




Abstract
Most neurodegenerative disorders, such as Alzheimer's (AD), Parkinson's, Huntington's and Creutzfeldt–Jakob disease, are characterised by the accumulation of insoluble filamentous aggregates known as amyloid. These pathologies share common pathways involving protein aggregation which can lead to fibril formation and amyloid plaques. The 4 kDa Aβ peptide (39–43 amino acids) derived from the proteolysis of the amyloid precursor protein is currently a validated target for therapy in AD. Both active and passive immunisation studies against Aβ are being trialled as potential AD therapeutic approaches. In this study, we have characterised engineered antibody fragments derived from the monoclonal antibody, WO-2 which recognises an epitope in the N-terminal region of Aβ (amino acids 2–8 of Aβ). A chimeric recombinant Fab (rFab) and single chain fragments (scFvs) of WO-2 were constructed and expressed in Escherichia coli. Rationally designed mutants to improve the stability of antibody fragments were also constructed. All antibody formats retained high affinity (KD ∼8 × 10−9 M) for the Aβ peptide, comparable with the intact parental IgG as measured by surface plasmon resonance. Likewise, all engineered fragments were able to: (i) prevent amyloid fibrillisation, (ii) disaggregate preformed Aβ1–42 fibrils and (iii) inhibit Aβ1–42 oligomer-mediated neurotoxicity in vitro as efficiently as the whole IgG molecule. These data indicate that the WO-2 antibody and its fragments have immunotherapeutic potential. The perceived advantages of using small Fab and scFv engineered antibody formats which lack the effector function include more efficient passage across the blood-brain barrier and minimising the risk of triggering inflammatory side reactions. Hence, these recombinant antibody fragments represent attractive candidates and safer formulations of passive immunotherapy for AD.
Introduction
Alzheimer's disease (AD) is the most prevalent neurodegenerative form of dementia (Selkoe, 2002; Masters and Beyreuther, 2006) and results in memory loss and behavioural problems including mood swings, confusion and breakdown of language in affected individuals. It is estimated that 26.6 million people worldwide were afflicted by AD in 2006 (Suh and Checler, 2002), which could potentially quadruple by 2050 (Brookmeyer et al., 2007). Currently there is no cure for AD, only treatments which can slow the progression of the disease and manage some of the symptoms. For example, if diagnosed and treated early with cholinesterase inhibitors, patients can retain cognitive and functional abilities for longer as well as experience behavioural improvements (McGleenon et al., 1999).
The Aβ protein has been identified as playing a major role in the pathogenesis of AD. This 4 kDa peptide (39–43 amino acids in length) is derived from the proteolysis of the amyloid precursor protein (APP) (Kang et al., 1987) and is one of the main constituents of the amyloid plaques associated with the disease (Selkoe, 1999). Although the Aβ peptide fibrillogenesis remains the subject of many studies, the precise mechanisms are not yet fully understood (Arimon et al., 2005). Nevertheless, it is known that the Aβ peptide has a tendency to self-associate and can adopt several forms including soluble oligomers (also referred to as Aβ-derived diffusible ligands or ADDLs), protofibrils and insoluble amyloid fibrils. Importantly, all these forms are neurotoxic in vitro (Lorenzo and Yankner, 1994) with soluble oligomers displaying the highest level of toxicity (Lambert et al., 1998; Walsh et al., 2002).
A number of approaches have been used to design therapeutic agents that can prevent or reverse the formation of these toxic Aβ species (Weiner and Frenkel, 2006). Many of these strategies involve small drug molecules which inhibit or interfere with Alzheimer's amyloidogenesis (Cherny et al., 2001; Yang et al., 2005; Zhao et al., 2008). Some of these compounds are now in clinical trials.
An alternative approach has focused on antibodies as potential therapeutics. Studies have now demonstrated that active immunisation with the Aβ peptide triggers the formation of antibodies that can clear and prevent the development of amyloid plaques in transgenic AD mice (Schenk et al., 1999; Janus et al., 2000). These data prompted human clinical trials to evaluate the efficiency of the AN1792 vaccine (aggregated Aβ). However, the trial was abandoned due to 6% of the participants developing sterile meningoencephalitis (Orgogozo et al., 2003). Consequently, research efforts were redirected towards developing a passive vaccine. The murine IgG monoclonal antibody 3D6, raised against the N-terminal 1–5 amino acids of Aβ, was tested for its ability to reduce pathology in AD transgenic mice. Although a reduction in diffuse amyloid deposits was observed (Bard et al., 2000), side effects such as cerebral amyloid angiopathy-related microhemorrhages still occurred (Racke et al., 2005). To reduce immunogenicity, the 3D6 mAb was humanised (Bapineuzumab or AAB-001) and is now in phase III clinical trials.
Side effects observed after administration of some therapeutic antibodies can be attenuated by deglycosylating whole IgG antibody (Wilcock et al., 2006) or removing the Fc portion by proteolysis to produce Fab'2 (Tamura et al., 2005). An alternative strategy, and the subject of this study, is to design recombinant Fab (rFab) or scFv (Holliger and Hudson, 2005; Levites et al., 2006). We constructed recombinant antibody fragments (scFv and chimeric Fab) derived from the well-characterised WO-2 hybridoma cell line (Ida et al., 1996) and demonstrated their immunotherapeutic potential compared with the intact IgG in targeting the Aβ peptide. The WO-2 derived Fab and scFv fragments retained the same high affinity as the intact IgG and can efficiently: (i) prevent amyloid fibrillisation, (ii) disaggregate preformed Aβ1–42 fibrils and (iii) inhibit soluble oligomer-mediated neurotoxicity.
Material and methods
Purification of WO-2 IgG and preparation of papain-digested WO-2 Fab
The WO-2 hybridoma (Ida et al., 1996) was grown and maintained in serum-free medium (Invitrogen, USA) for antibody production. WO-2 immunoglobulin was purified from culture supernatant by affinity chromatography on a Prosep-vA protein A column (Millipore, Billerica, USA). The bound WO-2 IgG was eluted with 0.2 M glycine/1 M NaCl, pH 2.0 into a neutralising solution of 1 M Tris, pH 8.0, then dialysed against PBS overnight at 4°C. WO-2 Fab fragment was generated by digestion with papain (Sigma, St Louis, MO, USA) (Gruen et al., 1993). Undigested IgG and the Fc portion were removed by affinity chromatography using protein A.
