





Abstract
DNA double-strand breaks (DSBs) are among the most serious types of DNA damage, causing mutations and chromosomal rearrangements. In eukaryotes, DSBs are immediately repaired in coordination with chromatin remodeling for the deposition of DSB-related histone modifications and variants. To elucidate the details of DSB-dependent chromatin remodeling throughout the genome, artificial DSBs need to be reproducibly induced at various genomic loci. Recently, a comprehensive method for elucidating chromatin remodeling at multiple DSB loci via chemically induced expression of a restriction enzyme was developed in mammals. However, this DSB induction system is unsuitable for investigating chromatin remodeling during and after DSB repair, and such an approach has not been performed in plants. Here, we established a transgenic Arabidopsis plant harboring a restriction enzyme gene Sbf I driven by a heat-inducible promoter. Using this transgenic line, we performed chromatin immunoprecipitation followed by deep sequencing (ChIP-seq) of histones H4K16ac and H2A.Z and investigated the dynamics of these histone marks around the endogenous 623 Sbf I recognition sites. We also precisely quantified DSB efficiency at all cleavage sites using the DNA resequencing data obtained by the ChIP-seq procedure. From the results, Sbf I–induced DSBs were detected at 360 loci, which induced the transient deposition of H4K16ac and H2A.Z around these regions. Interestingly, we also observed the co-localization of H4K16ac and H2A.Z at some DSB loci. Overall, DSB-dependent chromatin remodeling was found to be highly conserved between plants and animals. These findings provide new insights into chromatin remodeling that occurs in response to DSBs in Arabidopsis.
Introduction
Organisms are constantly exposed to the risk of DNA damage by ultraviolet rays, ionizing radiation and reactive oxygen species. DNA double-strand breaks (DSBs) are among the most serious types of DNA damage and can cause mutations and chromosomal rearrangements (Varga and Aplan 2005, Antunes et al. 2012, Shen et al. 2017). To maintain genomic integrity, both prokaryotes and eukaryotes, including plants and algae, have developed sophisticated DSB repair mechanisms. In eukaryotes, DSBs on the chromosomes are repaired in coordination with chromatin remodeling for the deposition of DSB-related histone modifications and variants (Price and D’Andrea 2013, Clouaire and Legube 2019). Such chromatin remodeling during DSB repair is required to facilitate the binding of DSB repair factors to the DSB loci and subsequent restoration of the DNA ends (Lukas et al. 2011, Shi and Oberdoerffer 2012, Price and D’Andrea 2013, Van and Santos 2018). In particular, the alterations of histone modifications and variants regulate DNA accessibility; promote the cohesiveness, flexibility, and mobility of chromatin within the nucleus and facilitate the recruitment of specific factors ensuring repair and its related transcription. Recently, key factors for the interaction between DSB repair mechanisms and the chromatin landscape have gradually been uncovered. Chromatin remodelers such as SWI/SNF complexes, Tip60, histone acetyltransferases (HATs) and histone deacetylases (HDACs) tightly control the acetylation levels of histone H3 and H4 tails to regulate chromatin relaxation in the vicinity of DSBs (Murr et al. 2006, Lee et al. 2010, Miller et al. 2010, Dhar et al. 2017). H4K16ac, H3K14ac and H3K56ac play particularly important roles in DNA damage response, open chromatin formation and repair factor recruitment (Chen et al. 2008, Kim et al. 2009, Li et al. 2010, Aricthota et al. 2022). In addition, the NuA4–Tip60 complex introduces H2A.Z, a variant of histone H2A, onto nucleosomes near the DSBs and promotes acetylation of histone H4 (Gursoy-Yuzugullu et al. 2015, Xu et al. 2012). H2A.Z accumulation after DNA damage is transient and rapidly removed by Anp32e in mammalian cells (Gursoy-Yuzugullu et al. 2015, Obri et al. 2014), but the counterpart (including homolog of Anp32e) in plants is unclear. Altogether, these specific chromatin marks not only establish open chromatin states for efficient DNA repair, but also directly contribute to activation of the DNA damage response, the recruitment of DSB repair machinery and the selection of repair pathway. Some histone marks have been reported to be deposited during DSB repair in plants (Lang et al. 2012, Hirakawa et al. 2019), but data on such chromatin remodeling are extremely limited and the elucidation of such mechanisms has lagged considerably behind equivalent mammalian research. Therefore, a comprehensive survey of the patterns and frequency of chromatin remodeling during DSB repair in plants is crucial to obtain a better understanding of DSB-dependent chromatin dynamics in eukaryotic genome.
To perform the genome-wide analysis to elucidate the details of DSB-induced chromatin remodeling, artificial DSBs need to be reproducibly induced at various genomic loci. Genotoxic agents (e.g. bleomycin, zeocin) and ionizing radiation, often used to induce DSBs, generate random breaks throughout the genome (thus heterogeneously across the cell population), and thus they are unsuitable for reproducible chromatin analyses. Meanwhile, DSB induction systems such as zinc finger nuclease (Kim et al. 1996), transcription activator-like effector nuclease (TALEN) (Christian et al. 2010) and clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9 (CRISPR-Cas9) (Cong et al. 2013, Mali et al. 2013) induce only one localized DSB, which is insufficient for comprehensive analysis. Recently, genome-wide and site-directed induction of DSBs followed by chromatin immunoprecipitation followed by deep sequencing (ChIP-Seq) analysis was performed and elucidated chromatin remodeling across the entire mammalian genome during the DSB repair process (Iacovoni et al. 2010, Iannelli et al. 2017, Clouaire et al. 2018). This DSB induction system is a very powerful tool to comprehensively investigate local chromatin remodeling at multiple DSB loci; however, such a DNA cleavage system uses a chemical inducer, and thus DNA cleavage enzymes continue to be produced sustainably until such inducers are removed from cells. Thus, this system is not ideal for tracking chromatin dynamics after DSB repair.
