





Abstract
Sexual differentiation is a fundamental process in the life cycles of land plants, ensuring successful sexual reproduction and thereby contributing to species diversity and survival. In the dioicous liverwort Marchantia polymorpha, this process is governed by an autosomal sex-differentiation locus comprising FEMALE GAMETOPHYTE MYB (FGMYB), a female-promoting gene, and SUPPRESSOR OF FEMINIZATION (SUF), an antisense strand-encoded long non-coding RNA (lncRNA). SUF is specifically transcribed in male plants and suppresses the expression of FGMYB, leading to male differentiation. However, the molecular mechanisms underlying this process remain elusive. Here, we show that SUF acts through its transcription to suppress FGMYB expression. Transgene complementation analysis using CRISPR/Cas9D10A-based large-deletion mutants identified a genomic region sufficient for the sex differentiation switch function in the FGMYB-SUF locus. Inserting a transcriptional terminator sequence into the SUF-transcribed region resulted in the loss of SUF function and allowed expression of FGMYB in genetically male plants, leading to conversion of the sex phenotype from male to female. Partial deletions of SUF had no obvious impact on its function. Replacement of the FGMYB sequence with that of an unrelated gene did not affect the ability of SUF transcription to suppress sense-strand expression. Taken together, our findings suggest that the process of SUF transcription, rather than the resulting transcripts, is required for controlling sex differentiation in M. polymorpha.
Introduction
Sexual differentiation is a crucial process for sexual reproduction, which is vital in enhancing genetic diversity and thus contributes to species survival. In land plants, a haploid spore produced by meiosis undergoes mitotic divisions to form a multicellular structure, called a gametophyte, which produces sexually dimorphic gametes—egg and sperm cells. During the evolution of vascular plants, the diploid sporophytic phase came to dominate the life cycle and gametophytes became significantly reduced into few-celled tissues: the embryo sac (the female gametophyte) and pollen (the male gametophyte) (Schmidt et al. 2015, Hisanaga et al. 2019b). Recently, taking advantage of its dominant gametophyte generation, studies using the revived model bryophyte Marchantia polymorpha have identified key regulators of sexual reproduction in the gametophytes, including those involved in sexual differentiation (Hisanaga et al. 2019b, Kohchi et al. 2021).
Marchantia polymorpha is a dioicous liverwort, female and male plants of which possess U and V sex chromosomes, respectively, in the dominant haploid phase. Under long-day conditions supplemented with far-red light, the female plants form broken-umbrella-shaped sexual branches, archegoniophores, harboring egg-bearing sexual organs, archegonia. The male plants form disk-shaped sexual branches, antheridiophores, with sperm-producing sexual organs, antheridia (Shimamura 2016, Kohchi et al. 2021, Cui et al. 2023). Our previous study showed that an autosomal locus encoding the R2R3 MYB-type transcription factor FEMALE GAMETOPHYTE MYB (FGMYB) and its antisense long non-coding RNA (lncRNA), named SUPPRESSOR OF FEMINIZATION (SUF), control sexual differentiation of the gametophytes in M. polymorpha (Hisanaga et al. 2019a). FGMYB is specifically expressed in female reproductive organs and promotes female differentiation. Genetically female fgmyb mutants showed an almost complete female-to-male sex conversion phenotype, but their sperm showed defective movement, likely due to the lack of the V chromosome–linked genes required for their motility. SUF is transcribed specifically in male plants from the promoter located in the 3ʹ flanking region of FGMYB and suppresses FGMYB expression, leading to male differentiation. Male suf mutants conversely gained FGMYB expression, resulting in male-to-female sex conversion phenotypes. However, the suf mutants lack egg cells, presumably due to the missing U chromosome–linked genes essential for female fertility. Collectively, the FGMYB-SUF locus acts as a toggle switch between female and male differentiation of M. polymorpha gametophytes (Hisanaga et al. 2019a). It has long been known that the sex of M. polymorpha is determined by a dominant U chromosome locus (Haupt 1932), although the responsible gene remained elusive for many years. Our recent study identified a sex-determination gene in the dominant U chromosome locus, BASIC PENTACYSTEINE ON THE U CHROMOSOME (BPCU), which encodes a member of the plant-specific transcription factor family and suppresses the expression of SUF in female plants. BPCU is likely to bind directly to the SUF locus and repress its expression through chromatin modification, thereby relieving FGMYB from suppression and determining feminization (Iwasaki et al. 2021).
lncRNAs are typically defined as transcripts that extend beyond 200 nucleotides in length and do not code for proteins. They play significant roles in modulating various biological processes, including chromatin organization, transcription and post-transcriptional regulation (Ponting et al. 2009, Guttman and Rinn 2012). Intriguingly, emerging evidence suggests that lncRNAs exhibit high or specific expression in the reproductive organs of both plants and animals, highlighting their potential importance in sexual reproduction across diverse organisms (Golicz et al. 2018). The modes of action of lncRNAs can be broadly divided into two categories: cis-acting, which influences genes in close proximity to their transcription sites, and trans-acting, which regulates distant genomic locations far from where they are transcribed (Wang and Chang 2011, Kornienko et al. 2013). Additionally, the regulation by cis-acting lncRNAs can be classified into at least three potential mechanisms: in the first, the mature lncRNA transcripts regulate the neighboring genes by recruiting regulatory factors through inherent specific motifs and/or by modulating their function; second, the act of transcription or subsequent RNA processing events themselves regulates gene expression, irrespective of the sequences of the resulting lncRNA transcripts; and last, the regulation relies on the DNA regulatory elements in the promoter or gene body of the lncRNAs, regardless of the RNA product or its synthesis (Kopp and Mendell 2018).
Our previous study showed that SUF expression as a transgene failed to complement the male-to-female sex conversion phenotype of the suf mutants, even though SUF was expressed, indicating that SUF acts only in cis to repress FGMYB expression in M. polymorpha (Hisanaga et al. 2019a). However, the molecular mechanisms underlying this process remained unclear. Here, we show that the transcription of SUF is essential for its role in suppressing FGMYB expression. Furthermore, deletion analyses of the SUF sequences suggest that the transcriptional process itself is crucial for its function. Our study provides insights into the mechanisms by which lncRNAs regulate sexual reproduction in land plants.