Generation of recombinant scFv and rFab
Total RNA, extracted from the WO-2 hybridoma cell line (Ida et al., 1996) using the Trizol Reagent (Invitrogen, Mount Waverley, VIC, Australia), was utilised for cDNA synthesis. The V genes were isolated from cDNA by RT–PCR using primers annealing to mouse light (mIgCκ-1: 5′-CTTCCACTTGACATTGATGTCTTTG-3′) and heavy constant region (mIgG2a-1: 5′-CAGGTCAAGGTCACTGGCTCAGG-3′). The VH and VL genes were sequenced and analysed by the IMGT databases.
To construct the WO-2 scFv, the VH (GenBank accession no. AM950178) and VL (GenBank accession no. AM950179) genes were amplified with primers WO-2 VHFOR (5′-GCCATTAACCATGGCCCAGGTTACTCTGAAAGAGTCT-3′), WO-2 VH BACK (5′-TGAACCGCCACCGCCACTACCGCCACCGCCTGAGGAGACTGTGAGAGTGGTGCCTTG-3′), WO-2 VLK FOR (5′-GGTAGTGGCGGTGGCGGTTCAGGCGGTGGCGGTAGCGATGTTTTGATGACCCAAACT-3′) and WO-2 VLK BACK (5′-GCCATTAAGGATCCAGCGGCCGCCCGCTTCAGCTCCAGCTTGGT-3′). The PCR products were assembled and cloned into the pGC expression vector (Fig. 1a) (Coia et al., 1997) as described by Gilliland et al. (1996). The chimeric rFab was generated using the same approach with primers WO-2 VH FOR/WO-2 VH Fab BACK (5′-TGAGGAGACGGTGACCGTGGTGCCTTGGCCCCA-3′) and WO-2 Fab K FOR (5′-CAGGCCGATATCGTGATGACCCAAACTCCA-3′)/WO-2 Fab K BACK (5′-TTTCAGCTCGAGCTTGGTCCCAGCACCGAA-3′). Amplified VH and VL fragments were digested with restriction enzymes (NcoI/BstEII for VH and EcoRV/XhoI for VL) and ligated sequentially into the pGC-Fab expression vector (Fig. 1b).
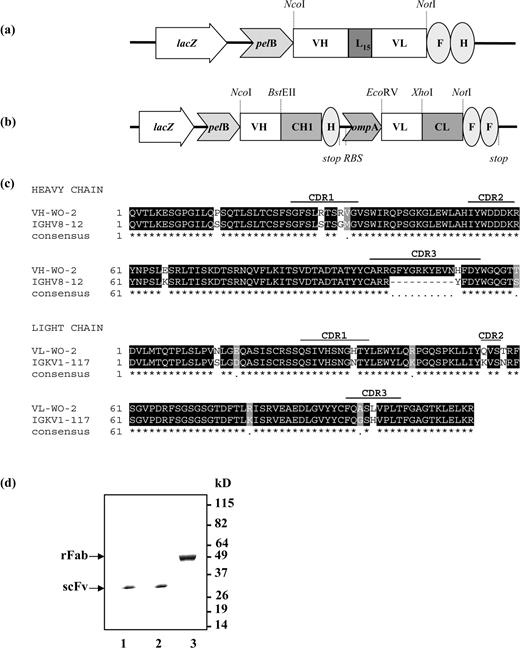
Schematic of recombinant WO-2 antibody constructs in pGC expression vector. (a) WO2 scFv construct, (b) Bicistronic WO-2 rFab construct. Abbreviations: lacZ, promoter of the bacterial lac operon; pelB and ompA, signal peptide sequences for secretion of recombinant antibody fragments into the periplasmic space; RBS, ribosome binding site; VH and VL, variable domain of antibody heavy chain and light chain, respectively; CH1 and CL, first human constant domain of antibody heavy chain and human constant domain of antibody light chain, respectively; L15, 15 residue linker peptide; F, FLAG-tag; H, polyhistidine tag; stop, stop codon terminating translation. The restriction sites used for cloning are indicated. (c) Sequence alignment of the WO-2 antibody heavy and light chain variable regions with mouse germline V region IGHV8-12 and IGKV1-117. (d) SDS–PAGE gel stained with Coomassie Blue. Lane 1, WO-2 scFv WT; lane 2, WO-2 S46P scFv; lane 3, WO-2 rFab.
Three-dimensional structure modelling of the antibody fragment
The molecular model of the WO-2 scFv was obtained by using the web antibody modelling (WAM) algorithm (Whitelegg and Rees, 2000) (http://antibody.bath.ac.uk/). Images of the model were generated using PyMOL (PyMOL version 0.82, http://pymol.sourceforge.net/) (DeLano Scientific LLC).
Mutagenesis of the antibody fragments
Site-directed mutagenesis of the WO-2 scFv was performed using the Quick Change® Site-directed Mutagenesis Kit II (Stratagene, La Jolla, CA, USA). Mutations were verified by sequencing the coding region of the antibody fragments on both strands.
Production, purification and biochemical characterisation of the recombinant antibody fragments
Recombinant proteins were expressed in the bacterial periplasm as described by Dolezal et al. (2000). Expression and isolation were monitored by gel filtration chromatography through a Superdex® 200 HR 10/30 column (Amersham Pharmacia Biotech, Uppsala, Sweden) and Coomassie staining. The presence of individual antibody fragments was confirmed by western blotting using a mouse anti-FLAG M2 antibody (Sigma) as previously described (Kortt et al., 1994).