Here, we established a transgenic Arabidopsis plant harboring a restriction enzyme gene Sbf I driven by a heat-inducible promoter. This DNA cleavage enzyme recognizes an 8 bp specific sequence at the endogenous 623 loci in the Arabidopsis nuclear genome. Using this transgenic plant, we performed a comprehensive analysis of the distribution patterns of histones H4K16ac and H2A.Z around Sbf I recognition sites during and after DSB induction. We also precisely quantified DSB efficiency at all cleavage sites by using the DNA resequencing data obtained by ChIP-seq procedures. The results revealed DSBs at 360 loci, and we then observed the localization of H4K16ac and H2A.Z in many of these regions. Our method makes it possible to combine high-resolution mapping of DSBs with ChIP-seq analyses for DSB-related histone marks and provide detailed profiles of the dynamics of DNA repair and chromatin remodeling in the plant genome.
Results
Heat shock induction of the restriction enzyme gene Sbf I in Arabidopsis thaliana
DNA DSBs need to be induced at a large number of targeted loci throughout the plant genome followed by estimation of DSB efficiency for their loci. To establish a model system to examine a DSB-related chromatin remodeling in plants, we introduced a gene encoding the restriction endonuclease Sbf I into the Arabidopsis genome. The recognition sequence of this enzyme is CCTGCA/GG, which is sparsely distributed at the endogenous 623 loci in the Arabidopsis nuclear genome. This number of recognition sites is smaller than those for other restriction enzymes, and thus physiological damage by the overaccumulation of Sbf I-dependent DSBs is expected to be relatively limited (Supplementary Fig. S1A, Supplementary Table S1). In addition, we adopted the Arabidopsis heat-inducible promoter HSP18.2 for transcription of the Sbf I gene (Fig. 1A), and showed that the transcription level of the introduced Sbf I gene is elevated up to approximately 6,000-fold after heat shock (HS) treatment at 37°C for 4 h, which then rapidly returned to its basal level over 2 additional days (Fig. 1B, C, Supplementary Fig. S1B, C). Sbf I protein also accumulated after HS treatment and was reduced to the basal level within 24 h (Fig. 1D, Supplementary Fig. S1D). These results indicate that these Sbf I-inducible plants could easily and quickly regulate Sbf I gene expression simply by HS treatment. On the basis of these results, we used the transgenic line Sbf I-7 for subsequent analyses.
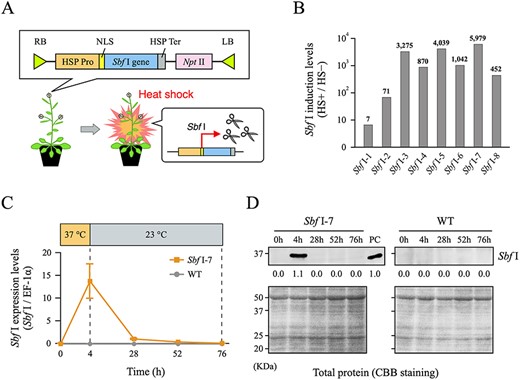
Establishment of heat-inducible Sbf I expression system in Arabidopsis thaliana. (A) Outline of T-DNA construct introduced into A. thaliana and Sbf I gene expression model by HS treatment. (B) Degrees of heat-induced transcription levels of the Sbf I gene by RT-qPCR in T1 plants. The values on the vertical axis show the intensity of heat-induced Sbf I expression levels after HS treatment (HS+) relative to that before HS treatment (HS−). Most transgenic lines significantly increased Sbf I transcription levels after HS treatment. In particular, the Sbf I-7 line showed no leaky Sbf I transcription before HS treatment, and had the highest heat-induced transcription level of approximately 6,000-fold (Supplementary Fig. S1B, C). (C) Transcription level of the Sbf I gene after HS treatment quantified by RT-qPCR. Wild-type (WT) sample was used as a negative control. Error bars are ±SD of three biological replicates. (D) Western blotting analysis of Sbf I after HS treatment. Sbf I enzyme (NEB, R0642) was used for western blots as a positive control (PC), and band intensity relative to that of PC (set as 1.0) was measured. In addition, WT samples were used as negative controls. CBB gel staining of the total protein was shown to indicate equal protein loading.
Genome-wide analysis of Sbf I-mediated cleavage activity in Arabidopsis nuclear genome
ChIP experiments are performed by chromatin DNA, whereas DSBs are detected by a different DNA sample. To investigate DSB-induced chromatin remodeling more precisely, it is appropriate to perform DSB detection and ChIP experiment using the same sample. In this study, we performed next-generation sequencing (NGS) analysis using sheared chromosomal DNA for the ChIP analysis (referred to as input DNA) from Sbf I-7 plants to quantify the DSB efficiency at the Sbf I recognition sites (Sbf I RS) in the Arabidopsis genome. If DSBs occurred at the Sbf I RS upon HS treatment, DNA fragments that include the Sbf I RS would be shortened or degraded and difficult to detect by NGS analysis. Thus, genomic regions with lower sequence coverage around the Sbf I RS in the HS-treated samples should reflect the cleavage of DNA by Sbf I. Furthermore, owing to this scheme, we could detect DSB sites from the ChIP-seq sequencing data, because ChIP sample and input sample (ChIP negative control) are both stemmed from the same chromatin DNA. We thus compared the mapped reads of input DNA from before and after HS treatment in the regions 1 kb upstream and downstream of all Sbf I RS in the Arabidopsis genome. As shown in Fig. 2A, the coverages of 100 bp around the Sbf I RS were substantially decreased in the HS samples at 360 of the 623 Sbf I RS. The amount of digested DNA differed depending on the cleavage site; approximately 40% of the DNA was digested at the loci where the DSB efficiency was the highest (Supplementary Fig. S2A, Supplementary Table S2). In the samples of 48 h after HS treatment (the 52 h samples), such low-coverage profile disappeared thus indicating successful artificial cleavage and subsequent DNA repair (Fig. 2A, B). In contrast, wild-type (WT) plants did not show a decrease in coverage at 360 DSB loci after HS treatment (Fig. 2A, Supplementary Fig. S2B, C).