Results
Transgene complementation analysis using CRISPR/Cas9D10A-based large-deletion mutants identifies a genomic region of FGMYB-SUF sufficient for sexual differentiation switch function
To understand the underlying mechanism of the FGMYB repression by SUF, we first identified the FGMYB-SUF genomic region sufficient for their function. Using CRISPR/Cas9D10A nickase technology (Hisanaga et al. 2019a, Koide et al. 2020), we generated large-deletion mutants that lack the entire FGMYB-SUF locus (∼24.5 kb), resulting in two independent lines each for genetically female (fgmyb-suf-18ld [U], fgmyb-suf-211ld [U], hereafter the sex chromosome background is indicated in brackets) and male (fgmyb-suf-46ld [V], fgmyb-suf-367ld [V]) plants (Fig. 1A; Supplementary Fig. S1A, B). The fgmyb-sufld mutants developed antheridiophores and antheridia regardless of their sex chromosomes (Fig. 1B; Supplementary Fig. S1C), consistent with the phenotypes of the fgmyb-suf double mutants in the previous study (Hisanaga et al. 2019a), indicating that male differentiation is the default genetic program of M. polymorpha. We then introduced a 19.6-kb genomic fragment containing FGMYB-SUF, spanning 4 kb upstream from the transcriptional start site (TSS) of FGMYB to 5.3 kb upstream from the TSS of SUF, into these mutants. The resulting transformants with the male mutant background (FGMYB-SUF fgmyb-suf-46ld [V]) still formed antheridiophores and antheridia, while those with the female mutant background (FGMYB-SUF fgmyb-suf-211ld [U]) formed archegoniophores and archegonia containing egg cells, suggesting that the transgene genetically complemented the female-to-male sex conversion phenotypes of the mutants (Fig. 1B; Supplementary Table S1). RT-PCR analysis showed that the expression patterns of FGMYB and SUF were also restored in these complemented plants (Fig. 1C), suggesting that this genomic fragment contains sufficient regions to regulate both FGMYB and SUF expression. Hereafter, we used the sequence of this 19.6-kb segment as the standard genomic locus for further analyses.
![Complementation experiments identify a genomic region sufficient for controlling sexual differentiation in the FGMYB-SUF locus. (A) Schematic diagram of the FGMYB-SUF loci in WT and CRISPR/Cas9D10A-based large-deletion (fgmyb-sufld) plants and a genomic fragment used for the complementation tests. Arrowheads indicate the position of guide RNAs (gRNAs) and primers used in RT-PCR (C). (B) Gametangiophores (top) and gametangia (bottom) of WT, fgmyb-sufld and complemented plants. Images of lines exhibiting representative sexual phenotypes among the independently obtained lines are shown. Arrowheads indicate egg cells. Scale bars are 5 mm in the top panels and 50 μm in the bottom panels. (C) RT-PCR analysis of FGMYB and SUF expression in immature gametangiophores (stages 1–2) of WT, fgmyb-sufld and complemented plants. Primer sets shown in (A) were used. The doublet bands of FGMYB and SUF likely represent spliced and unspliced variants. EF1α was used as an internal control. The sex chromosome backgrounds of the samples are given in brackets: [U] and [V] for females and males, respectively.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/pcp/65/3/10.1093_pcp_pcad170/1/m_pcad170f1.jpeg?Expires=1749050532&Signature=Tc2RMV~DdCgZlo2vDXWaq3HwGvHxL6cWuhPs3ypUCpG10uK078uYhTh0V49kEVp6c168S9ez5M9N~GwAl3kbTm3PqSiybxpPGSkF2VoDQwThlAdxGbU6MjOj6Rn8Vwr37GDlvB~hPnEsb1e4bbhM7ukaCFqcRR2nQ~kinH-ZTD7Gg-hUV46Ezps8fKjbXvFJrqdjR0dlCYiY6J1FV0cwLQ1qQYe5zYSYnLOKtNOLbqQdZ6EIEQhw-bx~IL7TUN7vHNA68YZSvG0YGQh9D9x5iIbO4ihvbNC4Ruo-O2Y8iTw1rmxEev5Mfv2o8Z1WEclPgtpAf~tpw96qSaH~SMtk8A__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Complementation experiments identify a genomic region sufficient for controlling sexual differentiation in the FGMYB-SUF locus. (A) Schematic diagram of the FGMYB-SUF loci in WT and CRISPR/Cas9D10A-based large-deletion (fgmyb-sufld) plants and a genomic fragment used for the complementation tests. Arrowheads indicate the position of guide RNAs (gRNAs) and primers used in RT-PCR (C). (B) Gametangiophores (top) and gametangia (bottom) of WT, fgmyb-sufld and complemented plants. Images of lines exhibiting representative sexual phenotypes among the independently obtained lines are shown. Arrowheads indicate egg cells. Scale bars are 5 mm in the top panels and 50 μm in the bottom panels. (C) RT-PCR analysis of FGMYB and SUF expression in immature gametangiophores (stages 1–2) of WT, fgmyb-sufld and complemented plants. Primer sets shown in (A) were used. The doublet bands of FGMYB and SUF likely represent spliced and unspliced variants. EF1α was used as an internal control. The sex chromosome backgrounds of the samples are given in brackets: [U] and [V] for females and males, respectively.
Additionally, to narrow down the regulatory region required for the FGMYB function, we made constructs with a series of truncations in the upstream region of FGMYB and introduced them into the fgmyb-sufld mutants (Supplementary Fig. S2A). Most of the transformants carrying 2- and 3-kb upstream regions complemented the female-to-male conversion phenotype. However, when testing a construct with just a 1-kb upstream region, five out of 11 genetically female lines displayed abnormal gametangiophores exhibiting mosaic patterns in sex differentiation and two did not show complementation at all (Supplementary Fig. S2B; Supplementary Table S1), suggesting that the 2-kb upstream region is sufficient, but 1 kb is insufficient for FGMYB expression.
