





Abstract
DNA methylation plays vital roles in repressing transposable element activity and regulating gene expression. The chromatin-remodeling factor Decrease in DNA methylation 1 (DDM1) is crucial for maintaining DNA methylation across diverse plant species, and is required for RNA-directed DNA methylation (RdDM) to maintain mCHH islands in maize (Zea mays). However, the mechanisms by which DDM1 is involved in RdDM are not well understood. In this work, we used chromatin immunoprecipitation coupled with high-throughput sequencing to ascertain the genome-wide occupancy of ZmDDM1 in the maize genome. The results revealed that ZmDDM1 recognized an 8-bp-long GC-rich degenerate DNA sequence motif, which is enriched in transcription start sites and other euchromatic regions. Meanwhile, 24-nucleotide siRNAs and CHH methylation were delineated at the edge of ZmDDM1-occupied sites. ZmDDM1 co-purified with Argonaute 4 (ZmAGO4) proteins, providing further evidence that ZmDDM1 is a component of RdDM complexes in planta. Consistent with this, the vast majority of ZmDDM1-targeted regions co-localized with ZmAGO4-bound genomic sites. Overall, our results suggest a model that ZmDDM1 may be recruited to euchromatic regions via recognition of a GC-rich motif, thereby remodeling chromatin to provide access for RdDM activities in maize.
Introduction
DNA methylation at the 5′-carbon of the cytosine is a conserved mark widespread in eukaryotes, which plays important roles in transposable element (TE) silencing, gene expression, and genome stability (Law and Jacobsen, 2010; Bucher et al., 2012; Kim and Zilberman, 2014; Zhang et al., 2018). Although DNA methylation occurs mainly in the CG context in mammals, it can occur in any cytosine context in plants (CG, CHG, and CHH, where H is A, T, or C; Law and Jacobsen, 2010). There are distinct epigenetic pathways that maintain different contexts of DNA methylation in plants. CG methylation is maintained by METHYLTRANSFERASE 1 (MET1; Finnegan et al., 1996; Ronemus et al., 1996), while CHROMOMETHYLASE 3 (CMT3) and CMT2 are responsible for maintaining CHG and CHH, respectively (Lindroth et al., 2001; Cao and Jacobsen, 2002; Cao et al., 2003; Du et al., 2012; Stroud et al., 2013, 2014).
De novo methylation mainly relies on the RNA-directed DNA methylation (RdDM) pathway, which is guided by small RNAs to specific genomic regions (Matzke and Mosher, 2014; Matzke et al., 2015; Zhang et al., 2018). The canonical RdDM pathway in the model plant Arabidopsis thaliana involves several steps that can be divided in two major arms. Briefly, in the first arm of the pathway, Pol IV transcripts are copied into double-stranded RNAs by RNA-DEPENDENT RNA POLYMERASE2 (RDR2) and diced by different DICER-like proteins into small interfering RNAs, which are then loaded into ARGONAUTE4 (AGO4) protein (Xie et al., 2004; Zilberman et al., 2004; Qi et al., 2006; Mi et al., 2008). In the second arm of the pathway, transcribing Pol V recruits siRNA-loaded AGO4, which in turn triggers the recruitment of the DNA methyltransferase DOMAINS REARRANGED METHYLTRANSFERASE2 (DRM2) to mediate methylation of cytosine in all sequence contexts (Pontes et al., 2006; Wierzbicki et al., 2009; Matzke and Mosher, 2014).
The basic unit-forming chromatin is the nucleosome. Canonical nucleosomes consist of an octamer of histone proteins, around which DNA is wrapped almost twice. The nucleosomes present natural barriers for transcription factor or chromatin binding protein to access DNA regulatory elements (Struhl and Segal, 2013). ATP-dependent chromatin remodeling factors could help nucleosomal translocation using the energy from ATP hydrolysis in order to provide increased access to DNA (Clapier et al., 2017). Decrease in DNA methylation 1 (DDM1) is a SWI/SNF2-like ATP-dependent chromatin remodeler that is crucial for maintaining DNA methylation, particularly at heterochromatin (Vongs et al., 1993; Jeddeloh et al., 1999; Zemach et al., 2013). Disruption of DDM1 in Arabidopsis causes a profound decrease in all contexts of DNA methylation in heterochromatic TEs and other DNA repeats, leading to a widespread transcriptional activation of TEs (Tsukahara et al., 2009; Zemach et al., 2013). Arabidopsis DDM1 can shift nucleosomes in vitro (Brzeski and Jerzmanowski, 2003) and allow DNA methyltransferases to access the histone H1-containing heterochromatin to methylate nucleosome core DNA in vivo (Zemach et al., 2013; Lyons and Zilberman, 2017). In addition, DDM1 and RdDM in Arabidopsis act as two separate pathways synergistically regulating all TE CHH methylation, in which DDM1 is required to maintain CHH methylation at the body of heterochromatic long TEs, while RdDM mediates CHH methyltion at euchromatic short TEs and the edges of long TEs (Zemach et al., 2013).
The maize (Zea mays) genome displays a distinct chromatin landscape compared with Arabidopsis: ∼80% of the maize genome is composed of TEs (Baucom et al., 2009; Schnable et al., 2009), which are scattered across the genome with an abundance of them located near protein coding genes. Although heavily methylated in the CG (>80%) and CHG (>60%) context, the majority of maize TEs contain extremely low levels of mCHH (<5%), which is proposed to occur due to the lack of a CMT2 homolog in the maize genome (Zemach et al., 2013; Bewick et al., 2017; Fu et al., 2018; Long et al., 2019). However, there exist thousands of specific regions proximal to genes with hyper-methylated CHH (>25%), and hence those regions were referred to as mCHH islands (Gent et al., 2013, 2014; Li et al., 2014, 2015; Fu et al., 2018; Long et al., 2019).
The mCHH islands often have depletion of H3K9me2 and are proposed to set up boundaries between euchromatin and heterochromatin (Gent et al., 2014; Li et al., 2015). In addition, the maintenance of mCHH islands in maize was demonstrated to primarily rely on RdDM (Alleman et al., 2006; Nobuta et al., 2008; Li et al., 2014, 2015). The disruption of maize DDM1 abolished the occurrence of mCHH islands, highlighting the involvement of maize DDM1 in RdDM (Fu et al., 2018; Long et al., 2019). Also, the interaction of DDM1 with RdDM has been reported in rice (Oryza sativa) and tomato (Solanum lycopersicum), although the precise relationship varied among different species (Tan et al., 2016, 2018; Corem et al., 2018). The mechanisms by which DDM1 is involved in RdDM remain elusive.
