector results in very low cloning efficiency. When the Promega T4 DNA ligase was used for ligation, we never observed the absence of the T residue in the recombinant plasmids (data not shown), suggesting that the 3′-T overhangs of the T-vectors generated by XcmI digestion were stable. Our cloning tests also indicated that the size of T-vectors, vector overdigestion, and the insert-to-vector molar ratios had a minimal impact on cloning efficiency.
In recent years, Gateway cloning technology has become increasingly popular for functional genomics in plants (Karimi et al., 2002, 2005, 2007a, 2007b; Curtis and Grossniklaus, 2003; Earley et al., 2006; Hilson, 2006; Himmelbach et al., 2007; Lei et al., 2007). In comparison to the Gateway system, ZeBaTA offers several significant advantages. First, it allows simple and highly efficient one-step cloning for PCR-amplified genes/fragments of interest into a specialized ZeBaTA expression vector. Second, the ZeBaTA constructs have a short vector-insert junction with only 8 bp (CCANNNNN), whereas the constructs generated by Gateway recombination contain a longer att linker sequence with 25 bp or even up to 125 bp (Lei et al., 2007). For example, protein-tagging constructs generated by Gateway cloning technology usually result in a long linker with at least eight or sometimes up to 20 amino acids (Dubin et al., 2008) between the tag and the protein that could interfere with protein function or expression. In contrast, ZeBaTA-based constructs for protein tagging result in a short linker with only three amino acids (Supplemental Fig. S1), thereby reducing the potential for interference with the tag-protein fusion. Last, the XcmI recognition sequences CCANNNNN/NNNNTGG, where N can be any pyrimidine or purine, is more flexible for designing special vectors for high-throughput functional analysis.
In this article, we demonstrate the flexibility of this system by developing a ZeBaTA-amiRNA vector for rice functional genomics (Fig. 5C; Supplemental Fig. S3). With the introduction of a single base mutation of GCA→CCA and TGC→TGG on the 5′ and 3′ stemloop backbones of an endogenous rice miRNA precursor Osa-MIR528 (Warthmann et al., 2008), respectively, the modified vector allows for generating an expression T-vector with preassembled 5′ and 3′ Osa-MIR528 stemloop backbone sequences. Thus, amiRNA expression constructs can be simply generated by cloning an amiRNA-amiRNA* fragment generated from a single-step PCR. Prediction of the original and modified Osa-MIR528 stemloop gave exactly the same secondary structures (Supplemental Fig. S4), suggesting that the modification of the stemloop backbone did not affect the structural features of the miRNA precursor. Further stable transformation using vectors based on the original or modified stemloop backbones gave a similar high efficiency for targeted OsPDS silencing, indicating that without changing structural features, the modified precursor stemloop backbone can functionally express a miRNA to trigger targeted gene silencing. Although we did not apply the approach to other miRNA precursors, our results suggest that this approach should be amenable to different amiRNA systems in other plants.
CONCLUSION
We developed the novel and versatile ZeBaTA cloning system for plant functional genomics studies. The newly developed 12 transient and 12 stable expression T-vectors in this study allow for direct cloning of PCR-amplified genes/fragments of interest for constitutive expression, gene silencing, protein tagging, protein subcellular localization detection, and promoter activity analysis in plants. We have demonstrated the usefulness of the system in rice and Arabidopsis. The ZeBaTA system can be easily adapted for the generation of more specialized expression vectors for other plants, as well as for E. coli, yeast, fungus, insect, and mammalian systems. In addition, the use of the basic ZeBaTA vector for PCR-fragment cloning will save a considerable amount of money for a laboratory to purchase commercial TA cloning kits. Therefore, our ZeBaTA system provides a general and cost-efficient molecular cloning platform for all molecular laboratories.
MATERIALS AND METHODS
ZeBaTA Vector Construction and Propagation
Standard molecular manipulations (Sambrook and Russell, 2001) were performed to make all the constructs. Details of plasmid construction are available in Supplemental Materials and Methods S1. The ZeBaTA vectors carry the ccdB gene, which inhibits growth of most Escherichia coli strains by expressing the CcdB protein to interfere with their DNA gyrase. All ZeBaTA vectors were propagated in the E. coli strain DB3.1, which contains a specific mutation in the gyrase gene, and thus insensitive to CcdB toxic protein (Hartley et al., 2000).
TA Cloning with ZeBaTA Vectors
The ZeBaTA vectors were digested with XcmI (New England Biolabs). PCR products were amplified using either a proofreading Pfu DNA polymerase (Stratagene) followed by the A-addition procedure (technical manual of pGEM-T and pGEM-T easy vector systems; http://www.promega.com/tbs/tm042/tm042.pdf) or a Platinum DNA polymerase (Invitrogen). The digested ZeBaTA plasmids and the PCR-amplified products were purified from agarose gels after electrophoresis using the Qiagen MinElute gel extraction kit (Qiagen), and product concentrations were then measured using a NanoDrop ND-1000 spectrophotometer (Nanodrop Technologies). T4 DNA ligases from Promega (catalog no. M1804; Promega), and USB Corporation (no. 70005) were tested for TA cloning efficiency. For ZeBaTA cloning, the ligation was carried out using the T4 DNA ligase from Promega (catalog no. M180; Promega) in a total volume of 10 μL containing 50 ng of T-vector and the corresponding volume of PCR product with a standard insert-to-vector molar ratio around 6:1. The ligation reaction mixture was transformed into E. coli strain DH 10B by electroporation. All recombinant plasmids identified from individual E coli colonies were verified by sequencing.