SUF transcription into the overlapping 3ʹ region represses FGMYB expression
SUF was shown to operate as a cis-acting lncRNA that suppresses the expression of FGMYB at the same locus (Hisanaga et al. 2019a). The regulation of gene expression by cis-acting lncRNAs may be attributed to the mature lncRNA transcripts, the process of transcription itself or the inherent function of the gene locus as a DNA regulatory element (Kopp and Mendell 2018). To test whether the transcription of SUF is required for its function, we generated a construct in which a polyadenylation signal from the cauliflower mosaic virus (CaMV) 35S terminator was inserted into the 5ʹ-half of the SUF-transcribed sequence (Fig. 2A). Polyadenylation of the 3ʹ end of RNA is one of the RNA processing events during RNA transcription. It is known that polyadenylation signals are required for transcription termination by RNA polymerase II (RNAPII) and intimately linked with the termination (Eaton and West 2020). The aforementioned construct was then introduced into fgmyb-suf-46ld [V]. The obtained genetically male plants (FGMYB-SUFpA fgmyb-suf-46ld [V], termed SUFpA [V] for brevity) developed archegonia without egg cells (Fig. 2B), consistent with the phenotypes of sufge [V] in the previous study (Hisanaga et al. 2019a). Transcriptome analyses using the immature gametangiophores (stages 1–2; Higo et al. 2016) of wild-type (WT) [U], WT [V] and SUFpA [V] suggested that the antisense transcription of SUF pre-terminated at the insertion site of the polyadenylation signal, and instead, FGMYB was expressed from the sense strand in SUFpA [V] (Fig. 2C). Notably, despite the overexpression of the SUF 5ʹ region (Fig. 2D; Supplementary Fig. S3A, B), FGMYB was expressed at a higher or comparable level in SUFpA [V] than in the WT female plants (Fig. 2E; Supplementary Fig. S3A, C), suggesting that SUF transcription beyond the polyadenylation signal insertion site is required for its function. Furthermore, combining our data with publicly available WT transcriptome data from thalli, mature gametangiophores (stage 4) and gametangia (Higo et al. 2016, Hisanaga et al. 2021, Iwasaki et al. 2021), we performed principal component analysis (PCA) and heat map analysis based on Spearman’s correlation coefficients and found that gametangiophores of SUFpA [V] showed almost the same gene expression profile as those of WT females (Fig. 2F; Supplementary Fig. S4A). Moreover, the expression pattern of sex-specific autosomal genes in SUFpA [V] resembled those of WT females regardless of the expression of sex chromosome genes (Supplementary Fig. S4B–D), suggesting that SUFpA [V] exhibited a male-to-female sex conversion, not only in the morphology but also in the gene expression profiles. Taken together, these results suggest that the SUF locus does not act as a DNA regulatory element but requires full-length transcription for its function and that SUF suppresses FGMYB expression through its transcription into the 3ʹ overlapping region.
![Premature termination of SUF transcription causes de-repression of FGMYB, leading to male-to-female sex conversion. (A) Schematic diagram of the polyadenylation signal inserted into the FGMYB-SUF locus in SUFpA [V] plants. (B) Gametangiophores (top) and gametangia (bottom) of SUFpA [V]. Scale bars are 5 mm (top panels) and 20 μm (bottom panels). (C–F) RNA-seq analysis of the immature gametangiophores of WT and SUFpA [V] (line #2). Read accumulation at the FGMYB-SUF locus (C), the expression levels of SUF (D) and FGMYB (E) and the PCA of various tissues in WT (F) are shown. In (C), the sense strand (+) and antisense strand (−) are indicated. Numbers in brackets indicate bins per million mapped reads. In (D) and (E), the horizontal bars and symbols indicate the means and grouping by P < 0.05 in a Tukey–Kramer test (n = 3), respectively. The sex chromosome backgrounds of the samples are given in brackets: [U] and [V] for females and males, respectively.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/pcp/65/3/10.1093_pcp_pcad170/1/m_pcad170f2.jpeg?Expires=1749050532&Signature=3ujg5FItrhxQq4owPrKg9vBnqLsLnT0dk1rlZcVQrgiAqh2jNi8g-AChsHksB~S58P4rZHCT4G5zq7SolKDRh~WuxR-usINjRFnD1k~9nZBPHJwfzh9YnJwcSHzCfhNuhfvInmhqYD1prBcumSEKc0XuPjLM4lSb9QOPqlJxBCr1v3W2zGo5D0eMsj8l-tDhS6OxlGVONB7ReJby7HCLqpfrjJHCqZkI~pBIlz4tXQcQzivB-TExoNKqIh~5rCKD5SsJEjeTxw23JAFuoIcJqzZx1eQQctEYZtikTDwy0g8JRuIiwHusp6COFYujlbQk5bMwTvmUVJMTHII~ZTztpg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Premature termination of SUF transcription causes de-repression of FGMYB, leading to male-to-female sex conversion. (A) Schematic diagram of the polyadenylation signal inserted into the FGMYB-SUF locus in SUFpA [V] plants. (B) Gametangiophores (top) and gametangia (bottom) of SUFpA [V]. Scale bars are 5 mm (top panels) and 20 μm (bottom panels). (C–F) RNA-seq analysis of the immature gametangiophores of WT and SUFpA [V] (line #2). Read accumulation at the FGMYB-SUF locus (C), the expression levels of SUF (D) and FGMYB (E) and the PCA of various tissues in WT (F) are shown. In (C), the sense strand (+) and antisense strand (−) are indicated. Numbers in brackets indicate bins per million mapped reads. In (D) and (E), the horizontal bars and symbols indicate the means and grouping by P < 0.05 in a Tukey–Kramer test (n = 3), respectively. The sex chromosome backgrounds of the samples are given in brackets: [U] and [V] for females and males, respectively.
5ʹ non-overlapping sequences of the SUF-transcribed region are dispensable for its repressive function
lncRNAs that function based on their transcription can be further classified into two types: those that require mature lncRNA transcripts and those that depend on the transcription process itself. In the former case, the sequence and secondary structure of the lncRNAs are crucial for their function, but not in the latter (Kopp and Mendell 2018). To examine these possibilities, we generated a series of constructs harboring partial deletions in the SUF-transcribed region (FGMYB-SUFΔ1 to Δ6; Fig. 3A). When these constructs were introduced into the fgmyb-suf-211ld [U] strain, the transformants (FGMYB-SUFΔ1 to Δ6 fgmyb-suf-211ld [U]) primarily formed archegoniophores similar to those of WT female plants (Fig. 3B; Supplementary Table S1). The fgmyb-suf-46ld [V] mutants carrying the same transgenes (FGMYB-SUFΔ1 to Δ6 fgmyb-suf-46ld [V]) formed antheridiophores, with the notable exception of FGMYB-SUFΔ1 fgmyb-suf-46ld [V], which formed archegoniophores or abnormal gametangiophores showing mosaic patterns between female and male differentiation (Fig. 3B; Supplementary Fig. S5A; Supplementary Table S1). Remarkably, no expression of SUF was observed in these genetically male plants lacking the Δ1 region, although SUF expression was detected in those lacking the Δ2 to Δ6 regions (Fig. 3C; Supplementary Fig. S5B). These results suggest that the Δ1 region downstream of the TSS of SUF is required for SUF transcription, while the remaining regions Δ2–Δ6 are dispensable for SUF transcription. Furthermore, the plants expressing the SUF mutants lacking a long region (Δ2–6) spanning from Δ2 to Δ6 (Fig. 3A) also exhibited female morphology or mosaic patterns in sex differentiation in the genetic female lines (FGMYB-SUFΔ2-6 fgmyb-suf-211ld [U]), whereas the genetic male lines (FGMYB-SUFΔ2-6 fgmyb-suf-46ld [V]) exhibited male morphology (Fig. 3B; Supplementary Table S1), supporting the findings that the Δ2-6 region is dispensable for the SUF function. Additionally, qRT-PCR analysis showed that the FGMYB expression levels in these genetically male plants were consistent with their sexual phenotypes (Supplementary Fig. S5C). Together, these results suggest that the structure and sequence of the SUF-specific 5ʹ-half region that does not overlap with FGMYB are dispensable for the gene suppression function of SUF and that SUF is a lncRNA that functions via the process of transcription itself rather than via its resultant transcripts.