In this study, taking advantage of the specific ZmDDM1 antibodies, we performed chromatin immunoprecipitation (IP) coupled with high-throughput sequencing (ChIP-seq) analysis to characterize the genome-wide occupancy of ZmDDM1 in developing maize embryos. The results revealed that ZmDDM1 bound to a GC-rich motif “GCYGCYGC”, and located primarily on transcription start sites (TSSs) or other intergenic regions. Surprisingly, rather than associating with heterochromatic features, the center of DDM1-targeted genomic sites was depleted of DNA methylation, while 24-nt siRNAs and CHH methylation accumulated at the edges of ZmDDM1-occupied sites. In addition, we found that ZmDDM1 interacted with the RdDM complex in vivo, and ZmDDM1-targeted regions largely overlapped with ZmDDM1 co-purified with Argonaute 4 (ZmAGO4)-bound genomic sites. Overall, these results suggest that ZmDDM1 may act as a component of RdDM to remodel chromatin to provide access for RdDM into euchromatin. Therefore, our findings provide mechanistic insights into how ZmDDM1 participates in RdDM in maize.
Results
Genome-wide characterization of ZmDDM1 occupancy in maize
The maize genome contains two homologous DDM1 genes, named ZmDDM1A/CHR101 (Zm00001d007978) and ZmDDM1B/CHR106 (Zm00001d033827), which are redundantly required for RdDM in mCHH islands (Fu et al., 2018; Long et al., 2019). To identify genomic sites targeted by ZmDDM1, we conducted ChIP-seq analysis utilizing the specific antibodies against ZmDDM1A and ZmDDM1B, which were developed in our previous study (Long et al., 2019). The developing embryos collected from wild-type, and the zmddm1a and zmddm1b mutant plants were used as materials, which were at the same stage of embryo development used in our previous study to produce whole-genome bisulfite sequencing dataset (Long et al., 2019). We obtained a total of ∼1.28 billion 2 × 150 paired reads, with an average of ∼80 million paired reads for each sample (Supplemental Table S1). These reads were mapped to the B73 genome (Jiao et al., 2017) with a unique mapping rate from 70% to 92%, corresponding to an average of ∼65 million mapped reads for each sample (Supplemental Table S1).
Using model-based analysis of ChIP-seq (MACS; v2.2.6), we identified a total of 36,255 ZmDDM1A concordant peaks and 41,613 ZmDDM1B peaks (Supplemental Figure S1A). In contrast, ChIP signals were hardly detectable in the mutant (Figure 1A), and only a trivial number of ZmDDM1A or ZmDDM1B peaks could be detected in zmddm1a (n = 346) or zmddm1b (n = 232) mutants, respectively (Supplemental Figure S1B), strongly corroborating the high fidelity of our antibodies and subsequent ChIP-seq data.
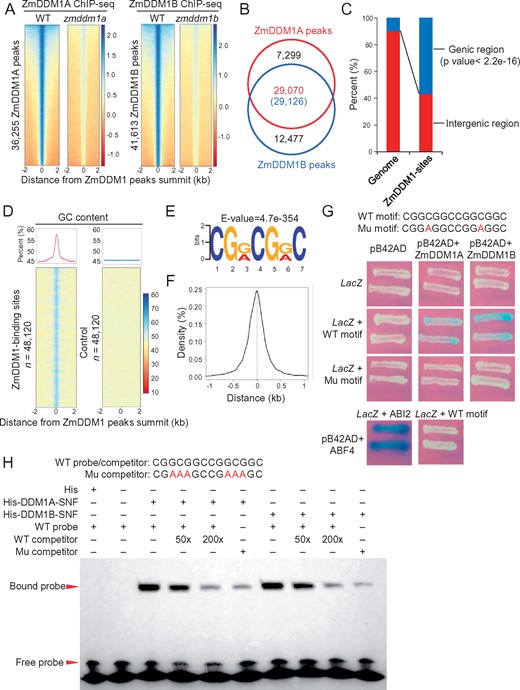
Genome-wide DDM1 occupancy in maize developing embryo. A, Heat maps of ChIP-seq signals at the 36,255 ZmDDM1A peaks (left) and 41,613 ZmDDM1B peaks (right) with flanking 2-kb regions between wild-type (WT) and the corresponding mutants. The regions were ranked according to the strength of ChIP signals from highest (top) to lowest (bottom). The strength of ChIP signals was measured as log2 (ChIP/input). B, The overlap of ZmDDM1A and ZmDDM1B peaks. The number in the parentheses indicated the number of ZmDDM1B peaks that overlap ZmDDM1A. C, Percentages of ZmDDM1 peaks located within genic or intergenic regions. The fraction of genic (defined from upstream 1kb of TSS to the downstream 1kb to TTS) and intergenic regions in the maize genome were shown for comparison. The asterisk indicated that ZmDDM1 peaks were significantly enriched at genic regions (binomial test; P <2.2e−16). D, Average plots and heat maps of GC content at the 48,190 ZmDDM1-binding sites and control loci. GC content was calculated as (G+C)/(A+T+C+G)*100% in nonoverlapping 50-bp window. E, The “CGRCGRC “motif was identified as the most prominent ZmDDM1 binding motif using MEME-ChIP program. F, Density plot of the distance of “CGRCGRC” motif to ZmDDM1 peak summit. G, Y1H showing the interactions between ZmDDM1 and a DNA sequence consisting of two copies of “CGRCGRC” motif. A motif with mutation of a “G” to “A” (highlighted as red character) was included to examine the signal. An empty vector without expressing ZmDDM1 was served as negative control while interaction between promoter of ABI2 and ABF4 was served as positive control (Wang et al., 2019). H, EMSA demonstrated the interactions between ZmDDM1 and the DNA motif. The recombinant protein 6× His-ZmDDM1 retarded the shift of probe. 50× and 200× unlabeled probes were used as competitors. “+” and “−” indicated presence and absence, respectively. The probe consisting of two 14-bp long motif with 3-bp mutation (highlighted in red; Mu competitor) was used to test the EMSA signal as well
The vast majority of ZmDDM1A (80.63%) and ZmDDM1B peaks (69.99%) overlapped (Figure 1B). It is noted that even for peaks called as either ZmDDM1A- or ZmDDM1B-specific, the enriched ChIP reads could be observed for the other protein but appeared only slightly below the peak-calling threshold (Supplemental Figure S1, C and D). These results indicate that ZmDDM1A and ZmDDM1B largely occupy the same genomic sites, further supporting the notion that ZmDDM1A and ZmDDM1B are functionally redundant (Li et al., 2014; Fu et al., 2018; Long et al., 2019). Certainly, we cannot rule out the possibility that ZmDDM1A or ZmDDM1B work specifically at some genomic sites at this point. To obtain a complete portrait of ZmDDM1 occupancy, we decided to merge ZmDDM1A and ZmDDM1B peaks together, and denoted the whole as ZmDDM1 peaks, which results in a total of 48,190 nonredundant peaks.