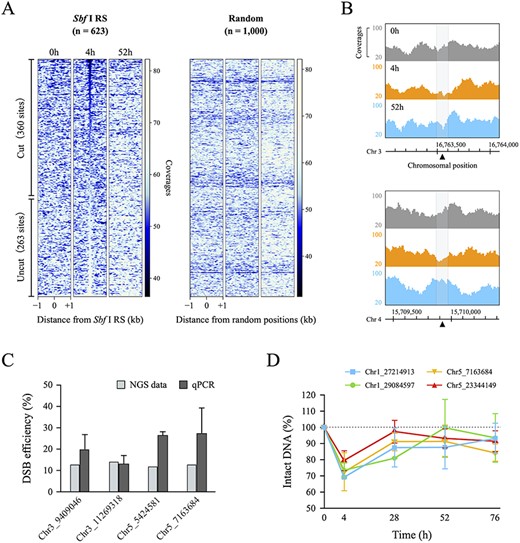
Quantification of DSB efficiency at 623 Sbf I recognition sites in the Arabidopsis nuclear genome. (A) Heatmap obtained from NGS data of input DNA in Sbf I-7 plants. These figures show the coverages around a 2 kb window surrounding the 623 Sbf I recognition sites (Sbf I RS) and 1,000 random positions. Left heatmaps were sorted in ascending order of the total coverages of 100 bp around the Sbf I RS for the 4 h sample. (B) Genome browser screenshots representing the coverages of input DNA around the two Sbf I RS (chromosomes 3 and 4). The gray area covers 100 bp around DSB loci (black triangle). (C) Comparison of the DSB efficiency between NGS data and qPCR. The values of NGS data (gray blocks) and qPCR (dark gray blocks) were calculated by the quotient of the total coverages of 100 bp around the Sbf I RS (4 h divided 0 h) and by the difference of the PCR-amplified values (0 h minus 4 h), respectively. (D) The percentage of intact DNA by qPCR analysis at different time points. Each value was quantified relative to the amount of intact DNA before HS treatment set as 100%. Error bars are ±SD of three biological replicates.
To validate our quantification of the DSB efficiency from NGS data of input DNA, we performed quantitative PCR analysis using primer sets designed to span each Sbf I RS, as shown in Supplementary Table S8. In principle, the difference in amplification efficiency by PCR between HS-treated and HS-untreated samples can be regarded as DSB efficiency. At the four Sbf I RS, DSB efficiencies quantified by qPCR were equal to or greater than those obtained from NGS analysis (Fig. 2C). Previously, Kozak et al. (2009) reported that most DNA ends were restored within hours to days when multiple DSBs were induced in the Arabidopsis genome. We also observed that DSBs at several cleavage sites were repaired up to 48 h after HS treatment; two and four sites were repaired by 24 and 48 h, respectively (Fig. 2D). These results indicated that, in most cells, DSBs were completely repaired within 48 h after their occurrence. Hence, we demonstrated that Sbf I-inducible plants could simultaneously cause transient DSBs at various loci in the Arabidopsis genome and are useful for comprehensive chromatin analysis during and after the DSB repair process. Hereafter, 360 loci where DSBs were observed were considered for ChIP analysis.
DNA cleavage activity by Sbf I did not change in a locus-dependent manner
DNA cleavage enzymes are substantially influenced by the genetic context and the higher-order chromatin structures at chromosomal positions (Chen et al. 2016, Chung et al. 2020). To examine the relationship between DSB efficiency and the genetic context around the Sbf I RS, 360 cut and 263 uncut sites were classified into four groups (Gene, Intergenic, Transposable element, and Others) based on the TAIR10 genomic annotations (https://www.arabidopsis.org), and those classified into the Gene group were further divided into four groups (5’ and 3’ Untranslated regions, Exon, and Intron). As shown in Fig. 3A, there are no distinguishable features between 360 cut and 263 uncut sites. Although we also examined the relationship between DSB efficiencies and genomic context, we could not find any significance. (Fig. 3A–C). We next queried whether the three-dimensional chromatin structure interferes with the DSB by Sbf I, particularly focused on the accessibility of DNA using PlantCADB (Ding et al. 2022), which included the data from various NGS analyses as follows: ATAC-seq, DNase-seq, FAIRE-seq and MNase-seq. However, Sbf I-based DNA cleavage was also not significantly affected by the higher-order epigenetic context of the target sequences (Fig. 3A, D). Therefore, DSB efficiency by Sbf I does not depend on DNA accessibility, and these results strongly suggest that Sbf I-inducible plants are versatile DSB induction tools in vivo.