Enzyme linked immunosorbent assays
Wells of 96-well Maxisorb Immuno plates (Nunc, Roskilde, Denmark) were coated with 5 µg/ml of purified MBP-Aβ1–42 (Caine et al., 2007) in carbonate-coating buffer. Control wells were coated with 50 µg/ml BSA or 50 µg/ml MBP overnight at 4°C. Wells were blocked with 200 µl of 2% MPBS for 2 h at 37°C. WO-2 scFv and rFab antibody fragments from crude periplasmic extracts and purified preparations (5 µg/ml) were incubated in wells with MBP-Aβ1–42. Control wells were incubated with the anti-FLAG M2 antibody for 1 h at RT. After washing, a 1/2000 dilution of anti-mouse IgG HRP-conjugate (GEB-Pierce, Rockford, IL, USA) was added to each well and incubated for a further 1 h at 20°C. The substrate 2,2′-Azino-bis(3-ethylbenzthiazoline-6-sulfonic acid) (ABTS, Roche, Indianapolis, USA) was added and absorbance was read at 405 nm.
Biosensor binding analysis
Surface plasmon resonance (SPR) measurements were performed using a Biacore T100 biosensor instrument (GE Healthcare, Uppsala, Sweden). All immobilisation and binding experiments were performed at 25°C in 1× HBS-EP+ buffer (10 mM HEPES, 150 mM NaCl, 3 mM EDTA, 0.05% surfactant P-20, pH 7.4). Analysis of the interaction between Aβ and WO-2 IgG was carried out using an indirect assay whereby WO-2 IgG was captured onto a sensor chip (CM5) surface via rabbit anti-mouse (RAM) IgG antibody. A standard amine coupling protocol was employed to immobilise the RAM IgG antibody onto a surface via exposed primary amines (Johnsson et al., 1991) in two separate flow cells (1 and 2). This procedure resulted in immobilisation of ∼12 000 RU (resonance units) of RAM IgG in both flow cells. Two different versions of Aβ were tested for binding to captured WO-2 IgG: (i) synthetic Aβ1–40 peptide and (ii) recombinant Im7-Aβ1–18 fusion protein (O.D., unpublished data) consisting of Aβ residues 1–18 fused to the C-terminus of Immunity protein 7—Im7 (Kleanthous et al., 1998). Aβ samples (2–64 nM) were injected over captured WO-2 and reference surfaces at 30 µl/min. Association and dissociation phases were each monitored for 5 min (300 s). At least one buffer only injection identical to Aβ injections was included for the purpose of double-referencing. RAM IgG surfaces were regenerated with a single 45 s injection (at 30 µl/min) of 10 mM Glycine, pH 1.5. Binding activities of various WO-2 antibody fragments to its Aβ antigen were evaluated using a recombinant Im7-Aβ1–18 fusion protein form. As was the case for WO-2 IgG, these measurements relied upon an indirect assay format whereby the Im7-Aβ1–18 fusion protein was captured onto a chip surface via the Im7 natural binding partner, colicin E7 endonuclease domain of Escherichia coli (Ko et al., 1999), herein referred to as DNaseE7. Basic principles and concepts of this capture assay will be more thoroughly described elsewhere (Hosse & Dolezal, manuscript in preparation). A standard amine coupling protocol was employed to immobilise DNaseE7 onto a CM5 chip in flow cells 3 and 4. This procedure resulted in immobilisation of ∼2500 RU of DNAseE7 protein. Im7-Aβ1–18 samples were diluted (∼0.1 µg/ml) and injected for 30 s at a flow rate of 30 µl/min over the DNaseE7 surface in flow cell 4 resulting in a capture of ∼40 RU of fusion protein for scFv measurements and 20 RU for Fab measurements. WO-2 antibody fragment preparations, typically spanning 2–128 nM in concentration, were injected over the captured Im7-Aβ1–18 at a flow rate of 30 µl/min. Association and dissociation phases were monitored for 3 and 5 min, respectively. At least one buffer only injection identical to WO-2 fragment injections was included for the purpose of double-referencing. DNaseE7 surfaces were regenerated with a single 1 min injection (at 30 µl/min) of Immuno-Pure™ gentle elution buffer (GEB-Pierce).
Preparation of Aβ1–42 peptide
An Aβ1–42 synthetic lyophilised peptide (W. M. Keck Laboratory, Yale University, New Haven, CT, USA) was dissolved in 1,1,1,3,3,3-hexafluoro-2-isopropanol (HFIP) (Sigma) and aliquoted. HFIP was removed by evaporation under a fume hood and residual traces of HFIP were removed by drying under vacuum in a SpeedVac (Savant Instrument). The resulting peptide film was stored at −80°C until required. For an Aβ1–42 monomeric stock solution preparation, the peptide film was dissolved in either DMSO (dimethyl sulfoxide) or 20 mM NaOH for 1 h, sonicated for 5 min and centrifuged at high speed for 10 min at 4°C, immediately prior to use. To prepare an oligomeric preparation, the monomeric Aβ1–42 stock solution was adjusted to 100 µM in ice-cold PBS, vortexed and incubated for 24 h at 4°C with continuous rotation.
Inhibition of Aβ1–42 fibrillisation
The monomeric Aβ1–42 stock solution was diluted to 20 µM in PBS, pH 7.4, and incubated with or without 4 µM purified WO-2 IgG, WO-2 rFab, WO-2 scFv or NC10 anti-neuraminidase scFv antibody control (Dolezal et al., 2000) for 5 days at RT. Reactions were monitored by transmission electron microscopy (TEM) and thioflavine T (ThT) fluorescence assay.
Disaggregation of preformed fibrils
Aliquots of the monomeric Aβ1–42 stock solution were diluted to 200 µM in PBS, pH 7.4, and incubated for 5 days at RT. Fibril formation was confirmed by TEM. The fibril solution (200 µM) was next diluted 1:10 with either PBS, NC10 scFv, mutant WO-2 S46P scFv, WO-2 rFab or WO-2 IgG (4 µM final concentration for each antibody). Reactions were monitored after 24 h by TEM and ThT fluorescence assay.