To examine the distribution of ZmDDM1 peaks in the maize genome, we mapped these regions relative to genomic annotations. Statistical analysis revealed that ZmDDM1 peaks were significantly overrepresented in genic regions (Figure 1C). To further ascertain genomic features underlying ZmDDM1 peaks, we analyzed the DNA nucleotide composition of the regions occupied by ZmDDM1. The GC content was much higher in ZmDDM1-binding sites than their flanking regions (Figure 1D). The enrichment of GC content within ZmDDM1-binding sites was not caused by stochastic effect because a control set of loci randomly selected from maize genome did not associate with elevated GC content (Figure 1D). Therefore, our results indicate that ZmDDM1 predominantly occupies the GC-rich sites. Consistent with this, an 8-bp-long GC-rich degenerate DNA sequence (CGYCGYCG, where Y is G or A; E = 4.7E−354) was identified as the most significant motif among ZmDDM1-binding sites (Figure 1E), and centered at the summit of ZmDDM1 peaks (Figure 1F).
To verify that ZmDDM1 can bind to this DNA sequence motif CGYCGYCG in vitro, we performed a yeast one-hybrid assay (Y1H). The Y1H results showed that the full-length ZmDDM1A or ZmDDM1B protein could bind to a 14-bp DNA sequence consisting of two copies of CGGCGGC motif (Figure 1G). In contrast, substitution of one core nucleoside inside this motif (C to A) could block the binding of ZmDDM1A or ZmDDM1B to this motif (Figure 1G). The binding of the ZmDDM1A or ZmDDM1B protein to the motif was further confirmed via electrophoretic mobility shift assays (EMSAs). The results revealed that the expressed ZmDDM1 domain can efficiently interact with the GC-rich DNA probe (Figure 1H). Taken together, our results indicate that ZmDDM1 targets GC-rich regions at a genome-wide scale.
Genic ZmDDM1-binding sites locate near transcription start sites and associate with gene transcriptional state
Given that 56.62% (n = 27,287) of ZmDDM1-binding sites occupy genic regions, we next sought to examine the distribution of genic ZmDDM1-binding according to distinct genic segments. Genic regions were split into 5′-UTR, 3′-UTR, exon, intron, 5′-flanking 1-kb region, and 3′-flanking 1-kb region. Many (46.35%) of the genic ZmDDM1-binding sites are located in the 5′- flanking 1-kb region (Supplemental Figure S2). Interestingly, plotting ZmDDM1-ChIP signals along gene body and flanking 2-kb region revealed that ZmDDM1 ChIP signals were highly enriched near TSSs, and to a less extent at transcription termination sites (TTSs), but much less in the gene body (Figure 2A).
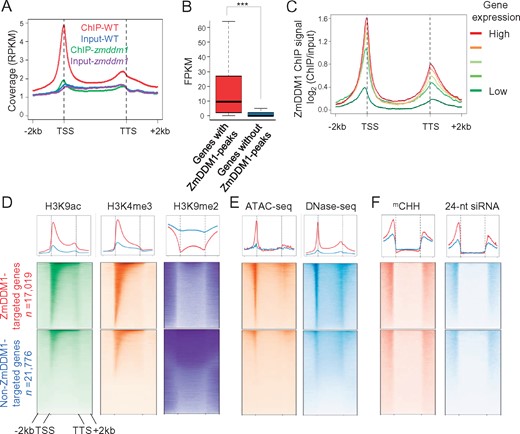
ZmDDM1 preferentially binds to transcription start sites with active gene expression. A, A metaplot exhibiting the average of reads coverage (normalized as reads per kilobase per million, RPKM) from ZmDDM1 ChIP-seq and input (ZmDDM1A and ZmDDM1B ChIP-seq libraries in WT and mutant were combined) across genes. The size of each annotated gene was scaled to 4 kb and normalized reads coverage was calculated in 50-bp window. B, Box plots showing expression levels of ZmDDM1-bound and ZmDDM1-unbound genes. The asterisk indicated a statistically significant difference using two-sided Wilcoxon test (P <2.2e−16). FPKM, fragments per kilobase per million. C, ZmDDM1 ChIP enrichment profile of different groups of genes classified by expression level from low expressed to high expressed in developing embryo. To produce enrichment relative to the genome average, ChIP-seq counts for the 50-bp intervals in each set of genes were normalized by read counts derived from input. D–F, Meta-analysis of H3K4me3, H3K9ac, H3K9me2 (D), ATAC-seq, DNase-seq (E), mCHH and 24-nt siRNA (F) at gene body and flanking 2 kb of ZmDDM1-occupied genes and non-ZmDDM1-occupied genes.
The enrichment of ZmDDM1 ChIP signal in TSSs prompted us to ask whether the binding of ZmDDM1 on TSSs is associated with gene transcription. To answer this question, we performed mRNA-seq analysis from wild-type developing embryos, which are the same tissues used for ChIP-seq experiments, as described above (Supplemental Table S1). As shown in Figure 2B, the expression levels of 17,019 genes (Supplemental Data Set S1) occupied by ZmDDM1 were significantly higher than the remaining 21,776 genes (Student’s t test, P < 0.001), which was not occupied by ZmDDM1 (Supplemental Data Set S2). In addition, gene expression increased with increasing ZmDDM1 occupancy after all genes were categorized into five classes according to their transcript abundances (Figure 2C; “Methods”). These results indicate that the extent of ZmDDM1 occupancy in genic DNA is intrinsically coordinated with the active state of gene transcription.
Based on the association between ZmDDM1 occupancy and gene transcription, we hypothesized that ZmDDM1 may access genic DNA associating with open and active chromatin. To test this hypothesis, we compared chromatin state and accessibility between ZmDDM1-occupied genes and non-ZmDDM1-occupied genes. By integrating publicly available datasets (Oka et al., 2017; Ricci et al., 2019; Supplemental Table S2), we found that the levels of two classically active chromatin marks, H3K4me3 and H3K9ac, were much higher for ZmDDM1-occupied genes than non-ZmDDM1-occupied genes (Figure 2D). In addition, we generated an in-house data set of H3K9me2, an inactive chromatin mark (Supplemental Table S1), and found that the level of H3K9me2 was much lower for ZmDDM1-occupied genes than non-ZmDDM1-occupied genes (Figure 2D). Moreover, the ATAC-seq and DNase-seq signals (Oka et al., 2017; Ricci et al., 2019; Supplemental Table S2), two methods to assay chromatin accessibility, were much stronger at the TSSs of ZmDDM1-occupied genes than that of non-ZmDDM1-occupied genes (Figure 2E). These results indicate that ZmDDM1 is enriched at genic regions with active and open chromatin.