![Partial deletions of the 5ʹ-half region of SUF-transcribed sequences confer no obvious impact on its repressive function. (A) Schematic diagram of the partially deleted FGMYB-SUF loci in the transgenic FGMYB-SUFΔ fgmyb-sufld plants. (B) Gametangiophores (top) and gametangia (bottom) of FGMYB-SUFΔ fgmyb-sufld. Images of lines exhibiting representative sexual phenotypes among the independently obtained lines are shown. Arrowheads indicate egg cells. Scale bars are 5 mm (top panels) and 50 μm (bottom panels). (C) qRT-PCR analyses of SUF expression in 10-day-old thalli of WT [U], WT [V], FGMYB-SUF fgmyb-sufld [V] and FGMYB-SUFΔ1 fgmyb-sufld [V]. EF1α was used as an internal control. The horizontal bars and symbols indicate the means and grouping by P < 0.05 in a Tukey–Kramer test (n = 3), respectively. The sex chromosome backgrounds of the samples are given in brackets: [U] and [V] for females and males, respectively.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/pcp/65/3/10.1093_pcp_pcad170/1/m_pcad170f3.jpeg?Expires=1749050532&Signature=XyS7yLExqcLH78MGKnO384Ka0c0~Itw2466pZEXmyL0z2VDFoO56TKmKvs4PkPKP~acSLp6-nWI2NI1mu-FCqkcpG4mVt8tfHAVOic9RlNIhHnmcO7a6BQLx76zxfHaEcOq9cNrB5Ey0vp9yo8sFFC7bjs0nuRWZCCpdCpKM72alUdbVNilNiQAC7kRSdWsqkQB6482towInMYMojsUjkXVNH2Idv-tHdzmD3IfRxsdojtgA4ZkFssE3ewTssM-txix5hicXqX3ItqdZDU0uGClEpHjlidVLXgzHXCqmLDk16zp0ob931yoA3Vn~iGSKRKGfQMByfX3KBp~wvWk5Qg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Partial deletions of the 5ʹ-half region of SUF-transcribed sequences confer no obvious impact on its repressive function. (A) Schematic diagram of the partially deleted FGMYB-SUF loci in the transgenic FGMYB-SUFΔ fgmyb-sufld plants. (B) Gametangiophores (top) and gametangia (bottom) of FGMYB-SUFΔ fgmyb-sufld. Images of lines exhibiting representative sexual phenotypes among the independently obtained lines are shown. Arrowheads indicate egg cells. Scale bars are 5 mm (top panels) and 50 μm (bottom panels). (C) qRT-PCR analyses of SUF expression in 10-day-old thalli of WT [U], WT [V], FGMYB-SUF fgmyb-sufld [V] and FGMYB-SUFΔ1 fgmyb-sufld [V]. EF1α was used as an internal control. The horizontal bars and symbols indicate the means and grouping by P < 0.05 in a Tukey–Kramer test (n = 3), respectively. The sex chromosome backgrounds of the samples are given in brackets: [U] and [V] for females and males, respectively.
Antisense transcription of SUF represses the expression of an artificial target gene
We then investigated whether the transcription of SUF specifically represses FGMYB transcription or whether it has an impact on gene transcription regardless of the sequence. We used a toxic gene [diphtheria toxin A (DTA)] (Pappenheimer 1977) under the control of a CaMV 35S promoter (pro35S:DTA) to test whether the SUF transcription is able to suppress sense-strand transcription regardless of the FGMYB-SUF sequence. We constructed a vector carrying a SUF sequence fused to the DTA gene in the reverse strand orientation (pro35S:DTA:SUF) (Fig. 4A). These vectors were introduced into germinating F1 spores, and the numbers of surviving transformants were counted. We expected that, while pro35S:DTA would yield a smaller number of transformants compared to the vector control due to its deleterious activity, pro35S:DTA:SUF would increase the survival rate of male transformants compared to those transformed with pro35S:DTA, due to the male-specific suppressive activity of the SUF transcription. As expected, the number of pro35S:DTA transformants was significantly lower than that of the vector control, while the number of the male transformants carrying pro35S:DTA:SUF was significantly higher than that of pro35S:DTA. However, this effect of pro35S:DTA:SUF was not observed in the female transformants (Fig. 4B, C; Supplementary Table S2), suggesting that the antisense transcription of SUF in male plants suppresses the sense DTA expression. Collectively, these results suggest that the antisense transcription of SUF could repress the sense-strand genes in a manner independent of the gene sequences.
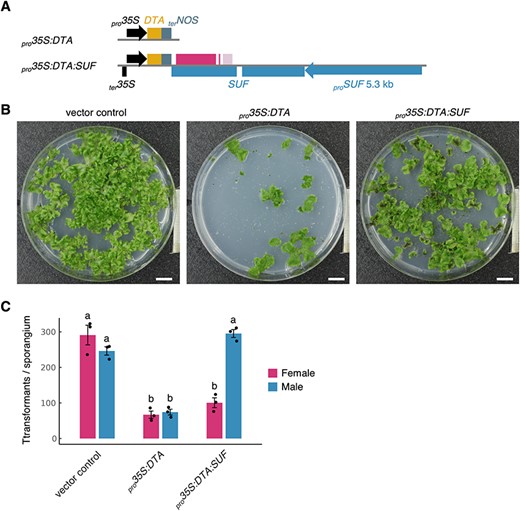
Antisense transcription of SUF represses expression of toxic gene DTA, an artificial target gene. (A) Schematic diagrams of pro35S:DTA and pro35S:DTA:SUF constructs. The CaMV 35S terminator (ter35S) in the bottom strand of the pro35S:DTA:SUF construct is included to facilitate the termination of the antisense transcription of SUF. (B) F1 germinating spores from pro35S:DTA and pro35S:DTA:SUF plants. pMpGWB100 was used as a vector control. The germinating spores were grown on selective media supplemented with antibiotics for 3 weeks under continuous white light conditions. Scale bars, 1 cm. (C) Survival rates of female and male pro35S:DTA and pro35S:DTA:SUF plants. The rates were estimated from the total number of transformants and the sex ratio determined by sex-ascertainment PCR performed on over 100 randomly selected individuals. Bars represent the mean ± SE from three independent experiments. Letters above the bars indicate grouping by P < 0.05 in a Tukey–Kramer test.