Transmission electron microscopy
Carbon-coated gold grids were glow-discharged under nitrogen. Protein samples (0.1–0.3 mg/ml) were applied to the grids and negatively stained with 2% potassium phosphotungstate, pH 6.8. Grids were air-dried and examined using an FEI Tecnai 12 transmission electron microscope operating at 120 kV. Images were recorded using an Olympus Megaview III CCD camera.
ThT fluorescence assay
Aβ1–42 fibrils were detected by incubating 5 µl of the reactions with 200 µl of 5 µM ThT solution (50 mM glycine, pH 9). The fluorescence intensity was measured at an excitation wavelength of 450 nm and an emission wavelength of 482 nm with a spectrofluorometer LSB-50 (Perkin Elmer, Norwalk, CT, USA) with both excitation and emission bandwidths of 5 nm as previously described (LeVine, 1999). Each data point represents the percentage of the maximum ThT fluorescence intensity of the Aβ1–42 fibrils in PBS after subtracting out the fluorescence signal from free ThT.
In vitro neuroprotection (MTT) assay against oligomer-mediated toxicity
Human neuroblastoma cell line M17 was maintained in serum-free OPTI-MEM medium (Invitrogen, Australia) containing 0.1 mM non-essential amino acids, 50 IU/ml penicillin, 50 µg/ml streptomycin (Gibco, Gaithersburg, MA, USA), 1 mM sodium pyruvate, 10% FCS in 5% CO2 at 37°C. Cells were seeded in 96-well tissue culture plates (Nunc) at ∼104 cells per well and incubated for 24 h. Aβ1–42 oligomers were incubated either alone or with purified WO-2 IgG, WO-2 rFab, WO-2 S46P scFv or NC10 scFv for 3 h at RT. These reactions (10 µl) were added to the cells (final concentration of peptide and antibodies of 15 and 0.5 µM, respectively). Plates were incubated for an additional 24 h at 37°C.
Cell viability was determined using MTT toxicity assays. Briefly, 10 µl of 5 mg/ml MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] was added to each well and incubated for a further 24 h at 37°C. Plates were centrifuged, the medium was removed and 100 µl of 0.1 M HCl in isopropanol was added to each well to dissolve the crystals. The absorbance was measured at 560 nm.
Results
Generation, expression and purification of the WO-2 rFab and scFvs
The monoclonal antibody WO-2 (IgG2a, κ) (Ida et al., 1996) recognises amino acid residues 2–8 of Aβ1–42 (Miles et al., 2008) in all forms (monomers, oligomers, protofibrils and fibrils) in different biochemical assays (Ida et al., 1996; Jensen et al., 2000). To construct the WO-2 scFv and chimeric Fab, RNA was extracted from WO-2 hybridoma cells and used as the VH and VL gene source. The VH (WO-2-H) and VL (WO-2-L) sequences (Fig. 1c) were analysed with IMGT/V-QUEST software (Giudicelli et al., 2004; Lefranc et al., 2005) and the IMGT/JunctionAnalysis tool (Yousfi Monod et al., 2004) to identify the immunoglobulin germ line V, (D) and J genes from which WO-2 variable regions are derived. The mouse germ line V, (D) or J alleles most similar to the WO-2-H nucleotide sequence by IMGT/V-QUEST are IGHV8-12*01, IGHD1-1*01 and IGHJ2*01, and those most similar to WO-2-L are IGKV1-117*01 and IGKJ5*01. The WO-2 scFv cassette (VH–VL oriented and separated by a 15 amino acids linker) was cloned into the pGC expression vector, whereas the WO-2-H and WO-2-L genes were cloned sequentially into the pGC Fab expression vector. Both constructs expressed protein in the periplasmic space of TOP10 F' E. coli cells and contained a C-terminal FLAG and 6-HIS tag for identification and affinity purification. SDS–PAGE profiles of the WO-2 scFv and rFab showed proteins of expected sizes at ∼30 and ∼50 kDa, respectively (Fig. 1d). The activity of these purified fragments was assessed by ELISA and western blot against an MBP-Aβ1–42 purified fusion protein. The fragments revealed high binding reactivity, similar to that of the purified parental IgG in both assays (data not shown).
Optimisation of WO-2 scFv stability by site-directed mutagenesis
The WO-2 scFv yield after affinity purification was low (75–100 µg/l of cell culture) compared with our previous production yields for anti-neuraminidase NC10 scFv (Dolezal et al., 2000). This was most likely the result of protein aggregation of WO-2 scFv given that the purified product precipitated during dialysis, which was not observed for the rFab. In an attempt to improve the scFv solubility, key amino acid residues in framework regions (FR) were mutated. The complementary determining regions (CDRs) were not altered so that the binding properties of the antibody Fv would be retained. The selected amino acids are highlighted on the WO-2 scFv wild-type (WT) model (Fig. 2a). Nine residues in the VH region (four in FR1, one in FR2 and four in FR3) and two residues in the VL region (one in FR1 and one in FR3) were selected based on an alignment of 500 mouse antibody sequences and mutated to the most conserved amino acid at that position. Initially, periplasmic extracts of the scFv variants were tested in ELISA for their reactivity against MBP-Aβ1–42 (Fig. 2b). Of the nine variants analysed (seven single mutants and two double mutants), six showed either an equivalent or a higher level of reactivity than that of the WO-2 scFv WT. The WO-2 S46P scFv variant that showed the best reactivity by ELISA was selected for large scale production. In comparison with the WT scFv, the WO-2 S46P scFv mutant did not form any detectable precipitate following affinity purification and the final yield of soluble product was increased ∼2-fold (200 µg/l). Hence, we have used the WO-2 S46P scFv mutant rather than the WO-2 scFv WT in subsequent experiments.

(a) Three-dimensional model of the WO-2 scFv and location of mutations. Residues highlighted in red were selected for site-directed mutagenesis. (b) ELISA of crude periplasmic extract of WO-2 scFv WT and mutants against Aβ fusion protein, MBP and BSA. The binding activity was measured at 405 nm. Experiments were performed in triplicate, and each sample was run in triplicate. Data were expressed as a mean ± SD.