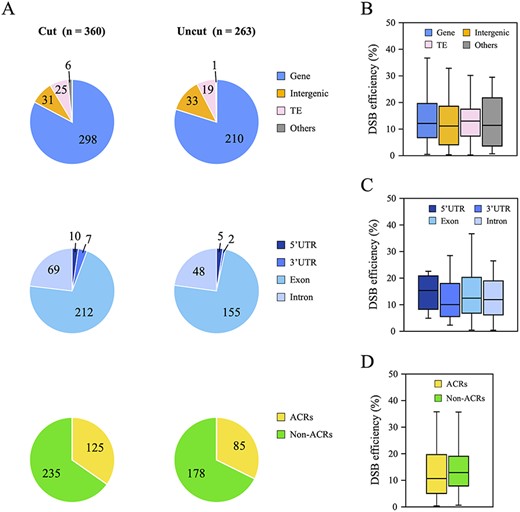
Relationship between DSB efficiency and DNA accessibility at 623 Sbf I RS. (A) Classification of 360 cut sites and 263 uncut sites based on the genetic context and the accessible chromatin regions (ACRs). First, these cleavage sites were classified into four groups (Gene, Intergenic, TE and Others) based on the TAIR10 genome annotation, and then the Gene group (Cut: 298 loci and Uncut: 210 loci) was further divided into four categories (5ʹ and 3ʹ UTR, Exon, and Intron). ACRs and non-ACRs were determined in accordance with the method described in Materials and Methods. (B–D) Boxplots representing the correlation between DSB efficiency and the genetic context (B and C), or the higher-order chromatin structure (D) at 360 cut sites.
DSBs induced transient deposition of histones H4K16ac and H2A.Z around multiple cleavage sites
Studies have reported that, in human and yeast cells, both histones H4K16ac and H2A.Z are involved in DSB repair and transiently deposited on the damaged chromatin (Li and Wang 2017, González-Bermúdez et al. 2022, Gursoy-Yuzugullu et al. 2015, Xu et al. 2012); however, to the best of our knowledge, no such report has yet been published on plant cells. To evaluate the evolutional conservation of such chromatin remodeling mechanism/dynamics via DSB repair in plants, we performed ChIP-seq analyses of Sbf I-7 plants before and after HS treatment with each antibody against H4K16ac and H2A.Z (Fig. 4A), and presented H4K16ac and H2A.Z profiles 1 kb upstream and downstream at 360 DSB and 263 non-DSB loci (Fig. 4B). We analyzed the data with particular care on the following two points; (1) to obtain ‘genuine’ DSB-dependent/prone deposition of histones; (2) to discriminate those from HS-induced ones. First, we found that occupancies of both H4K16ac and H2A.Z increased over only approximately 500 bp around the DSBs compared with the level at non-DSB loci and random positions, and the majority of them were restored to their steady-state levels after 52 h(Fig. 4B, C). For H2A.Z, we observed the deposition of this histone mark before HS-treatment. To validate whether H2A.Z was newly deposited due to DSB, we classified the H2A.Z profiles of the 360 DSB loci shown in Fig. 4B into four groups based on the ratio of their H2A.Z occupancy at 4 h compared to that at 0 h: (1) 1.2- to 3.6-fold, (2) 0.6- to 1.2-fold, (3) 0.3- to 0.6-fold and (4) 0.015- to 0.03-fold. As a result, the H2A.Z levels of groups 3 (0.3- to 0.6-fold) and 4 (0.015- to 0.03-fold) showed H2A.Z deposition after HS treatment without preexisting levels, and groups 1 and 2 also exhibited an increase in occupancy (Supplementary Fig. S3C). The occupancy of these histone marks increased locally in the vicinity of most of the 360 DSB loci; however, the extent of such histone deposition continued up to 1 kb and did not spread over a wide range (Fig. 4C, D, Supplementary Fig. S3A). Second, deposition of neither H4K16ac nor H2A.Z was caused by HS treatment but was instead due to Sbf I-based DNA cleavage (Fig. 4B, Supplementary Fig. S3B). These increases in the levels of both histone marks were not observed in WT plants under the same condition; therefore, the majority of observed chromatin remodeling in Sbf I-7 plants was considered HS-independent. Altogether, these results indicated that DSBs induced H4K16ac and H2A.Z recruitment into cleavage sites in the Arabidopsis genome.
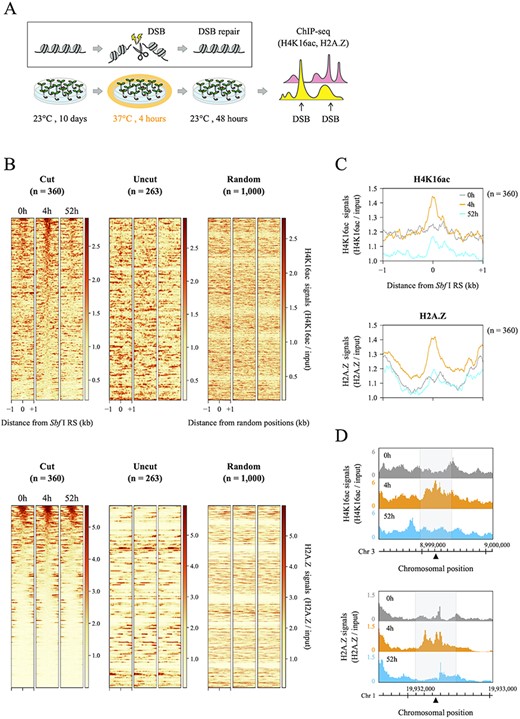
Distributions of histones H4K16ac and H2A.Z before and after DSB induction around the 360 DSB loci. (A) Experimental flow chart for DSB induction followed by ChIP-seq analysis. (B) Average profiles and heatmaps showing the occupancies of H4K16ac and H2A.Z in Sbf I-7 plants. These graphs and figures indicate the signal intensity of each histone mark around a 2 kb window surrounding 360 cut sites, 263 uncut sites and 1,000 random positions. All ChIP-seq data were normalized by input data, and two heatmaps of the 360 DSB loci were sorted in descending order of the total coverages of 500 bp around Sbf I RS for a 4 h sample. (C) Average profiles of H4K16ac and H2A.Z ChIP-seq signals around 360 cut sites shown in Fig. 4B. (D) Genome browser screenshots representing the distributions of H4K16ac and H2A.Z around the two DSB loci (located on chromosomes 1 and 3). The gray area covers 500 bp centered on a DSB locus (black triangle).