Discussion
In this study, we showed that the male-specific antisense lncRNA SUF acts through its transcription to suppress FGMYB, a female-promoting gene, leading to male differentiation in M. polymorpha (Fig. 5). The SUF function was abrogated by the insertion of a transcriptional terminator sequence downstream of the SUF TSS (Fig. 2). Furthermore, the deletion of the 5ʹ-half of the SUF-transcribed region, which does not overlap with FGMYB, did not affect the SUF repressive function (Fig. 3). These observations suggest that the SUF function is not dependent on any DNA regulatory elements within the SUF locus and that the transcriptional process of SUF itself, rather than the resulting transcripts, is crucial for the repression of the FGMYB expression. Moreover, the replacement of the FGMYB sequence with the DTA gene sequence did not impair the gene suppression function of SUF (Fig. 4), further supporting the idea that repression by SUF is mediated by its transcription, irrespective of the sequence.
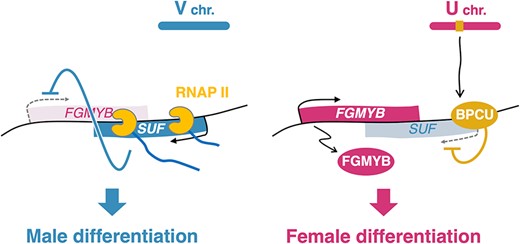
Model of the regulatory mechanism of sex differentiation via the FGMYB-SUF module in M. polymorpha. In males (left), the antisense lncRNA SUF is transcribed from the antisense strand of the female-promoting gene FGMYB. The antisense transcription itself, rather than the transcript, represses FGMYB expression in cis, leading to differentiation into the ‘default’ male. In females (right), the sex-determining factor BPCU on the U chromosome directly represses SUF expression, leading to the de-repression of FGMYB expression and differentiation into female.
Our transcriptomic study also provides a comprehensive insight into the genome-wide functions of the FGMYB-SUF module in sexual differentiation (Fig. 2F; Supplementary Fig. S4). In the SUFpA [V] strain, which shows a male-to-female conversion phenotype due to the loss of SUF function, the expression patterns of most autosomal sex-specific genes are similar to those in WT female plants (Supplementary Fig. S4B), suggesting that the FGMYB-SUF module governs the expression of sex-specific genes across the genome. A distinct subset of genes appears to be correlated with sex chromosomes (Supplementary Fig. S4B), suggesting that their regulation is likely independent of the FGMYB-SUF module and governed by sex chromosome–linked genes. Notably, certain V chromosome genes (MpVg00090, MpVg00320, MpVg00700 and MpVg00785), whose functions are currently unknown, were not expressed in the SUFpA [V] line (Supplementary Fig. S4C), implying that these genes are candidate targets for the suppression by FGMYB.
The study of lncRNAs in gene regulation is increasing in plant biology (Fonouni-Farde et al. 2021, Wierzbicki et al. 2021). Yet, only a few plant antisense lncRNAs have been demonstrated to act in cis, affecting the alleles from which they are produced. For example, in Arabidopsis thaliana, COOLAIR, a set of lncRNAs with conserved secondary structures, directly bind to the RNA-binding protein FCA, which in turn recruits PRC2 subunit CURLY LEAF to modulate histone modifications at the FLOWERING LOCUS C (FLC) locus, thereby resulting in FLC silencing and accelerated flowering (Hawkes et al. 2016, Tian et al. 2019). These findings highlight the critical role of the transcripts in the COOLAIR function. Another lncRNA in A. thaliana, SVALKA (SVK), triggers RNAPII collisions at the 3ʹ-end of the sense gene, CBF1, through read-through transcription, thereby reducing its mRNA levels and promoting cold acclimation (Kindgren et al. 2018). Our study suggests that the repression activity of SUF is independent of sequence specificity, which is similar to SVK. However, we are yet to exclude the possibility that the 3ʹ-half of the SUF-transcribed region, which is primarily reverse-complementary to FGMYB exons, alone may be sufficient for its function as an lncRNA, demanding further research to reach a definitive conclusion.
The extension of SUF transcription into the 3ʹ-half, overlapping with FGMYB, is required for its suppressive activity (Fig. 2; Supplementary Fig. S3). A possible underlying mechanism could involve direct RNAPII transcriptional interference in cis, where antisense transcription interferes with the transcription of the corresponding sense gene (Pelechano and Steinmetz 2013). This mechanism is well-illustrated by the studies of a mouse lncRNA, Airn, which silences its overlapping sense gene Igf2r by perturbing the recruitment of the transcriptional machinery to the Igf2r promoter (Latos et al. 2012, Santoro et al. 2013). Similarly, in humans, the read-through transcription of LUC7L, which extends into the downstream HBA2 gene, alters the epigenetic states of the promoter, leading to the silencing of HBA2 (Tufarelli et al. 2003). Notably, our previous research found no significant changes in the repressive histone marks at the FGMYB locus between the thalli of SUF-expressing males and non-expressing females (Iwasaki et al. 2021), although the state of histone modifications in reproductive organs remains to be examined. Additionally, SUF could suppress the expression of an unrelated sense-oriented gene in cis (Fig. 4), which does not conflict with the theory of transcriptional interference. Therefore, our findings suggest that sequence-independent mechanisms, such as transcriptional interference, might play a role in controlling sexual differentiation in M. polymorpha. Intriguingly, in the SUFpA [V] plants, where SUF transcription is prematurely terminated, a significant accumulation of short 5ʹ SUF transcripts was observed (Fig. 2D; Supplementary Fig. S3B). This potentially hints that in the absence of transcriptional interference, the higher promoter activity of SUF becomes apparent, possibly surpassing that of FGMYB. Such a scenario could explain the production of complete SUF transcripts in WT male plants, consequently leading to the repression of FGMYB expression. Further investigation is needed to clarify the detailed mechanisms.
Emerging evidence indicates that lncRNAs are highly expressed in reproductive tissues, particularly in male reproductive organs, across both plant and animal kingdoms, suggesting their critical role in sexual reproduction and underlying cellular processes such as chromatin modification and cell fate determination (Golicz et al. 2018). In M. polymorpha, another male-specific antisense lncRNA, SUPPRESSOR OF KNOX1 (SUK1), has been identified (Dierschke et al. 2021). SUK1 is likely to suppress its sense-strand counterpart, KNOX1, which is predominantly expressed in female gametes, potentially through a non-sequence-specific, transcriptional overlap mechanism. This observation and our results collectively raise the possibility that a similar mode of antisense lncRNA-mediated gene regulation could be widely employed in the control of the development of sexually dimorphic reproductive organs in M. polymorpha. Further investigations will elucidate the underlying mechanism of lncRNA-mediated gene regulation during sexual reproduction in land plants.