Our previous study demonstrated that the formation of mCHH islands, which generally occur at the 5′- and 3′-ends of expressed genes, relies on ZmDDM1 (Long et al., 2019). To better understand the mechanism by which ZmDDM1 is required for the establishment of mCHH islands, we investigated the occurrence of CHH methylation between ZmDDM1-occupied genes and non-ZmDDM1-occupied genes. Interestingly, we found that the ZmDDM1-occupied genes exhibited much higher levels of CHH methylation and 24-nt siRNA at flanking regions than those of non-ZmDDM1-occupied genes (Figure 2F;Supplemental Figure S3), indicating that ZmDDM1-mediated RdDM operates more actively for ZmDDM1-occupied genes than non-ZmDDM1-occupied genes. These results further corroborate that ZmDDM1 is required for the formation of mCHH islands. Meanwhile, it is worth noting that although mCHH and 24-nt siRNA were more enriched at ZmDDM1-occupied genes, the elevated levels of mCHH and 24-nt siRNA were present and enriched flanking non-ZmDDM1-occupied genes (Figure 2F), potentially suggesting the existence of another RdDM pathway or alternatively suggesting that a subset of the non-ZmDDM1 occupied genes may actually have ZmDDM1 peaks that were not strong enough to be called as significant.
Intergenic ZmDDM1-binding sites are still associated with active chromatin and delineate mCHH at their edges
The vast majority of the maize genome consists of various TEs, which are scattered throughout the genome and highly DNA methylated to safeguard genome stability (Baucom et al., 2009; Schnable et al., 2009; Regulski et al., 2013). Out of 20,785 ZmDDM1-binding sites located in intergenic regions, 39.76% (n = 8,266) overlapped with TEs, mainly represented by Helitron and LTR retrotransposons (Supplemental Figure S4; “Methods”). Since the majority of intergenic regions are highly methylated, we thought that the chromatin state of these intergenic ZmDDM1-binding sites would be repressive. However, the center of intergenic ZmDDM1-binding sites exhibited typical features of active chromatin with high levels of H3K4me3 and H3K9ac (Figure 3A) but low levels of H3K9me2, CG and CHG methylation (Figure 3B). In addition, ATAC-seq and DNase-seq signals were also significantly elevated at ZmDDM1-binding sites than their flanking regions (Figure 3C). Moreover, the active chromatin state of intergenic ZmDDM1-binding sites remained even when considering peaks overlapped with TEs (Supplemental Figure S5). In contrast, a control set of loci randomly shuffled from maize intergenic regions with the equal number and length of intergenic ZmDDM1 peaks yielded a completely distinct pattern, as expected based on the general repressive and inaccessible characters of intergenic regions in maize genome (Figure 3, A and B). Overall, these results indicate that ZmDDM1 could also bind to intergenic regions with active and open chromatin.
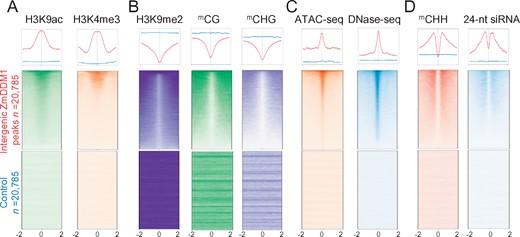
ZmDDM1 still binds to active chromatin in intergenic regions. A–D, Meta-analysis of H3K9ac, H3K4me3 (active chromatin mark, A), H3K9me2, mCG, mCHG (repressive chromatin mark, B), ATAC-seq, DNase-seq (chromatin accessibility, C), mCHH and 24-nt siRNA (RdDM mark, D) at 20,785 intergenic ZmDDM1 peaks and control loci, respectively. Shown were ±2 kb from summit of ZmDDM1 peaks or control peaks.
To assess whether intergenic ZmDDM1-binding sites are associated with elevated mCHH, we plotted ZmDDM1-ChIP signals and mCHH together. As shown in Figure 3D, highly elevated levels of mCHH and 24-nt siRNA were positioned at both edges of ZmDDM1-binding sites (Figure 3D). Taken together, these results suggest that ZmDDM1 targets active and open chromatin irrespective of genic and nongenic regions, consequently promoting the formation of mCHH neighboring its binding sites.
ZmDDM1 copurified with ZmAGO4 in planta
The involvement of ZmDDM1 in forming mCHH prompts us to explore whether ZmDDM1 may directly interact with the components of RdDM. To address this, we pursued an IP approach using the anti-ZmDDM1B antibody. It is noted that the anti-ZmDDM1B antibody was used in this analysis since ZmDDM1B expressed at a higher level than ZmDDM1A (Long et al., 2019). After the IP product was separated in a polyacrylamide gel, we observed two major visible bands that were present in wild-type following silver staining but absent from the zmddm1b mutant (Figure 4A). These two bands were excised from the gel and subjected to mass spectrometry (MS) analysis. One of the bands corresponded to the ZmDDM1B protein (Supplemental Figure S6, A and B and Supplemental Table S3), whereas the other one contained peptides covering 38% of the protein sequence corresponding to ZmAGO4A (Zm00001d008249; Supplemental Figure 6C and Supplemental Table S3). To confirm the association of ZmDDM1B with ZmAGO4, co-IP (Co-IP) assay was performed using a polyclonal antibody raised against ZmAGO4, whose specificity was validated by ZmAGO4-IP followed by mass-spectrometric analysis (Supplemental Figure S7).
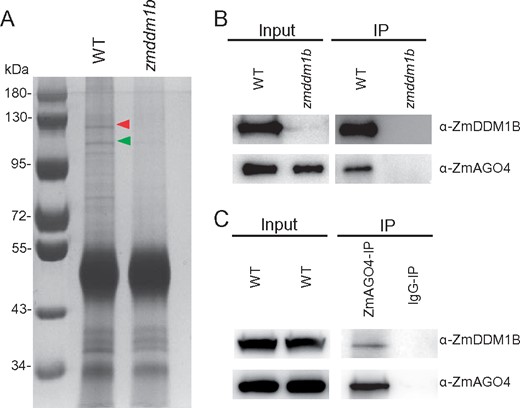
ZmDDM1B co-purify with ZmAGO4 in maize developing embryos. A, The protein complex from WT maize developing embryos was immunopurified using specific antibody against ZmDDM1B, and separated on 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis. zmddm1b mutant embryos were used for the control sample. The proteins were visualized by silver staining. The positions of protein size markers were shown at the left of gel. The ZmAGO4 protein band was indicated by red arrowhead and the ZmDDM1B protein band was indicated by green arrowhead. B, Co-IP of ZmDDM1B with ZmAGO4. ZmAGO4 was able to be detected from ZmDDM1B-immunoprecipitates from WT embryos. ZmAGO4 failed to be detected from ZmDDM1B-IP product from zmddm1b embryo. C, Co-IP of ZmAGO4 with ZmDDM1B. ZmDDM1B was able to be detected from ZmAGO4-immunoprecipitates from WT embryos. IgG purified from pre-immune serum was used for IP in WT embryo as a negative control.
There are two homologs of AGO4 in the maize genome, which share 95% of identity (Supplemental Figure S8), making it impractical to raise specific antibodies against individual ZmAGO4A or ZmAGO4B. The ZmAGO4 proteins could readily be detected in ZmDDM1B-IP products from wild-type plants but not from the zmddm1b mutant (Figure 4B). Vice versa, ZmDDM1B protein could also be identified in ZmAGO4-IP products (Figure 4C). These results indicate that ZmDDM1B could be co-purified with ZmAGO4 in vivo, further supporting the association of ZmDDM1 with RdDM.