Kinetics measurement by SPR
Kinetic binding parameters and overall affinities of various WO-2 antibody formats binding to Aβ were determined by SPR. Initially, two different Aβ forms (Aβ1–40 synthetic peptide and recombinant Im7-Aβ1–18 fusion protein) were evaluated for binding to captured WO-2 IgG. Binding analysis that adopted 1:1 binding interaction model revealed no significant difference in kinetic parameters for the two Aβ formats (Table I) thus demonstrating that Im7-Ab1–18 fusion protein could be used as an alternative Aβ antigenic source. Binding analysis of WO-2 Fab (papain-digested), WO-2 rFab, WO-2 WT scFv and WO-2 S46P scFv were subsequently performed using Im7-Aβ1–18 fusion protein. To allow for easy and comparable binding analysis of these antibody fragments, the SPR assay format was inverted such that the fusion protein was captured onto a CM5 chip via its binding partner, DNAseE7, followed by antibody fragment injection. The results for each interaction between antibody fragment and captured this Im7-Aβ1–18 were evaluated by globally fitting the data to a 1:1 interaction model. Calculated association (ka) and dissociation (kd) rate constants as well as the overall KD values are shown in Table I. No significant affinity differences were observed between recombinant and parental monoclonal antibody formats. Thus, the WO-2 rFab, the WO-2 WT scFv and the WO-2 S46P scFv bound to Aβ antigen with comparable affinity to that of the parental IgG and the papain-digested Fab.
Rate and equilibrium constants determined by SPR (Biacore) at 25°C
| ka × 104 (M−1 s−1) | kd × 10−4 (s−1) | KD (nM) | |
|---|---|---|---|
| Captured WO-2 IgG | |||
| Aβ1–40 | 8.2 ± 1.4 | 6.0 ± 0.5 | 7.4 ± 0.7 |
| Im7-Aβ1–18 | 10.0 ± 0.7 | 5.2 ± 0.8 | 5.2 ± 1.2 |
| Captured Im7-Aβ1–18 | |||
| WO-2 Fab | 10.9 ± 0.7 | 8.8 ± 0.3 | 8.0 ± 0.8 |
| WO-2 rFab | 12.3 ± 0.4 | 12.5 ± 0.1 | 10.2 ± 0.3 |
| WO-2 wt scFv | 14.7 ± 0.9 | 11.2 ± 0.1 | 7.6 ± 0.5 |
| WO-2 S46P scFv | 9.1 ± 1.4 | 11.5 ± 0.6 | 12.9 ± 2.7 |
| ka × 104 (M−1 s−1) | kd × 10−4 (s−1) | KD (nM) | |
|---|---|---|---|
| Captured WO-2 IgG | |||
| Aβ1–40 | 8.2 ± 1.4 | 6.0 ± 0.5 | 7.4 ± 0.7 |
| Im7-Aβ1–18 | 10.0 ± 0.7 | 5.2 ± 0.8 | 5.2 ± 1.2 |
| Captured Im7-Aβ1–18 | |||
| WO-2 Fab | 10.9 ± 0.7 | 8.8 ± 0.3 | 8.0 ± 0.8 |
| WO-2 rFab | 12.3 ± 0.4 | 12.5 ± 0.1 | 10.2 ± 0.3 |
| WO-2 wt scFv | 14.7 ± 0.9 | 11.2 ± 0.1 | 7.6 ± 0.5 |
| WO-2 S46P scFv | 9.1 ± 1.4 | 11.5 ± 0.6 | 12.9 ± 2.7 |
Rate and equilibrium constants determined by SPR (Biacore) at 25°C
| ka × 104 (M−1 s−1) | kd × 10−4 (s−1) | KD (nM) | |
|---|---|---|---|
| Captured WO-2 IgG | |||
| Aβ1–40 | 8.2 ± 1.4 | 6.0 ± 0.5 | 7.4 ± 0.7 |
| Im7-Aβ1–18 | 10.0 ± 0.7 | 5.2 ± 0.8 | 5.2 ± 1.2 |
| Captured Im7-Aβ1–18 | |||
| WO-2 Fab | 10.9 ± 0.7 | 8.8 ± 0.3 | 8.0 ± 0.8 |
| WO-2 rFab | 12.3 ± 0.4 | 12.5 ± 0.1 | 10.2 ± 0.3 |
| WO-2 wt scFv | 14.7 ± 0.9 | 11.2 ± 0.1 | 7.6 ± 0.5 |
| WO-2 S46P scFv | 9.1 ± 1.4 | 11.5 ± 0.6 | 12.9 ± 2.7 |
| ka × 104 (M−1 s−1) | kd × 10−4 (s−1) | KD (nM) | |
|---|---|---|---|
| Captured WO-2 IgG | |||
| Aβ1–40 | 8.2 ± 1.4 | 6.0 ± 0.5 | 7.4 ± 0.7 |
| Im7-Aβ1–18 | 10.0 ± 0.7 | 5.2 ± 0.8 | 5.2 ± 1.2 |
| Captured Im7-Aβ1–18 | |||
| WO-2 Fab | 10.9 ± 0.7 | 8.8 ± 0.3 | 8.0 ± 0.8 |
| WO-2 rFab | 12.3 ± 0.4 | 12.5 ± 0.1 | 10.2 ± 0.3 |
| WO-2 wt scFv | 14.7 ± 0.9 | 11.2 ± 0.1 | 7.6 ± 0.5 |
| WO-2 S46P scFv | 9.1 ± 1.4 | 11.5 ± 0.6 | 12.9 ± 2.7 |
Inhibition of Aβ1–42 fibrillisation by WO-2 IgG and its recombinant fragments
In order to investigate the ability of different antibody formats to inhibit Aβ1–42 fibrillisation, fibril formation was initiated in the absence or presence of the WO-2 IgG, WO-2 rFab, WO-2 S46P scFv and monitored using TEM (Fig. 3) and the ThT fluorescence assay (Fig. 5a) over a period of 5 days. As a negative control, the anti-neuraminidase NC10 scFv was evaluated under identical conditions. Without any added antibody fragments, the Aβ1–42 peptide did not form any aggregate structures after 24 h (Fig. 3-1a), but after 3 days, protofibrils were observed (Fig. 3-1b) which polymerised into amyloid fibrils by day 5 (Fig. 3-1c). Similarly, incubating