Materials and Methods
Plant materials and growth conditions
WT female and male accessions of M. polymorpha ssp. ruderalis, Takaragaike-2 (Tak-2) and Takaragaike-1 (Tak-1), respectively (Ishizaki et al. 2015b, Bowman et al. 2017, Kohchi et al. 2021), were used in this study. Plants were cultured on a half-strength Gamborg’s B5 medium (Gamborg et al. 1968) containing 1% (w/v) agar, or on vermiculite, under continuous white light (50–60 µmol photons m−2 s−1) at 22°C. For induction of sexual reproduction, plants grown more than 10 d from gemmae were transferred to continuous white light conditions supplemented with continuous far-red light (30 µmol photons m−2 s−1) as described previously (Kubota et al. 2014).
Vector construction
Plasmids were constructed as follows, using the primers listed in Supplementary Table S3. The sequences were confirmed by Sanger sequencing.
pMpGE018-FGMYBgRNA5,6-SUFgRNA6,7 (for complete deletion of the FGMYB-SUF locus).
To generate larger deletion mutants of the FGMYB-SUF locus than the complete deletion mutant in the previous study (11-kb deletion) (Hisanaga et al. 2019a), four pairs of oligonucleotides (FGMYBgRNA5F/FGMYBgRNA5R, FGMYBgRNA6F/FGMYBgRNA6R, SUFgRNA6F/SUFgRNA6R and SUFgRNA7F/SUFgRNA7R) were designed at 6.5 kb upstream of the TSS of FGMYB and 7.7 kb upstream of the TSS of SUF (expected 24.5-kb deletion). The four annealed duplex gRNA sequences were subcloned into the BsaI sites of pMpGE_En04, pBC-GE12, pBC-GE23 and pBC-GE34 (Hisanaga et al. 2019a, Koide et al. 2020), respectively, and the latter three vectors were further integrated into the first vector to generate pMpGE_En04-FGMYBgRNA5,6-SUFgRNA6,7. The resultant entry vector was transferred into pMpGE018 carrying CRISPR/Cas9D10A (Hisanaga et al. 2019a, Koide et al. 2020) using Gateway LR Clonase II Enzyme mix (Thermo Fisher Scientific, Waltham, MA) to create pMpGE018-FGMYBgRNA5,6-SUFgRNA6,7.
pMpGWB101-FGMYB-SUF (for complementation).
In a previous study, pENTR1a-FGMYB-SUF was created by subcloning a genomic region spanning 4 kb upstream of the TSS of FGMYB to 5.3 kb upstream of the TSS of SUF (Cui et al. 2023). This entry clone was transferred into pMpGWB101 (Ishizaki et al. 2015a) using the Gateway LR reaction to generate pMpGWB101-FGMYB-SUF.
pMpGWB101-proFGMYB3/2/1kb:FGMYB-SUF (for complementation with truncated FGMYB promoters).
The FGMYB sequences containing the promoter regions 3, 2 and 1 kb upstream of the TSS were amplified from Tak-1 genomic DNA using primer pairs consisting of FGMYB3kbup_Cloning_fwd, FGMYB2kbup_Cloning_fwd or FGMYB1kbup_Cloning_fwd, with FGMYBPmeI-R, respectively, and then introduced into the AscI and PmeI sites of pENTR1a-FGMYB-SUF using a SLiCE reaction (Zhang et al. 2012) to generate pENTR1a-proFGMYB3/2/1kb:FGMYB-SUF. These entry vectors were all subcloned into pMpGWB101 using Gateway LR Clonase II to yield pMpGWB101-proFGMYB3/2/1kb:FGMYB-SUF.
pMpGWB101-FGMYB-SUFpA (for premature termination of SUF transcription).
To insert a polyadenylation signal into the 5ʹ-half region of the SUF-transcribed sequence, the SUF sequence was amplified from Tak-1 genomic DNA using the primer pair FGMYB_Eco105I-F_D-TOPO and SUFMreI-R and cloned into the pENTR/D-TOPO vector (Thermo Fisher Scientific, Waltham, MA) to generate pENTR/D-TOPO-SUF. Subsequently, this SUF plasmid was digested with XmaI, and a fragment of the polyadenylation signal amplified from pMpGWB140 (one of the pMpGWB vector series sharing the same basic structure as described in Ishizaki et al. 2015a), using the primer pair SUF_35Ster_Xmal_IFFwd and SUF_35Ster_XmaI_IFRev, was introduced using the In-Fusion system (Takara Bio, Shiga, Japan). The resultant vector pENTR/D-TOPO-SUFpA, along with pENTR1a-FGMYB-SUF, was digested with both Eco105I and MreI, and the pertinent gel-purified fragments were ligated to create pENTR1a-FGMYB-SUFpA. The FGMYB-SUFpA cassette was then transferred into pMpGWB101 using a Gateway LR reaction to yield pMpGWB101-FGMYB-SUFpA.
pMpGWB101-FGMYB-SUFΔ (for partial deletions of the SUF-transcribed region).
To create plasmids with partial deletions (Δ1–Δ6 regions) of SUF (Fig. 3A), the SUF sequences downstream of the deletion regions were amplified from Tak-1 genomic DNA using the primer pairs FGMYB_Eco105I-F and SUF_del-1/2/3/4/5/6_R, respectively. Similarly, the SUF sequences upstream of the deletion regions were amplified from Tak-1 genomic DNA using the sets of primer pairs SUF_del-1/2/3/4/5/6_F and SUFMreI-R, respectively. These respective PCR fragments were cloned into Eco105I and MreI digested pENTR/D-TOPO-SUF using a SLiCE reaction to generate plasmids pENTR/D-TOPO-SUFΔ1 to Δ6. The resultant vectors and pENTR1a-FGMYB-SUF were both digested with Eco105I and MreI, and the pertinent gel-purified fragments were ligated to yield plasmids pENTR1a-FGMYB-SUFΔ1 to Δ6.
To construct a plasmid with a comprehensive deletion of the Δ2 to Δ6 region of SUF (Fig. 3A), the SUF sequences downstream and upstream of the deletion region were amplified from pENTR1a-FGMYB-SUF using the primer pairs FGMYB_Eco105I-F and SUF_del-6_2 R, and SUF_del-2_2 F and SUFMreI-R, respectively. These PCR fragments were introduced into Eco105I and MreI digested pENTR1a-FGMYB-SUF using a SLiCE reaction to yield pENTR1a-FGMYB-SUFΔ2-6.
The resultant entry vectors were recombined with pMpGWB101 using Gateway LR Clonase II to create plasmids pMpGWB101-FGMYB-SUFΔ1 to Δ6 and pMpGWB101-FGMYB-SUFΔ2-6.
Plasmids for the DTA experiments.
To create a plasmid for the constitutive expression of the DTA gene, the coding sequence (CDS) of DTA was amplified from the pJHY-TMp1 vector (Ishizaki et al. 2013a) using the primer pair DTA_Cloning_F_D-TOPO and DTA_Cloning_R and cloned into the pENTR/D-TOPO vector. The resultant pENTR/D-TOPO-DTA was recombined into pMpGWB102 (Ishizaki et al. 2015a) using a Gateway LR reaction to yield pMpGWB102-DTA (pro35S:DTA).