ZmDDM1 channels ZmAGO4 into active chromatin
The co-purification of ZmDDM1 with ZmAGO4 prompted us to investigate the genome-wide occupancy of ZmAGO4-involved RdDM complex. ChIP-seq utilizing the ZmAGO4 antibody was conducted using the same wild-type samples as ZmDDM1 ChIP-seq experiment (“Methods”). Using the identical MACS2 parameters for ZmDDM1-ChIP peak calling, we identified a comparable number of ZmAGO4 peaks between two biologically independent replicates with high degree of overlap (>90%; Supplemental Figure S9A), suggesting that our ChIP-seq results are reliable. Similarly, only concordant peaks shared by two biological replicates were kept for the subsequent analyzes, yielding a total of 89,989 ZmAGO4 peaks (Supplemental Figure S9A).
To further assess the relationship between ZmAGO4 and RdDM in maize, we monitored the level of mCHH within ZmAGO4-ChIP peaks. A meta-plot of CHH methylation across ZmAGO4 peaks and its flanking 2-kb regions revealed CHH sites were hypermethylated in the center of ZmAGO4 peaks (Figure 5A). In addition, 24-nt siRNAs, but not 21- or 22-nt sRNAs, were enriched in the summit of ZmAGO4 peaks, confirming the role of ZmAGO4 in carrying 24-nt siRNA into RdDM loci in maize (Figure 5B). In addition, there was a strong positive relationship between CHH methylation level, 24-nt siRNA abundance, and ZmAGO4 ChIP signal (Supplemental Figure S9B). Meta-gene analysis of ZmAGO4-ChIP signals revealed that ZmAGO4 mainly localized slightly upstream of TSS and downstream of TTS (Figure 5C), resembling the pattern of CHH methylation over genes (Gent et al., 2013; Li et al., 2014, 2015; Secco et al., 2015; Song et al., 2015; Long et al., 2019). Taken together, these lines of evidence not only support the reliability of our ZmAGO4-ChIP analysis, but also agree that ZmAGO4 is the important component of RdDM and responsible for the mCHH establishment in maize, which has been previously well documented in Arabidopsis (Zilberman et al., 2004; Duan et al., 2015) and rice (Wu et al., 2010).
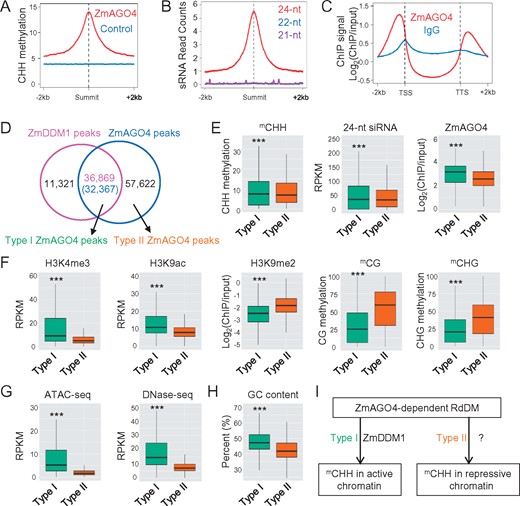
ZmDDM1 directs the occurrence of ZmAGO4-dependent RdDM on active chromatin. A, Profile of CHH methylation in the ZmAGO4 peaks summit-centered genomic regions (red line). Profile of CHH methylation at a set of control loci was also shown (blue line). B, Abundance of 21-, 22-, and 24-nt siRNA in the ZmAGO4 peaks summit-centered genomic regions. C, Meta-plots showing average ZmAGO4 ChIP signals across all of annotated protein-coding genes. lgG-ChIP signal was shown in the same plot as a negative control. D, Venn diagram showing the overlap between ZmDDM1 and ZmAGO4 peaks. E, Comparisons of CHH methylation, 24-nt siRNA levels and ZmAGO4 occupancy between types I and II ZmAGO4 peaks. Triple asterisks indicated P < 2.2e−16 (two-sided Kolmogorov–Smirnov test). F, Comparisons of H3K4me3, H3K9ac, H3K9me2, CG, and CHG methylation level between types I and II ZmAGO4 peaks. Triple asterisks indicated P <2.2e−16 (two-sided Kolmogorov–Smirnov test). G, Comparisons of ATAC-seq and DNase-seq abundance between types I and II ZmAGO4-binding sites. Triple asterisks indicated P <2.2e−16 (two-sided Kolmogorov–Smirnov test). H, Comparisons of the GC content between types I and II ZmAGO4-binding sites. Triple asterisks indicated P <2.2e−16 (two-sided Kolmogorov–Smirnov test). I, A diagram depicting the likely existence of two ZmAGO4-involved RdDM pathways in maize. The first pathway is dependent on ZmDDM1 activity and occurs preferentially in active and open chromatin. The second pathway is independent of ZmDDM1 activity and associate with relatively repressive chromatin. It is noted that the other chromatin remodeler involving in the type II pathway remains elusive at this point. RPKM, reads per million per kilobase.
As the genome-wide localization of both ZmDDM1 and ZmAGO4 were ascertained, we examined their spatial correlation. As shown in Figure 5D, 76.5% of ZmDDM1 peaks overlapped with ZmAGO4 peaks (“Methods”), further supporting that ZmDDM1 acts as a component of RdDM in maize. However, only ∼35.97% of ZmAGO4 peaks overlapped with ZmDDM1 peaks (Figure 5D), suggestive of the probable existence of a ZmDDM1-independent RdDM pathway managing the other 64.13% of ZmAGO4-targeted RdDM sites. Thereby, for simplicity, we called those ZmAGO4-binding sites that overlapped with ZmDDM1 peaks as type I (n = 32,367), and the other ZmAGO4-binding sites without overlapping with ZmDDM1 peaks as type II (n = 57,622; Figure 5D;Supplemental Figure S9C).
The finding that there were both ZmDDM1-dependent and ZmDDM1-independent pathways generating ZmAGO4 targets raises the question of how these two pathways contribute to the establishment of mCHH. To address this, we first compared the CHH methylation level and 24-nt siRNA abundance between both types of ZmAGO4-binding sites. As shown in Figure 5E, although the 24-nt siRNA abundance is slightly higher within type I ZmAGO4 peaks than type II, the overall levels of CHH methylation were comparable between both types, suggesting that ZmDDM1-involved RdDM pathway operates as a similar extent with the other RdDM pathway in producing mCHH.