the Aβ1–42 peptide with an NC10 scFv antibody did not affect the Aβ1–42 fibrillisation. On day 1, no visible structures were observed (Figs 3-2a). By day 3, oligomers and small fibrils were seen (Fig. 3-2b) and on day 5, amyloid fibrils were observed (Fig. 3-2c). In contrast, fibril formation did not occur when WO-2 IgG (Fig. 3-3a, 3b and 3c), WO-2 rFab (Fig. 3-4a, 4b and 4c) or WO-2 S46P scFv (Fig. 3-5a, 5b and 5c) were incubated with the Aβ1–42 peptide. However, in all instances, Aβ1–42 aggregates were observed by day 3. At this resolution, it was not possible to determine if these were Aβ1–42 oligomers of a defined size or random aggregates clustered by antibodies. Indeed, by day 5, in the presence of WO-2 rFab (Fig. 3-4c) or WO-2 S46P scFv (Fig. 3-5c), clusters ranging from 3 to 20 nm were observed. Interestingly, a lattice-like structure was observed in the sample containing the WO-2 IgG (Fig. 3-3c) and Aβ1–42 peptide but not in those with the WO-2 rFab or the WO-2 S46P scFv, following a 5 day incubation period. This observation can be explained by the bivalency of the IgG molecule which allows the formation of the lattice-like structure by cross-linking Aβ1–42 aggregates, whereas the monovalent WO-2 rFab or WO-2 S46P can only bind a single Aβ1–42 at a time.
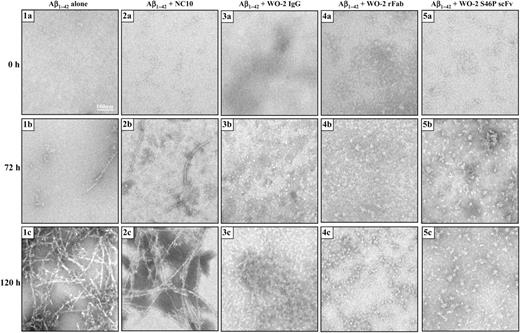
TEM analysis of Aβ1–42 fibril formation with WO-2 IgG and its recombinant fragments. Aβ1–42 was incubated alone (1a, 1b and 1c) or in the presence of an NC10 scFv (2a, 2b and 2c), WO-2 IgG (3a, 3b and 3c), WO-2 chimeric rFab (4a, 4b and 4c), WO-2 S46P scFv (5a, 5b and 5c). Images were captured by TEM at time 0 (1a, 2a, 3a, 4a and 5a), 72 h (1b, 2b, 3b, 4b and 5b) and 120 h (1c, 2c, 3c, 4c and 5c). Scale bar, 100 nm.
To confirm these results, the degree of fibrillisation was assessed over 6 days using the ThT fluorescence assay in the presence or absence of the antibody fragments (Fig. 5a). In this assay, the fluorescence intensity is directly related to the number of amyloid fibrils. Co-incubation of NC10 scFv (irrelevant antibody) with Aβ1–42 did not prevent the formation of ThT-positive fibrils. However, incubation of the Aβ1–42 peptide with WO-2 IgG, WO-2 rFab and WO-2 S46P scFv (molar ratio 1:5 antibody: Aβ1–42) produced a significantly lower level of fluorescence suggesting that this suite of antibody fragments can all inhibit the nucleation mechanism leading to the formation of amyloid fibrils. These results are consistent with the TEM data, and together indicate that WO-2 as a whole antibody or as engineered fragments is an effective inhibitor of fibrillisation.
Disintegration of preformed Aβ1–42 fibrils by WO-2 IgG and its recombinant fragments
The ability of the WO-2 antibody formats to disaggregate preformed amyloid fibrils was evaluated by TEM and the ThT fluorescence assay. Fibrils were formed over a period of 7 days (Fig. 4a) and then incubated with either PBS (Fig. 4b), NC10 scFv (Fig. 4c), WO-2 IgG (Fig. 4d), WO-2 rFab (Fig. 4e) or WO-2 S46P scFv (Fig. 4f) for 24 h. TEM images revealed that all the WO-2 antibody formats disrupted the fibril morphology, whereas the sample incubated with the NC10 scFv did not show any difference in fibril morphology compared with the Aβ1–42 fibrils alone (Fig. 4b and c). Unexpectedly, very few protofibrils were observed in the WO-2 S46P scFv sample, whereas more were present in the WO-2 IgG and WO-2 rFab samples. To confirm these observations, a ThT fluorescence assay was performed (Fig. 5b). In presence of the WO-2 antibodies, the fluorescence signal decrease to 30%, 25% and 23% for the whole IgG, rFab and S46P scFv, respectively. These results indicate that after 24 h, WO-2 can disaggregate ThT-positive amyloid-β-fibrills at substoichiometric concentration.
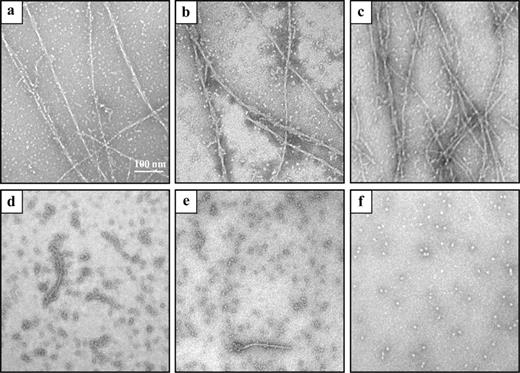
TEM analysis of the disaggregation of Aβ1–42 fibrils by WO-2 IgG and its recombinant fragments. Aβ1–42 preformed fibrils were confirmed by TEM (a) and then incubated alone (b) or in the presence of an NC10 scFv (c), WO-2 IgG (d), WO-2 chimeric rFab (e) and WO-2 S46P scFv (f) at RT for 24 h.