H4K16ac and H2A.Z dynamics during and after DSB repair
To understand how often these histone marks were deposited or removed, the dynamics of H4K16ac and H2A.Z at each DSB locus were classified based on their variation patterns. First, we categorized alteration patterns of both H4K16ac and H2A.Z into 3 categories (increased, decreased and unchanged shown in Fig. 5A) based on their relative enrichment around each DSB site before/after HS treatment (Supplementary Table S6). The classification of these histone marks was performed as described in the Materials and Methods using the total coverages within 500 bp around cleavage sites. Second, we compared the alteration patterns of these histone marks between Sbf I-7 and WT plants and classified them based on their consistency. If the category of Sbf I-7 is inconsistent with that of WT, alteration in the deposition of these histone marks at each DSB site is DSB dependent. If the categories of both Sbf I-7 and WT are consistent, it is regarded as DSB-independent change. In the end, the dynamics of H4K16ac and H2A.Z were divided into four patterns. Patterns A and B were defined as DSB-dependent new deposition (DSBincreased) and DSB-dependent removal (DSBdecreased), respectively. Patterns C and D were integrated as a group of DSB-independent fluctuations and defined as “Others” (Fig. 5A). As described above, we have also shown that the mapping rate within 100 bp around the Sbf I RS in the Sbf I-7 chromosomes was reduced by the DSBs (Fig. 2A, B). To test whether this effect negatively influenced the H4K16ac and H2A.Z levels, we excluded the coverage of each histone mark within 100 bp around DSBs from the calculation of these histone levels; however, the patterns of variation of these histone marks did not change significantly (Supplementary Fig. S3D, E).
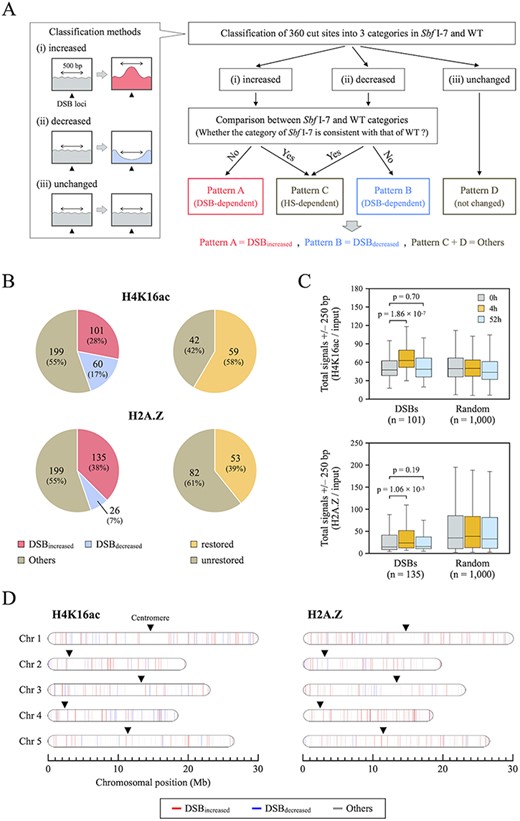
Alteration and restoration rates of H4K16ac and H2A.Z after DSB induction. (A) Overall framework of the classification by H4K16ac and H2A.Z distribution patterns to identify DSB-dependent chromatin remodeling. After classifying 360 DSB loci into three categories (increased, decreased and unchanged) in Sbf I-7 and WT plants, Patterns A and B were defined as DSB-dependent new deposition (DSBincreased) and DSB-dependent removal (DSBdecreased), respectively. Patterns C and D were integrated as DSB-independent fluctuations and defined as “Others”. (B) Pie charts showing the rates of alteration (left) and restoration (right) of H4K16ac and H2A.Z in Sbf I-7 plants. (C) Boxplots representing the totals of H4K16ac and H2A.Z read counts at 250 bp upstream and downstream of DSBincreased loci or at 1,000 random positions (used as a negative control). Wilcoxon rank sum test was used to calculate P values (P < 0.05). (D) Alterations of H4K16ac and H2A.Z after DSB induction mapped on the Arabidopsis chromosomes. The colored bars indicate individual cleavage sites and corresponding the changes in H4K16ac and H2A.Z distributions (DSBincreased: red, DSBdecreased: blue, and Others: gray).
Fig. 5B and Supplementary Fig. S3F show that the levels of both histones H4K16ac and H2A.Z were altered at almost half of the DSB loci after HS treatment. For H4K16ac, there was an increase at 101 loci (28%) and a decrease at 60 loci (17%), whereas for H2A.Z, there was an increase at 135 loci (38%) and a decrease at 26 loci (7%). Fig. 5C also shows significant increase in the averaged occupancy of these histone marks at DSBincreased loci. DSBincreased loci for both histones H4K16ac and H2A.Z were distributed throughout the Arabidopsis genome and partially suggest that increased occupancy of both histone marks is not influenced by genomic position (Fig. 5D). Under our experimental condition, the repair of cleaved DNA is expected to be completed within 48 h (Fig. 2A, D). However, within that time, H4K16ac and H2A.Z were only completely restored to their original state at 59 of 101 (58%) and 53 of 135 sites (39%), respectively (Fig. 5B). These results may reflect a slight delay between the restoration of damaged DNA and the eviction of DSB-related histone marks. These results suggest that H4K16ac and H2A.Z are localized in a DSB-dependent manner and play important roles during DSB repair in the plant genome.