To integrate the antisense SUF sequence downstream of the DTA gene, a CaMV 35S terminator sequence was amplified from pMpGWB102 using the primer pair HindIII_35Ster_F and HindIII_35Ster_R. The SUF sequence, covering a region from the promoter to upstream of the putative transcription termination signal, was amplified from pENTR1a-FGMYB-SUF using the primer pair AscI_SUF+5933_F and AscI_SUFpro-5270_R. These PCR fragments were then inserted into the HindIII and AscI sites of pMpGWB102, respectively, in the reverse strand orientation. pENTR/D-TOPO-DTA was also recombined with this vector using Gateway LR Clonase II to yield pro35S:DTA:SUF.
Generation of transgenic lines
Transformations of M. polymorpha were performed using Agrobacterium-mediated methods on sporelings (Ishizaki et al. 2008) or regenerating thalli (Kubota et al. 2013). Plasmids used for the large deletion of the FGMYB-SUF locus and the DTA experiments were introduced into WT F1 sporelings (Tak-2 × Tak-1). Other plasmids were introduced into thalli of fgmyb-suf-211ld and fgmyb-suf-46ld for genetic female and male lines, respectively. Transformants were selected on agar media containing 0.5 μM chlorsulfuron or 10 μg ml−1 hygromycin.
To confirm the mutations of the large-deletion lines, the mutated regions of fgmyb-suf-18ld, −211ld and −367ld were amplified from their genomic DNAs using the primer pair LD_Seq_F2 and LD_Seq_R3. The sequences of these PCR fragments were directly checked by Sanger sequencing using LD_Seq_F2 and LD_Seq_R3 primers. For fgmyb-suf-46ld, the PCR fragment amplified in the same way was subcloned into pGEM-T Easy Vector (Promega, Madison, WI), and the mutated sequence was checked by Sanger sequencing using LD_Seq_F3 and LD_Seq_R4 primers.
Ascertainment of genetic sex
Thalli were cut into approximately 3-mm squares and crushed in extraction buffer containing 100 mM Tris-HCl pH 9.5, 1 M KCl and 10 mM EDTA as described previously (Ishizaki et al. 2013b). The extracted genomic DNAs were used as templates for sex-ascertainment PCR using KOD FX Neo DNA polymerase (Toyobo Life Science, Osaka, Japan) and primer sets for rhf73 (U chromosome marker) and rbm27 (V chromosome marker) (Fujisawa et al. 2001) listed in Supplementary Table S3.
Observation of reproductive organs
After cutting the stalk of the gametangiophore, the receptacle was placed on agar media and photographed with an EOS Kiss X3 digital camera (Canon, Tokyo, Japan). The receptacle was then transferred onto a microscope slide, and the inner gametangia were gently removed using a needle in a droplet of water for observation under a microscope Axiophot (ZEISS, Oberkochen, Germany).
RT-PCR
Total RNA was extracted from immature gametangiophores (stages 1–2; Higo et al. 2016) using TRIzol reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions. DNA was digested with RQ1 RNase-Free DNase (Promega). One microgram of total RNA was reverse transcribed using oligo dT20 primers and ReverTra Ace (Toyobo Life Science, Osaka, Japan) to synthesize first-strand cDNA. RT-PCR was performed using KOD One PCR Master Mix (Toyobo Life Science) with the primer pairs listed in Supplementary Table S3. FGMYB (Mp1g17210) was amplified under the following conditions: initial incubation at 94°C for 2 min, followed by 33 cycles of 98°C for 10 s, 60°C for 5 s and then 68°C for 8 s. SUF (Mp1g17205) was amplified under the following conditions: initial incubation at 94°C for 2 min, followed by 39 cycles of 98°C for 10 s, 60°C for 5 s and then 68°C for 1 s. EF1α (Mp3g23400) was amplified under the following conditions: initial incubation at 94°C for 2 min, followed by 27 cycles of 98°C for 10 s, 58°C for 5 s and then 68°C for 1 s.
Strand-specific RT-PCR
Strand-specific RT-PCR of SUF expression was performed as previously described (Hisanaga et al. 2019a) with slight modifications. Total RNA was extracted from 10-day-old gemmalings grown under continuous white light or immature gametangiophores (stages 1–2) using an RNeasy Plant Mini Kit (Qiagen, Hilden, Germany) with an RNase-Free DNase Set (Qiagen) according to the manufacturer’s instructions. One microgram of total RNA was reverse transcribed using a mixture of oligo dT20 primers and SUF-specific primers carrying adapter sequences listed in Supplementary Table S3 with ReverTra Ace to synthesize first-strand cDNA. RT-PCR was performed using KOD FX Neo DNA polymerase with the primer pairs listed in Supplementary Table S3. SUF was amplified under the following conditions: initial incubation at 94°C for 2 min, followed by 38 cycles of 98°C for 10 s, 65°C for 15 s and then 68°C for 5 s. EF1α was amplified under the following conditions: initial incubation at 94°C for 2 min, followed by 27 cycles of 98°C for 10 s, 58°C for 15 s and then 68°C for 30 s.
Quantitative real-time RT-PCR
Total RNA was extracted from 10-day-old gemmalings grown under continuous white light using TRIzol reagent and RQ1 RNase-Free DNase and from immature gametangiophores (stages 1–2) using an RNeasy Plant Mini Kit with an RNase-Free DNase Set, according to the manufacturer’s instructions. First-strand cDNA was generated as described earlier for RT-PCR. Quantitative real-time PCR was performed using homemade Taq DNA polymerase (Pluthero 1993) and SYBR Green I Nucleic Acid Gel Stain (Lonza, Basel, Switzerland) with the primer pairs listed in Supplementary Table S3. PCR reactions were carried out with a CFX96 real-time PCR detection system (Bio-Rad Laboratories, Hercules, CA) under the following conditions: initial incubation at 95°C for 30 s, followed by 45 cycles of 95°C for 5 and 60°C for 30 s. Three independent biological replicates were analyzed. EF1α was used as an internal control.
Transcriptome analysis
Total RNA was extracted from immature gametangiophores (stages 1–2) using an RNeasy Plant Mini Kit with an RNase-Free DNase Set according to the manufacturer’s instructions. The amount and quality of RNA were assessed using an Agilent 2100 Bioanalyzer with RNA 6000 Pico Kit (Agilent Technologies, Santa Clara, CA). Libraries were prepared using a NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (New England Biolabs, Ipswich, MA), where mRNA was enriched via polyA selection, and were sequenced using the NextSeq 500 platform (Illumina, San Diego, CA).