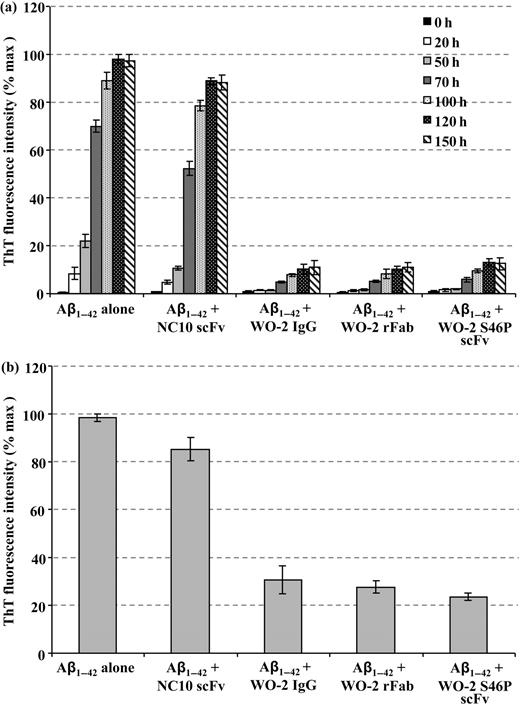
The effect of WO-2 IgG and its recombinant fragments on Aβ1–42 fibril formation and preformed Aβ1–42 fibrils. Fibril formation was monitored by ThT fluorescence as a function of time in the absence or presence of the WO-2 IgG and its recombinant fragments (a). Aβ1–42 preformed fibrils were analysed by ThT fluorescence after incubation for 24 h with NC10 scFv, or WO-2 IgG and its recombinant fragments (b). Experiments were performed in duplicate, and each sample was run in triplicate.
Protection of Aβ1–42 toxicity by WO-2 recombinant antibodies
Numerous studies have identified soluble oligomers (also called ADDLs) as the most toxic form of Aβ. Such structures can be obtained by incubating Aβ1–42 monomers in PBS for 24 h at 4°C (Fig. 6a). To test whether the WO-2 antibody fragments could protect cells against the toxic effect of these oligomers, an MTT assay was used. Cell viability of a human neuronal line was significantly decreased (38% of cell survival) when incubated for 24 h with Aβ1–42 oligomers compared with cells alone (100% of cell survival) (Fig. 6b). Likewise, cell viability was affected to the same degree (40% of cell survival) if cells were incubated with Aβ1–42 oligomers that were pre-incubated with NC10 scFv. In contrast, if Aβ1–42 oligomers were pre-incubated with either WO-2 IgG, WO-2 rFab or WO-2 S46P scFv (molar ratio 1:5 antibody:Aβ1–42), cell viability was only reduced to ∼70%. These data suggest that WO-2 can protect the cells from the neurotoxic effect of Aβ1–42 oligomers. Most importantly, the IgG, the rFab and the scFv formats performed equally well in this function.
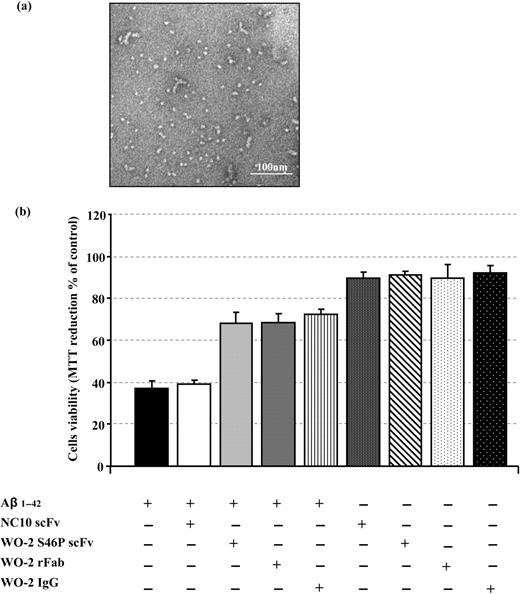
The effect of WO-2 IgG and its engineered fragments on Aβ1–42 oligomer-mediated neurotoxicity. Aβ1–42 monomers incubated for 24 h at 4°C form oligomers (a). M17 neuroblastoma cells were incubated with Aβ1–42 oligomers alone or with Aβ1–42 oligomers pre-incubated with NC10 scFv, WO-2 IgG, WO-2 chimeric rFab or WO-2 S46P scFv. The MTT assay was used to measure cell survival (b). Experiments were performed in duplicate, and each sample was run in triplicate. Data were expressed as a percentage of control (M17 neuroblastoma cells only) ± SD.
Discussion
Passive immunisation with certain anti-Aβ monoclonal antibodies has been proposed as a safer immunotherapeutic approach for treating AD compared with active Aβ vaccination (Lichtlen and Mohajeri, 2008). Four monoclonal antibodies against different domains of Aβ are currently in development: Bapineuzumab (humanised version of 3D6 from ELAN/WYETH) is in phase III, LY2062430 (humanised version of m266) from Eli Lilly is in phase II, RN-1219 from Pfizer/Rinat is in phase I and R-1450, a human antibody, from Hoffman-LaRoche/MorphoSys is in phase I (Brody and Holtzman, 2008). In the case of human phase I clinical trials with Bapineuzumab, six patients demonstrated signs of cognitive improvement in the Mini Mental State Examination (MMSE) test compared with eight placebos (http://www.elan.com/News/2006/20060504.asp).