Co-localization of histone H4K16ac and H2A.Z after DSB induction
The incorporation of both histones H4K16ac and H2A.Z onto damaged chromatin alters the nucleosome properties and creates open, relaxed chromatin domains that extend around cleavage sites (Li et al. 2010, Lukas et al. 2011, Xu et al. 2012). Furthermore, H2A.Z exchange is essential for the subsequent acetylation and ubiquitination of the damaged chromatin (Li and Wang 2017, González-Bermúdez et al. 2022, Xu et al. 2012, Gursoy-Yuzugullu et al. 2015). Our results and these findings evoked a hypothesis that H4K16ac and H2A.Z are co-localized on the same nucleosomes with respect to the DSB response. However, to the best of our knowledge, no such reports have been published. To verify such interaction, we integrated the profiles of H4K16ac and H2A.Z in 360 DSBs and investigated the co-localization of these histone marks by DSB induction. Interestingly, these histone marks increased together at 56 of 180 DSB loci (Fig. 6A, B). In addition, we examined the restoration rate of H4K16ac and H2A.Z at these co-localized sites after HS treatment, and found that both histone marks were restored at only 15 of the 56 sites (Fig. 6A). These results suggest that both H4K16ac and H2A.Z are often recruited at DSB loci for efficient repair but are not always co-localized and evicted simultaneously.
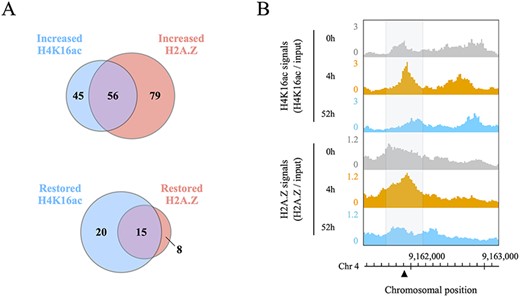
Co-localization of histone H4K16ac and H2A.Z during and after DSB repair. (A) Venn diagram of increased H4K16ac and H2A.Z peaks around 180 DSB loci, and the restoration rate of H4K16ac and H2A.Z in 56 regions where H4K16ac and H2A.Z were co-localized. (B) Genome browser screenshots showing the co-localization of H4K16ac and H2A.Z signals around a DSB locus (black triangle) located on chromosome 4. All data were normalized by input signals. Each gray area covers 500 bp centered on a DSB locus.
In summary, we established restriction enzyme-based DSB-inducible plants and showed that the DSBs induced the deposition of both histones H4K16ac and H2A.Z for accurate DSB repair in Arabidopsis thaliana. Considering that our results in A. thaliana plants are in line with those reported in animals (Li et al. 2010, Xu et al. 2012), such DSB-repair and associated chromatin remodeling mechanisms/dynamics are highly conserved among eukaryotic genomes.
Discussion
DNA double-strand breaks (DSBs) not only are a form of physical DNA damage but also affect the epigenetic landscape. To understand the mechanisms of DSB-related chromatin remodeling in detail, comprehensive and quantitative experiments must be performed that combine genome-wide chromatin analysis with a DSB induction system at various genomic loci. In this study, we established Sbf I heat-inducible plants in which transient DSBs can simultaneously occur at various positions in the Arabidopsis genome (Fig. 1). In addition, the combination of high-resolution mapping of DSBs and ChIP-seq analysis enabled us to quantitatively analyze the patterns of chromatin remodeling before and after DSB induction. We actually identified 360 Sbf I-dependent DSBs (Fig. 2A) and elucidated the spatial and temporal distribution patterns of histones H4K16ac and H2A.Z at their cleavage sites. We found that H4K16ac and H2A.Z increased at approximately 30% and 40% of the 360 DSB loci, respectively, and such deposition was observed throughout the Arabidopsis genome (Fig. 5). Intriguingly, some of these DSB loci showed co-localization of H4K16ac and H2A.Z and/or a slow rate of recovery of these histone marks (Fig. 5B, 6A). On the basis of these results, we characterized the patterns and extent of dynamic changes in the distribution of H4K16ac and H2A.Z, and elucidated that these histone marks altered everywhere throughout the Arabidopsis genome.