Rice Protoplast Transient Expression
Rice (Oryza sativa) protoplasts were isolated from 2-week-old seedlings of rice cv Nipponbare. Protoplast preparation and transfection were carried out by following previously described procedures (Chen et al., 2006b). After transfection, protoplasts were incubated in darkness for about 16 h at room temperature.
GUS, Western-Blot, and Fluorescence Microscopy Assays
GUS, western-blot, and fluorescence microscopy assays of transiently transfected rice protoplast samples were carried out following previously described procedures (Chen et al., 2006b). For GUS assays, protoplasts were cotransfected with the GUS construct and a 35S-LUC construct as the internal control. GUS assays were performed using 4-methylumbelliferyl-β-d-glucuronide (MUG; Sigma) as the substrate according to Jefferson et al. (1987). LUC assays were performed by using the Promega luciferase assay system (Promega). The GUS activity in each sample was expressed relative to LUC activity to normalize data for variation in transfection efficiency and cell viability. For western-blot analysis, total protein isolated from transfected protoplast samples was subjected to dodecyl sulfate-PAGE. A rat monoclonal anti-HA antibody (Roche Molecular Biochemicals) was used to detect the HA-tagged GFP protein in protoplasts. Fluorescence microscopy was performed with a Nikon Eclipse 80i fluorescence microscope (Nikon). Excitation and emission filters Ex450-490/BA520-560 and Ex540-580/BA600-660 were used for GFP and DsRed, respectively.
Plant Transformation and Analysis
For Arabidopsis (Arabidopsis thaliana) transformation, the ZeBaTA vectors were first introduced into Agrobacterium tumefaciens GV3101 by electroporation. Arabidopsis cv Col-0 plants were transformed via the floral-dip method as described by Clough and Bent (1998). Arabidopsis seeds obtained after transformation were plated on one-half-strength Murashige and Skoog medium containing 30 mg L−1 hygromycin and 100 mg L−1 carbenicillin for selection. GUS histochemical staining was carried out as described by Jefferson et al. (1987).
For rice transformation, the ZeBaTA vectors were introduced into A. tumefaciens LBA4404 by electroporation. Calli of rice cv Nipponbare were used for transformation via the Agrobacterium-mediated procedure as described previously (Hiei et al., 1994; Yin and Wang, 2000). Semiquantitative reverse transcription (RT)-PCR was used to examine the level of RNA accumulation of control plants and transgenic albino plants. Total RNA was isolated from leaf tissues by using TRIzol reagent (Invitrogen) and was treated with RNase-free DNase I (Ambion) to remove DNA contamination. One microgram of DNase I-treated total RNA was subjected to RT using the Promega RT system (Promega). RT-PCR was carried out under standard conditions with specific primers for OsPDS (Miki and Shimamoto, 2004), and the internal control ubiquitin genes.
Sequence data from this article can be found in the GenBank data library under accession numbers FJ905202 to FJ905213 (for pXSN, pXUN, pXSN-FLAG, pXUN-FLAG, pXSN-HA, pXUN-HA, pXSN-Myc, pXUN-Myc, pX-DG, pX-DR, pXGUS-P, and pXGFP-P, respectively), and FJ905214 to FJ905225 (for pCXSN, pCXUN, pCXSN-FLAG, pCXUN-FLAG, pCXSN-HA, pCXUN-HA, pCXSN-Myc, pCXUN-Myc, pCX-DG, pCX-DR, pCXGUS-P, and pCXGFP-P, respectively).
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Figure S1. Sequence structure of the XcmI-ccdB-XcmI cassette in ZeBaTA vectors.
Supplemental Figure S2. TA cloning test of ZeBaTA-based A. tumefaciens binary vector.
Supplemental Figure S3. Schematic representation of single-step PCR generation of amiRNA construct for gene silencing in rice.
Supplemental Figure S4. Predicted secondary structure of the mutated osa-MIR528 stemloop.
Supplemental Table S1. Primers used in this study.
Supplemental Materials and Methods S1. Construction of ZeBaTA vectors and plant expression vectors for proof of concept testing.
ACKNOWLEDGMENTS
We are grateful to Dr. Michael M. Goodin, University of Kentucky, Lexington, KY, for kindly providing the pGDG and pGDR plasmids; Dr. Ko Shimamoto, Nara Institute of Science and Technology, Ikoma, Japan, for kindly providing the pANDAmini vector; and Dr. Philippe Herve, International Rice Research Institute, Metro Manila, Philippines, for kindly providing the pNW55 plasmid containing rice miRNA precursor Osa-MIR528. We thank Dr. II-Pyung Ahn and Dr. Mao-Feng Chai for helping with Arabidopsis transformation, and Dr. Bruce Jaffee and Dr. Tom Mitchell for editing of the manuscript.
LITERATURE CITED
Lei Z, Zhao P, Cao M, Cui R, Chen X, Xiong L, Zhang Q, Oliver DJ, Xiang C (
Mead DA, Pey NK, Herrnstadt C, Marcil RA, Smith LM (
Sambrook J, Russell D (
Author notes
This work was supported by the National Science Foundation-Plant Genome Research Program (grant no. 0605017).
Corresponding author; e-mail [email protected].
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (www.plantphysiol.org) is: Guo-Liang Wang ([email protected]).
Some figures in this article are displayed in color online but in black and white in the print edition.
The online version of this article contains Web-only data.
Open access articles can be viewed online without a subscription.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}