Given that ZmDDM1 is associated with active and open chromatin, we suspect that there may be differences in chromatin state and accessibility between types I and II ZmAGO4 peaks. Indeed, we found that the type I ZmAGO4-binding sites possessed higher levels of H3K4me3 and H3K9ac, but lower level of H3K9me2, CG, and CHG methylation than the type II ZmAGO4-binding sites (Figure 5F). In addition, the type I ZmAGO4-binding sites showed much higher levels of ATAC-seq and DNase-seq signals than the type II ZmAGO4-binding sites (Figure 5G). Moreover, the type I ZmAGO4-binding sites had much higher GC contents than type II (Figure 5H). Taken together, these results indicate that ZmDDM1-dependent ZmAGO4-binding sites tend to be more euchromatic and accessible than ZmDDM1-independent ZmAGO4 binding sites (Figure 5I).
Discussions
As an ATP-dependent chromatin remodeler, the widespread and critical roles of DDM1 in regulating the methylation of all cytosine contexts have been demonstrated in plants and mammals (Tao et al., 2011; Zemach et al., 2013; Yu et al., 2014; Lyons and Zilberman, 2017). However, the specific functional impacts of DDM1 seem to vary in different organisms. Especially, the role of DDM1 in RdDM to establish mCHH appears fairly divergent and species-specific. In Arabidopsis, DDM1 and RdDM work in two parallel pathways to synergistically mediate mCHH at all TE contexts (Zemach et al., 2013). In rice, DDM1 represses the transcription of noncoding RNAs and DRM2-mediated CHH methylation in heterochromatic regions, while it facilitates DMR2-mediated CHH methylation and the production of siRNAs in euchromatin regions (Tan et al., 2016, 2018). In tomato, the disruption of DDM1 resulted in the redistribution of CHH methylation and 24-nt siRNAs at a genome-wide scale (Corem et al., 2018). Our recent study demonstrated that dysfunction of DDM1 in maize abolished the global production of 24-nt sRNAs and the formation of mCHH, particularly in mCHH islands (Fu et al., 2018; Long et al., 2019). However, it is worth noting that the former conclusions about the role of DDM1 derived from observations of the altered methylation pattern after disrupting the ddm1 gene, raising the possibility that the multifaceted changes occurring in mutants may not be the direct output, but complicated by unintended pleiotropic effects. In this study, we profiled the genome-wide occupancy of ZmDDM1, and aimed to address in vivo how and where DDM1 binds to chromatin. Our current findings provide two lines of solid evidence supporting the action of ZmDDM1 as a component of RdDM in maize. First, ZmDDM1 could be co-purified with ZmAGO4, which is well-known as an essential factor in RdDM. Second, the majority of ZmDDM1-binding sites were co-localized with ZmAGO4-dependent RdDM genomic loci.
Illustrating the genome-wide landscape of ZmDDM1-binding sites allows us to ascertain two unprecedented chromatin features shaping ZmDDM1 localization in maize. First, ZmDDM1 binds to GC-rich chromatin and displays the autonomous ability to recognize a GC-rich motif “GCYGCYGC”. It has been known in mammals and yeast that GC- and AT-rich chromatin differ in their structure and state, with more active and more inactive in the two types of genomic regions, respectively (Dekker, 2007; Fenouil et al., 2012). This is consistent with the second nature of ZmDDM1-binding regions, which are marked by high levels of active histone marks. Therefore, ZmDDM1 functions to define the occurrence of RdDM pathway in active chromatin. This is in good agreement with the fundamental role of ZmDDM1 in maintaining mCHH islands, which are predominantly situated at both sides of the highly expressed genes (Gent et al., 2013; Li et al., 2015). The marked enrichment of ZmDDM1 in TSSs and the positive relationship of ZmDDM1 occupancy with transcript abundance suggest that ZmDDM1 may be especially active within open chromatin regions. The relationship between gene transcription, ZmDDM1 localization and mCHH formation seem intrinsically intercorrelated. In maize, the genic mCHH islands act to preserve the silencing state of transposons from activity of nearby genes (Li et al., 2015). In this scenario, the greater activity of gene transcription during maize embryo development may require high levels of ZmDDM1 activity to generate elevated levels of mCHH in flanking TEs. This raises an interesting issue regarding how ZmDDM1 coordinates with gene transcription. As ZmDDM1 tends to occupy TSSs, one possibility we speculate is that ZmDDM1 may interact with components in transcriptional machinery or simply be able to access more open chromatin more easily. Alternatively, considering the activity of DDM1 to move and remodel nucleosomes in vitro (Brzeski and Jerzmanowski, 2003), it is also possible that increased ZmDDM1 activity will facilitate the access of transcriptional machinery to TSSs by positioning +1 nucleosome to promote transcription.
The nature of ZmDDM1 targeting of active chromatin is also evident in intergenic regions, which are commonly inactive due to the massive presence of TEs. However, it appears that the sectors of active chromatin intergenic regions targeted by ZmDDM1 are still surrounded by heavily inactive chromatin, somehow analogous to oases dispersed in desserts. This opens an intriguing question of whether ZmDDM1 activity creates the active state of these intergenic regions, or alternatively, the active state of these regions is a prerequisite for ZmDDM1 localization. No matter which possibility is correct, ZmDDM1 is responsible for the formation of ZmAGO4-mediated mCHH in these intergenic regions. Nevertheless, the ZmDDM1-occupied positions appear next to sites of ZmAGO4-mediated RdDM. This delineation of ZmDDM1 and ZmAGO4 on chromatin may denote the spatial organization of ZmDDM1 with other RdDM components, and suggests that ZmDDM1-directed RdDM occurs not locally but at a restricted distance. In addition, the finding of ZmDDM1 particularly functional in mCHH formation at active chromatin seems inconsistent with the outcomes after disrupting ZmDDM1, where the drastic reduction in mCHH was observed at a genome-wide scale (Fu et al., 2018; Long et al., 2019). In zmddm1 mutants, 24-nt siRNA was largely lost, the phenomena used to explain the overall reduction of mCHH (Fu et al., 2018; Long et al., 2019). Therefore, we speculate that either ZmDDM1 has other unexplored role in the biogenesis of 24-nt siRNA, or alternatively, the defect in the biogenesis of 24-nt siRNA is simply derived from the indirect effects. After all, as there were thousands of genes occupied by ZmDDM1, it is likely that the disruption of ZmDDM1 may cause the mis-regulation of genes involved in the biogenesis of 24-nt sRNA, and consequently resulted in the global loss of 24-nt sRNA and mCHH in mutant. Alternatively, as proposed by Fu et al., the absence of ZmDDM1 may compromise heterochromatin, causing at least one critical RdDM component diluted and unable to be enriched at RdDM sites across the genome (Fu et al., 2018). Moreover, the specific-binding of ZmDDM1 in active chromatin points out the existence of a second RdDM pathway in parallel with ZmDDM1 to take charge of mCHH formation in repressive chromatin. We anticipate that this pathway is mediated by another unexplored chromatin remodeling factor. Furthermore, the nature of ZmDDM1 being predominantly located in euchromatin rather than heterochromatin raises the other intriguing question that whether ZmDDM1 underwent neofunctionalization compared with other plant species. If this is the case, we suspect that this phenomenon might be related to distinct features of chromatin landscape and lack of CMT2 gene in maize.