Three possible mechanisms have been proposed to account for the beneficial effects of antibodies; these include (i) direct disaggregation of amyloid fibrils by the antibody (Solomon et al., 1996, 1997), (ii) Fc receptor-mediated phagocytosis and removal of Aβ by microglial cells (Bard et al., 2000) and (iii) a change in the equilibrium between the central nervous system and the plasma causing efflux of Aβ from the brain (peripheral sink hypothesis) (DeMattos et al., 2001). Regardless of the exact cellular mechanisms, it seems that one important aspect is the epitope recognised by the antibodies on the Aβ molecules (Lichtlen and Mohajeri, 2008). Indeed, it appears that antibodies directed against the central regions of Aβ (such as m266) may act by clearing amyloid in the periphery (DeMattos et al., 2001), whereas antibodies against the immunodominant epitope (EFRH) in the N-terminal part of Aβ (Bard et al., 2003; Esposito et al., 2008) have been reported to cross the blood-brain barrier and act on Aβ in brain tissue (Bard et al., 2000). Unfortunately, passive administration of these anti-Aβ antibodies directed against the N-terminal part of Aβ to AD mice models have also been reported to trigger inflammatory processes such as cerebral microhemorrhages (Pfeifer et al., 2002). This has prompted the generation of engineered antibody fragments including scFvs (Frenkel et al., 2000; Liu et al., 2004; Fukuchi et al., 2006; Solórzano-Vargas et al., 2008; Yue et al., 2008), rFabs and V-domains (Habicht et al., 2007) which lack the Fc region. Given that IgG molecules have limited access to the brain (Banks et al., 2002), it is anticipated that due to their smaller size, these antibody fragments may enter more efficiently into this area.
In this study, we constructed and characterised antibody fragments of the monoclonal antibody WO-2 recognising the N-terminal region of the Aβ peptide (amino acids 2–8). Although other anti-Aβ scFvs have been described for their immunotherapeutic potential, this work is the first example in which three different formats have been compared. Moreover, to our knowledge, this study is the first to describe a recombinant chimeric Fab fragment displaying anti-aggregating, disaggregating and neuroprotective activities.
Given that the amount of soluble functional WO-2 WT scFv (100 µg/l) produced was not sufficient to carry out biochemical assays, our first step was to improve stability by mutating WO-2 WT scFv in the VH and VL FR using rational design and site-directed mutagenesis. Of the variants generated, the WO-2 S46P scFv with a point mutation in the VH FR2 region produced the best results including a 2-fold increase in final yield of soluble product (100 versus 200 µg/l) and no precipitate. This finding was consistent with other studies which demonstrated not only that the VH FR2 is an important region for scFv stability (Masuda et al., 2006) but similar substitutions (any amino acid to a proline) resulted in increased stability (Matthews et al., 1987). Likewise, reformatting the WO-2 scFv into an rFab further enhanced the stability which probably resulted from the additional binding forces provided by the constant regions (Quintero-Hernández et al., 2007).
Binding kinetics of the three fragments (WO-2 scFv WT, WO-2 S46P scFv and WO-2 rFab) revealed that the affinities were essentially the same as the parental IgG and as the papain-digested Fab. Furthermore, all were in the nanomolar range required for therapeutic applications. These data suggest that the three-dimensional structure of the binding domains of the parental IgG is not affected by the insertion of a linker for the scFv constructs or the grafting of human constant region for the rFab. Our current work involves generating a humanised recombinant antibody by CDR-grafting and further improvement of the expression yield will be done on these humanised versions of WO-2.
Next, we determined in biochemical assays in vitro the immunotherapeutic potential of the three formats. The results presented here suggest that this suite of antibodies shows the ability to inhibit Aβ1–42 fibril formation. One possible mechanism for inhibition involves the binding of antibody to Aβ1–42 molecules preventing the filament nucleation process (Chirita et al., 2004). In our case, WO-2 appears to lead to the formation of amorphous aggregates suggesting that the antibodies cannot inhibit the early stage of aggregation as previously described by Mamikonyan et al. (2007) with a polyclonal antibody preparation. Interestingly, the co-incubation of WO-2 IgG with Aβ1–42 molecules leads to a lattice-like structure which has not been previously reported in similar assays (Legleiter et al., 2004; Mamikonyan et al., 2007). Presumably, the bivalency of the IgG molecules allows cross-linking of small amorphous aggregates presenting multiple epitopes, resulting in the lattice structures that are not observed with the monovalent binders, WO-2 scFv and WO-2 rFab. Moreover, these antibody fragments can also disaggregate preformed fibrils. Again, the exact mechanism remains unclear. It has been proposed that the high affinity antibody binding to soluble Aβ1–42 species disturbs the equilibrium between fibrillar and non-fibrillar Aβ1–42 and thus causes the amyloid fibrils to disaggregate (Chirita et al., 2004).
The antibodies were also tested in the context of cellular neurotoxicity in the M17 human neuroblastoma cell line. In all instances, WO-2 IgG, WO-2 rFab and WO-2 S46P scFv reduced the neurotoxicity caused by Aβ1–42 oligomers. These results are promising as oligomers have been suggested to be the most toxic form of Aβ and are involved in the early stage of AD pathogenesis (Lambert et al., 1998; Walsh et al., 2002).
Recently, it has been demonstrated that expression of intrabodies directed against the EFRH motif of Aβ can decrease and prevent the generation of Aβ peptide by shielding the β-secretase cleavage site of APP (Paganetti et al., 2005). Therefore, it will be worth testing the in situ expression of WO-2 S46P scFv as an intrabody in a similar system. In addition, testing current WO-2 antibody formats in an AD mouse model will allow a direct comparison of the efficiency on amyloid load and cognitive function of such molecules in vivo.
It is likely that all the advantages of engineered antibody fragments (smaller size, low cost production in microbial expression systems, possibility to express as intracellular protein and so on) will make these molecules attractive tools for safer immunotherapy of AD and other neurodegenerative diseases.
Acknowledgements
We thank Pr K. Beyreuther for providing us the WO-2 hybridoma cell line, Dr Qiao-Xin Li for the Aβ1–40 peptide sample and Dr J. Caine for the Aβ-MBP fusion protein. We are very grateful to Lesley Pearce for her technical assistance. We thank Dr S.D. Nuttall and Dr N.M. McKern for critical reading of the manuscript.
References
Edited by Anna Wu

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}