In studying the dynamics of the epigenome through the DSB repair process, Sbf I heat-inducible plants in which DSBs can temporarily occur at specific positions throughout the plant genome are very powerful analytical tools. Previously, DSB induction systems with CRISPR-Cas9 and restriction enzymes have been established for the analysis of the yeast and human cells; however, these methods could not elucidate the distribution of histone marks on the repaired chromosomal DNA because the induction of such nucleases was achieved by chemical inducers, and thus was not reversible (Iacovoni et al. 2010, Iannelli et al. 2017, Clouaire et al. 2018). Our expression system using the HSP18.2 promoter can very easily control the expression of the target gene by changing the environmental temperature; the transcription of the target gene is activated at 37°C and deactivated at 23°C. This heat-inducible promoter is highly compatible with many restriction enzymes including Sbf I because there are many alternatives with the optimum reaction temperature at 37°C. In addition to these properties for the promoter and restriction enzyme, the plasmid used in this study is a fully customizable vector that contains two unique restriction sites at the Sbf I gene sequence and promoter/terminator junction (Supplementary Fig. S6). Therefore, this plasmid can easily replace the Sbf I gene portion with other restriction enzyme gene sequences, which is expected to expand the DSB region for analysis. Also, we assessed the impact of DNA accessibility, which depends on the genetic context and the higher-order chromatin structures, on the DSB efficiency, and elucidated that this feature did not correlate with DNA cleavage activity by Sbf I, unexpectedly (Fig. 3). In this regard, we considered that the molecular size of the Sbf I protein is an important factor. Sbf I protein is composed of 323 amino acid (AA) residues, which is much smaller than other DNA-cleaving enzymes (e.g. CRISPR-Cas9: 1,368 AA residues, TALEN: 2,500–3,500 AA residues). In addition, DNA cleavage enzymes such as CRISPR-Cas9 and TALEN have been reported to be significantly affected by the higher-order epigenetic context of their target sequences (Chen et al. 2016, Chung et al. 2020). From such difference in the protein size, we speculated that the conformation of chromosomal DNA could not have had much influence on the DNA accessibility of Sbf I. From our results, at least for the 360 cut sites, those sites represent the DSB-based chromatin remodeling irrespective of the genetic context or inherent chromatin configuration, although underlying molecular mechanism with respect to the different cleavage efficiency between 360/263 sites still unclear. Possible involvement of homologous recombination (HR)-repair and resultant underestimation of DSB-sites warrants further analysis.
In this study, we could identify DSBs at 360 of 623 Sbf I recognition sites using Sbf I-inducible plants (Fig. 2A). Our DSB induction system and detection method enable us to quantify the DSB efficiency as a combination of the sites (1) just cleaved and (2) already repaired via non-homologous end joining (NHEJ) pathways of which sequences harbor mutations enough to make them unmappable. However, it is difficult to detect HR-mediated repair activity which restored DSBs completely without mutations on the double-strand DNA. Thus, in this study, we could not fully discuss relative contributions of HR and NHEJ repair pathways in our experimental system and detection scheme. In order to monitor the pathway-specific DSB repair, the introduction of direct-repeat GFP and pEJ assays to our experimental system could be one of the solutions (Pierce et al. 1999, Mansour et al. 2008, Pierce and Jasin 2014).
We found that most of the damaged DNA was restored within 48 h after HS treatment, but there were some regions where DSBs remained at 48 h after HS treatment (Fig. 2A). For the reasons given below, we ascribed this slow DSB repair to differences among cells and genomic loci in both the timing of DSB formation and the rate of DSB repair in each cell and cleavage site. The expression level and activity of introduced restriction enzymes in plant cells vary in different tissues and developmental stages. Some cells may have high levels of both, while others may have low levels. Thus, cells with high cleavage efficiency require more time for DSB repair. Furthermore, plants and other eukaryotes have two major DSB repair pathways, which are known as HR and NHEJ. These pathways differ in the way in which the two DNA ends are joined and the time taken for them to be completely repaired (Kozak et al. 2009, Davis and Chen 2013, Jasin and Rothstein 2013). Therefore, we also considered that the difference in the repair rate at each DSB locus observed in our analysis is due to these repair pathway choices.
Histone modifications and variants not only act independently but also interact with each other in a cooperative manner (Suganuma and Workman 2008, Li and Wang, 2017, Lu et al. 2015). For instance, γ-H2A.X (phosphorylated H2A.X) is well known to play a crucial role in DNA damage response, and is recruited at DSBs within minutes to hours after DSB induction (Kinner et al. 2008). This response serves as priming for interaction with other histone modifications and variants to promote DSB-associated signaling and repair processes. In particular, in the DNA repair process of mammalian and yeast cells, studies have reported that H4K16ac and H2A.Z are transiently deposited at DSB loci to improve repair efficiency (Li et al. 2010, Xu et al. 2012, Gursoy-Yuzugullu et al. 2015). According to studies on the mechanism depositing these histone marks in DSB regions, MOF and KAT5 (Tip60) are known to be the major enzymes catalyzing the acetylation of H4K16, and in particular, KAT5 is recruited to the genomic locus where the DSB occurred (Li et al. 2017). Meanwhile, H2A.Z exchange onto DSB loci is mediated by the NuA4 complex, including Tip60 and p400 subunits (Xu et al. 2012). Notably, the NuA4 complex can deposit both H4K16ac and H2A.Z. In this study, H4K16ac and H2A.Z levels around DSBs were increased at almost half of the 360 DSB loci after HS treatment (Fig. 4), and increases in both histones occurred simultaneously at 56 DSB loci (Fig. 6). These results indicate that the DSB-dependent localization of these histone marks is a common event, at least among eukaryotes, including plants, and imply that both H4K16ac and H2A.Z are deposited via the NuA4 complex. However, whether both H4K16ac and H2A.Z are really contained in single nucleosomes remains unclear because, in this study, we only examined the localization patterns of these histone marks independently. As the solution to this problem, co-immunoprecipitation experiment for elucidating the dynamics of these histone marks at more time points and identification of reader/writer proteins that participate in H4K16 acetylation and H2A.Z incorporation are necessary.
In conclusion, we introduced a heat-inducible DNA cleavage system into Arabidopsis thaliana that generates a sufficient number of endogenous, sequence-specific DSBs to analyze the DSB-induced chromatin landscape. This tool allowed us to analyze DSB-associated mechanisms at several positions over time and we actually discovered the new biological aspects that H4K16ac and H2A.Z localized or co-localized at DSB loci in plants. Restriction enzyme-inducible plants represent powerful tools for monitoring chromatin remodeling during DSB repair, so that events that take place under different chromatin contexts can be investigated at an unprecedentedly large scale and statistically.