Similar to all the other documented plant species, the zmddm1 mutation also caused a global reduction in mCG and mCHG, supporting the critical roles of ZmDDM1 in maintaining CG and CHG methylation. However, the main occupancy of ZmDDM1 on active chromatin regions seems exclude the probability that ZmDDM1 functions locally to create access for DNA methyltransferases catalyzing mCG and mCHG. There are two possibilities we hypothesize to reconcile this incongruity. First, a substantial amount of ZmDDM1 is actually in heterochromatin, whereas it likely gets distributed over huge regions to form peaks. In contrast, when ZmDDM1 gets into euchromatin, it can arrange into peaks at nucleosomes with specific histone variants or modifications that form more rigid interactions between nucleosomes and GC-rich DNA. Second, ZmDDM1 may utilize an unexplored mechanism in maintaining mCG and mCHG in heterochromatin, which deserve extensively further studied.
Last but not least, it is intriguing to see that mutation of DDM1 caused distinct phenotypes in different organisms, from healthy plant in the first generation of Arabidopsis ddm1 mutant (Kakutani et al., 1996), to multifaced phenotypes in rice and tomato ddm1 mutants (Tan et al., 2016; Corem et al., 2018), to defective embryogenesis in maize zmddm1 mutants (Fu et al., 2018; Long et al., 2019) and mouse lsh mutants, which disrupt a gene closely related to DDM1 (Geiman et al., 2001; Sun et al., 2004). These results support the notion that the severity of effects of ddm1 on DNA methylation may be related to genome size and complexity. Therefore, we are looking forward to learning whether the critical requirement for DDM1 in euchromatin holds true not only in maize, but also in other genomes.
Materials and methods
Plant materials and genetic stocks
The maize (Zea mays) mutant lines used in this study were the same as in our previous report (Long et al., 2019). All plants were grown in the field from 2016 to 2018 in Beijing, China. For all experiments, 18 d after pollination developing maize embryos were used as the source tissues, which were spilt from the whole kernel using a razor blade, then washed three times with sterile water to collect the embryos after removing any potential endosperm contamination.
ChIP assay and data analysis
ChIP was performed as previously described with minor modifications (Yang et al., 2016). Briefly, embryos were cross-linked with 1% formaldehyde in GB buffer (0.4-M sucrose, 10-mM Tris–HCl, pH 8.0, and 1-mM EDTA). Fixation was done with two rounds of 15 min of vacuum infiltration at room temperature and was stopped by adding glycine to 0.17-M final concentration followed by 5 min of vacuum infiltration at room temperature. Fixed samples were washed three times with water at 4°C, dried with towels, frozen, and ground to powder in liquid nitrogen. Nuclei pellets were suspended in a buffer containing 0.25-M sucrose, 10-mM Tris–HCl pH 8, 10-mM MgCl2, 1% Triton X-100, 5-mM β-mercaptoethanol, 0.1-mM PMSF (Sigma-Aldrich), and protease inhibitors (one mini tablet per milliliter; Roche). The suspensions were transferred to microfuge tubes and centrifuged at 12,000g for 10 min. The pellets were suspended in 1.7-M sucrose, 10-mM Tris–HCl, pH 8, 2-mM MgCl2, 0.15% Triton X-100, 5-mM β-mercaptoethanol, 0.1-mM PMSF, and protease inhibitors (one tablet in 30-mL solution; Roche), and centrifuged through a layer of the same buffer in microfuge tubes. The nuclear pellets were lysed in a buffer containing 50-mM Tris–HCl, pH 8, 10-mM EDTA, 1% SDS, and protease inhibitors (one tablet in 30-mL solution; Roche). The lysed nuclei were sonicated with a Bioruptor (UCD-200) in a water bath at 4°C for 12 cycles with 30 s on and 30 s off. IP was performed using about 10 µg of chromatin, following a previously described protocol (Locatelli et al., 2009). Typically, 10 μL of affinity-purified antibody was used for IP. The precipitated DNA was dissolved in 50-μL 10-mM Tris–HCl pH 8, 1-mM EDTA and treated with RNase (DNase-free).
The NEBNext Ultra II DNA Library Prep Kit for Illumina (NEB) was used to prepare ChIP-seq libraries following the manufacturer’s instructions. The HiSeq 2500 sequencing system was used to sequence the libraries. Finally, ∼20 GB high-quality 150-bp paired-end reads were generated from each library. ChIP-seq reads were mapped to the B73 reference genome (Jiao et al., 2017; release AGPv4) using Bowtie2 (Langmead and Salzberg, 2012; version 2.2.4; parameters: -no-unal -I 0 -X 800 -no-discordant), and mapped reads with MAPQ ≥5 were kept for downstream analysis. Duplicated reads are removed by Picard version v.2.16.0 (http://broadinstitute.github.io/picard/). Peaks were called by MACS2-v.2.1.2 (Zhang et al., 2008; Parameter setting: -g 2.3e9 -B -SPMR -q 0.01) and other parameters were set to default values. Only peaks with 50% or larger overlap between biological replicates were kept for analysis.
The ZmDDM1-targeted TEs or genes were defined as any gene/TE having a ZmDDM1 peak summit within it using BEDtools (Quinlan and Hall, 2010). TE and genes were annotated according to B73v4.TE. filtered. gff3 and B73_RefGen_v4.43. gff3 (Jiao et al., 2017), respectively. The IntersectBed, subcommand contained in BEDTools, was used to identify the overlapping ChIP peaks between biological replicates (an overlapping peak was counted if at least 50% of the peak in Rep1 overlapped with that of Rep2; IntersectBed; parameter: -f 0.5), to identify the overlapped ZmDDM1 peaks with ZmAGO4 peaks and to define ZmDDM1 ChIP peaks in the different genomic features.
mRNA sequencing
Total RNA was isolated with two biological replications using TRIzol reagent (Invitrogen) according to the manual instruction. Sequencing libraries were prepared with a NEBNext Ultra II RNA Library Prep Kit for Illumina (NEB) and sequenced on the HiSeq2500 (Illumina) according to the manufacturer’s instructions. Approximately 5 Gb high-quality 100 single-end reads were generated from each library. Low-quality reads and portions of reads with adaptor contamination were removed using trim_galore (bioinformatics.babraham.ac.uk) with default parameters. Reads were aligned to the maize reference genome using TopHat2 and Cufflinks (Trapnell et al., 2012; Kim et al., 2013) with a transcriptome index built from the complete set of protein-coding gene annotations in Zea_mays.AGPv4.43.gff3 with default parameters. Genes were then grouped into expression categories based on fragments per kilobase of exon model per million mapped fragments (FPKM) values. Those genes with FPKM values ≤1 were put into the silent expression category, and the remaining genes were split into quartiles based on FPKM values: low, mid-low, mid-high, and high.