RNA-seq reads were pre-processed to remove low-quality reads using fastp v0.20.4 (Chen et al. 2018) and mapped to the MpTak_v6.1r2 genome (MarpolBase, https://marchantia.info; Iwasaki et al. 2021) using HISAT2 v2.2.1 (Kim et al. 2019) with default parameters. The mapped reads were sorted using Samtools v1.11 (Danecek et al. 2021) and counted using the ‘featureCounts’ function in the Subread package v2.0.1 (Liao et al. 2014) with default parameters. Bigwig files were generated using the ‘bamCoverage’ command in the deepTools package v3.5.1 (Ramírez et al. 2016) with ‘–binSize 10 –normalizeUsing BPM –filterRNAstrand forward/reverse’ flags. The effective genome size was calculated using faCount (https://github.com/ucscGenomeBrowser/kent/tree/master/src/utils/faCount).
Transcriptome profiles of each sample were visualized by PCA based on variance stabilizing transformation –normalized read counts of autosomal genes, using the ‘plotPCA’ function in the DESeq2 package v1.36.0 (Love et al. 2014). Transcriptome profiles for WT thalli, mature gametangiophores (stage 4) and gametangia were also included in the analysis. The corresponding data were obtained from the Sequence Read Archive under the following accession numbers: female thalli: SRR15603446, SRR15603447 and SRR15603448 (Iwasaki et al. 2021); male thalli: SRR15603449, SRR15603450 and SRR15603451 (Iwasaki et al. 2021); mature archegoniophores: DRR050351, DRR050352 and DRR050353 (Higo et al. 2016); mature antheridiophores: DRR050346, DRR050347 and DRR050348 (Higo et al. 2016); archegonia: DRR209029, DRR209030, DRR209031 and DRR209032 (Hisanaga et al. 2021); and antheridia: DRR050349 and DRR050350 (Higo et al. 2016).
To evaluate the similarity of gene expression across samples, Spearman’s correlation coefficients were calculated based on normalized gene expression values [transcripts per million (TPM)] of autosomal genes using the ‘cor’ function in the stats package v4.2.1 (R Core Team 2022). Hierarchical clustering and visualization were subsequently performed using the pheatmap package v1.0.12 (Kolde 2019).
Fold changes (FCs) and adjusted P-values (Padj) between immature gametangiophores of WT female and male plants were calculated by a Wald test based on raw read counts using the ‘DESeq’ function in the DESeq2 package v1.36.0 (Love et al. 2014). Differentially expressed genes (DEGs) were identified with |log2FC| > 1 and Padj < 0.01, except for low-expression genes (mean TPM < 1). For the resulting 2,582 autosomal and 60 sex chromosomal genes, z-scores of log-transformed expression values (TPM) were calculated and visualized with hierarchical clustering using the pheatmap package v1.0.12 (Kolde 2019). A heatmap was generated similarly for sex-specific genes in the previous studies (Hisanaga et al. 2019a; Iwasaki et al. 2021), but without hierarchical clustering.
Statistical analysis
All statistical analyses were performed using R v4.2.1 (R Core Team 2022). Tukey–Kramer tests and two-sided Dunnett’s tests, following an analysis of variance, were conducted using the ‘glht’ function in the multcomp package v1.4.25 (Hothorn et al. 2008). For expression values (TPM), the log(TPM + 1) values were used in the Tukey–Kramer tests.
DTA experiment
pMpGWB100 (for control) (Ishizaki et al. 2015a), pro35S:DTA and pro35S:DTA:SUF were each independently transformed into WT F1 sporelings derived from one sporangium, following a previously described method (Ishizaki et al. 2008). After co-cultivation with Agrobacterium, the sporelings were split into eight parts and individually cultured on separate plates with the hygromycin-containing agar media for 3 weeks. The resulting transformants were photographed using the Canon EOS Kiss X3 digital camera. Subsequently, the total number of transformants for each vector was counted, and sex-ascertainment PCR using genomic DNAs extracted from more than 100 randomly selected individuals was performed according to the method described earlier. Based on both the total number of transformants and the sex ratio, the number of female and male transformants was estimated. Each transformation procedure, as well as the calculation of the number of female and male individuals, was independently performed three times.
Supplementary Data
Supplementary Data are available at PCP online.
Data Availability
The nucleotide sequences reported in this paper have been submitted to the Sequence Read Archive at the DNA Data Bank of Japan at https://www.ddbj.nig.ac.jp/dra/ and can be accessed with BioProject number PRJDB16522 (DRR499935-DRR499945).
Funding
Japan Society for the Promotion of Science (JSPS) Grant-in-Aid for Scientific Research (S) (17H07424 to T.Ko.); JSPS Grant-in-Aid for Scientific Research (A) (22H00417 to T.Ko.); JSPS Grant-in-Aid for Challenging Research (Exploratory) (22K19345 to T.Ko.); JSPS Grant-in-Aid for International Leading Research (22K21352 to T.Ko.); JSPS Grant-in-Aid for JSPS Fellows (21J22429 to T.Ka.); The Ministry of Education, Culture, Sports, Science and Technology (MEXT) Grant-in-Aid for Scientific Research on Innovative Areas (19H04860 to S.Y.; 19H05675 to T.Ko.); MEXT Grant-in-Aid for Transformative Research Areas (20H05780 to S.Y.; 23H04744 to Y.Ya.); and JST FOREST Program (JPMJFR2256 to Y.Ya.).
Acknowledgments
We thank Keitaro Okahashi for his initial contribution to generating the plasmid for the complete deletion of the FGMYB-SUF locus and designing the experiment of the polyadenylation signal insertion into SUF; Hirotaka Kato for proposing the DTA experiment; Takefumi Kondo and Yukari Sando for RNA-seq library preparation and sequencing; and Miyuki Iwasaki, Noriyuki Suetsugu, Keiji Nakajima, Tetsuya Hisanaga, Takashi Araki, Keisuke Inoue, Katsuyuki T. Yamato and Masaki Shimamura for helpful discussions. For transcriptome data analysis, we used the supercomputer of Academic Center for Computing and Media Studies (ACCMS), Kyoto University, and the NIG supercomputer at Research Organization of Information and Systems (ROIS), National Institute of Genetics.
Author Contributions
T.Ka. and T.Ko. designed and conceived the project. T.Ka. performed the experiments with some assistance provided by M.M., under the supervision of M.M., S.Y., Y.Yo., Y.Ya., R.N. and T.Ko. T.Ka. analyzed RNA-seq data. T.Ka. and T.Ko. wrote the manuscripts with the support of S.Y., Y.Yo., Y.Ya. and R.N. All authors contributed to reviewing and editing the manuscript.
Disclosures
The authors have no conflicts of interest to declare.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}