Heatmap and meta-plot analysis
About 200, 400, and 200-scaled bins were created for 2-kb upstream of TSS, gene body and 2-kb downstream of TTS, respectively. For ZmDDM1 ChIP-seq and ZmAGO4 ChIP-seq-identified peak summits, 200 10-bp bins were created. For MethylC-seq, weighted methylation levels were computed for each bin. For published ChIP-seq datasets and siRNA-seq analysis, the number of reads per bin was normalized by total mapped reads in each library. H3K9me2 ChIP-seq generated in this study was further normalized by dividing the input signal from the ChIP signal (log2 (ChIP/input)). The average value of each bin was used to construct meta-plots by Deeptools (Ramirez et al., 2016).
Protein expression and antibody preparation
The sequence encoding the N-terminal region of ZmAGO4 (1–1194 bp) was cloned into the pET28a expression vector (Novagen). The primers used for cloning are listed in Supplemental Table S4. The constructs were expressed in the Escherichia coli strain BL21 (DE3). Recombinant proteins were purified with the Midi PrepEase kit (EMD Biosciences Inc.) using His tags. The purified recombinant proteins (1 mg) were used for raising polyclonal antibody in rabbits. Ten microliters of ZmAGO4 antibody were used for ZmAGO4 ChIP-seq assay for each replicate. ZmDDM1A and ZmDDM1B antibodies were developed in our previous study (Long et al., 2019). Ten microliters of H3K9me2 (ABclonal, cat # A2359) antibody were added for H3K9me2 ChIP-seq assay.
Co-IP assays
Embryos were ground in liquid nitrogen. Protein extracts were prepared in IP buffer (2.7-mM KCl, 137-mM NaCl, 10-mM Na2HPO4, 2-mM KH2PO4, 0.5-mM β-mercaptoethanol, and 1-mM PMSF) and 1% protease cocktail (Roche). The suspension was homogenized with a homogenizer for 2 min on ice and filtered through two layers of Miracloth (Merck). The soluble protein fraction was collected by two rounds of centrifugation at 13,000g for 20 min at 4°C. The supernatant was precleared by adding 20 μL of protein A-decorated Dynabeads (Invitrogen #10001D) at 4°C for 2 h. The tube was placed on the magnet to remove the beads. The solution was incubated with 10 μL of ZmDDM1 antibody (∼1 mg·µL−1) at 4°C for 2 h and then 100 μL of Protein A Dynabeads (Invitrogen) was added to the solution and incubated at 4°C for 2 h. The beads were collected and washed three times for 10 min with 1 mL IP buffer. Beads were heated for 10 min at 95°C with 40 μL 1× loading buffer (50-mM Tris–HCl, pH 6.8, 10% glycerol, 2% SDS, and 5% β-mercaptoethanol). Purified proteins were separated by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis. The gels were stained by silver staining, and all visualized bands were digested with trypsin (final concentration 0.067 µg·µL−1) and identified using liquid chromatography–tandem MS analysis as previously described (Nallamilli et al., 2013).
Yeast one-hybrid assays
The full-length coding sequence of ZmDDM1A or ZmDDM1B was cloned into the pB42AD vector (Clontech), while 14 bp with two copies of CGGCGGC sites were cloned into the pLacZi2µ vector. The fusion plasmids and the empty vector were transformed into the EGY48 yeast cells by the PEG/LiAc method, and yeast cells were plated onto the first selective medium without Ura or Trp. Transformants were cultured on the second selective medium (without Ura and Trp) containing galactose (20%), raffinose (20%), buffered (BU) salt (50 mL 1.95 g of Na2HPO4, 1.855 g of NaH2PO4·2H2O), and X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) for blue color development as described in the Matchmaker One-Hybrid System (Clontech). The primers used for cloning are listed in Supplemental Table S4.
EMSA
Sequences encoding the N‐terminal region of ZmDDM1A or ZmDDM1B including the SNF domain and its flanking regions (ZmDDM1A-SNF: 219-541 aa; ZmDDM1B-SNF: 283-575 aa) was cloned into the pET-32a vector (Novagen) using EcoR-I and Xho-I restriction enzyme (New England BioLabs). Constructs were expressed in E. coli strain BL21 (DE3). The recombinant proteins were purified with the Midi PrepEase kit (EMD Biosciences Inc.) using His tags. The oligonucleotide probes for EMSAs were synthesized by Invitrogen (Beijing). Labelled probes with biotin at the 5′-end were used as binding probes, whereas unlabeled probes were used as competitors. EMSA was carried out using a LightShift Chemiluminescent EMSA Kit (Thermo Fisher Scientific) according to the manufacturer’s instruction. The primer and probe sequences are shown in Supplemental Table S4.
Accession numbers
All sequence data are available at the National Center for Biotechnology Information Sequence Read Archive under accession number GSE162678. zmddm1a (mu1044815) and zmddm1b (mu1021319) used in this study were obtained from the Maize Stock Center.
Supplemental data
The following materials are available in the online version of this article.
Supplemental Figure S1. Characterization of ZmDDM1-binding peaks.
Supplemental Figure S2. ZmDDM1 preferentially occupies TSS of protein coding genes.
Supplemental Figure S3.mCHH and 24-nt siRNA were enriched at the edge of ZmDDM1 occupied regions.
Supplemental Figure S4. Distribution of intergenic ZmDDM1 peaks.
Supplemental Figure S5. Active chromatin state of ZmDDM1-binding sites in intergenic regions.
Supplemental Figure S6. Proteins identified by ZmDDM1B IP-MS.
Supplemental Figure S7. Proteins identified by ZmAGO4 IP-MS.
Supplemental Figure S8. Amino acid sequence alignment of ZmAGO4A and ZmAGO4B.
Supplemental Figure S9. Characterization of ZmAGO4-binding sites in maize embryo.
Supplemental Table S1. Summary of sequencing datasets used in this study.
Supplemental Table S2. Published datasets used in this study.
Supplemental Table S3. The proteins identified in the MS analysis from two bands in ZmDDM1-IP.
Supplemental Table S4. List of primers used in this study.
Supplemental Data Set S1. List of genes bound by ZmDDM1.
Supplemental Data Set S2. List of genes not bound by ZmDDM1.
These authors contributed equally to this work (J.C.L., J.H.L., A.A.X.).
N.M.S and Y.H. conceived and supervised the project; J.C.L., J.H.L., and A.A.X. conducted the experiments; J.C.L. performed bioinformatics and statistical analyses; manuscript was prepared by J.C.L. and Y.H. All authors read and approved the final manuscript.
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (https://dbpia.nl.go.kr/plcell) is: Yan He ([email protected]).
Acknowledgments
We thank members of our laboratories for helpful discussions and assistance during this project. We greatly appreciate Jonathan I Gent at University of Georgia for his critical reading and comments on the manuscript.
Funding
This work was supported by grants from National Natural Science Foundation of China (31970524 and 31471172) to Y.H.
Conflict of interest statement. None declared.
References
Author notes
Senior author.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}