Materials and Methods
Plant materials and growth conditions
Arabidopsis thaliana (Columbia ecotype) was grown at 23°C with continuous light (30–50 μmol m–2 s–1). For the genetic transformation of Arabidopsis, HSP18.2 promoter (Yoshida et al. 1995), SV40 nuclear localization signal (MAPKKKRKVI), full coding region of the Sbf I gene (EMBL accession number: DQ026300) and the HSP terminator sequence (Dansako et al. 2003) were subcloned into the pGreenII-MH2 vector (Hellens et al. 2000, Hirashima et al. 2006). In the following order, the constructs contained the HSP18.2 promoter, the SV40 nuclear localization signal, the Sbf I coding region and the HSP terminator. The resulting plasmids were introduced into Agrobacterium tumefaciens and the Arabidopsis WT plant background was transformed. Among the T1 seeds obtained, we screened the homozygous T4 plants and used them in this study.
Methods for HS treatment of plants
Sbf I-inducible plants were stratified at 4°C in the dark for 2 days, and then grown on MS medium under continuous light (30–50 μmol m–2 s–1) at 23°C for 10 days. These plants underwent HS treatment as whole plants in an NK-type artificial weather chamber (Nippon Medical and Chemical Instruments Co., Ltd.). In accordance with the procedures of Yoshida et al. (1995), Tsukaya et al. (1993), and Takahashi et al. (1992), the optimum temperature and time to promote transcription induction of the HSP18.2 promoter were 37°C and 4 h. Therefore, HS treatment was performed under the same conditions in this study. HS-treated plants were subjected to Sbf I gene expression and ChIP analysis.
RNA isolation and gene expression analysis
Total RNA was extracted from the approximately 20 individual seedlings of 10-day-old plants with the RNeasy Plant Mini Kit (QIAGEN) and recombinant DNase (Macherey-Nagel) and reverse-transcribed with ReverTra Ace® (TOYOBO) and random hexamer. The resultant cDNA samples were used as templates for the expression analysis by quantitative RT-PCR (RT-qPCR). RT-qPCR reactions were performed using the Thunderbird® SYBR qPCR mix (TOYOBO) and Eco Real-Time PCR System (Illumina). The PCR primers used are listed in Supplementary Table S8. Relative RNA levels of the Sbf I gene were calculated and normalized to internal controls, the AT5G60390 (EF-1α) gene-encoded GTP-binding elongation factor and the AT4G05320 (UBQ10) gene-encoded polyubiquitin protein (Czechowski et al. 2005, Udvardi et al. 2008).
Protein extraction and western blotting analysis
Total proteins were extracted from approximately 5 individual seedlings with a buffer containing 50 mM Tris-HCl (pH 6.8), 2% SDS, 6% 2-mercaptoethanol and 10% glycerol. Proteins were separated by SDS-polyacrylamide gel electrophoresis with 12% polyacrylamide gel, and the resolved proteins were transferred onto the Immun-Blot PVDF membrane (BIO-RAD). Sbf I-specific antibody (1:2,000 dilution) was used for western-blot analysis. To generate the Sbf I-specific antibody, a synthetic peptide (CDPDHLIHLNGSRFLGPYER) corresponding to N-terminal sequence of Sbf I protein was used for the immunization of rabbits (Eurofins).
Detection of DNA double-strand breaks
Genomic DNA was extracted from the same plant samples derived from RNA extraction using the DNeasy Plant Mini Kit (QIAGEN) and 0.2 ng of total DNA was used as template DNA. qPCR analysis was performed with primer sets designed to span the Sbf I recognition sites shown in Supplementary Table S8. Data values represent means from more than four technical replicates. The percentage of DNA DSBs at each cleavage site was determined using the PCR amplification of intact DNA from the untreated samples as the standard, and the difference between the HS-treated samples (HS+) and the untreated samples (HS−) was calculated as the percentage of DSBs remaining. The detailed calculation method is shown below.
ΔCt was obtained by subtraction of the threshold cycle Ct in HS− samples from the Ct in HS+ samples: ΔCt = Ct (HS−) − Ct (HS+).
Chromatin immunoprecipitation sequencing (ChIP-seq) library preparation
Approximately 150 individual seedlings of 10-day-old plants (1.0 g) were used for chromatin fixation, isolation and fragmentation, followed by a ChIP experiment as described previously (Zhao et al. 2020, Hata et al. 2021, Kudo et al. 2021), with the following modifications. Chromatin was sonicated to 50–500 bp with a peak of 200 bp using a UD-201 ultrasonic disruptor (TOMY), with the following conditions: Output 2, Duty cycle 60%, 20 s × 18 times (interval 1 min). ChIP experiments were performed basically in accordance with the above procedure using 1 μg of an anti-H4K16ac antibody (Millipore, 07–329) and 2.4 μg of an anti-H2A.Z antibody (Kudo et al. 2021), respectively. The ChIP DNA obtained was purified using a QIAquick PCR Purification Kit (QIAGEN). We performed ChIP-qPCR to test whether the ChIP experiments were successful. ChIP-qPCR revealed that the two antibodies efficiently immunoprecipitated chromatin (Supplementary Fig. S5A). ChIP-seq libraries were prepared with sheared DNA (0.5 ng of ChIP DNA or 5.0 ng of input DNA) using ThruPLEX® DNA-seq Kit (Clontech). These obtained libraries were size-selected at 300–700 bp using ×1.0 AMPure XP beads (Beckman Coulter). NGS was performed using a 151 bp paired-end protocol on an Illumina HiseqX Ten platform.
ChIP-seq data processing
The quality of each raw sequencing file is shown in Supplementary Table S7. Adapter sequences and low-quality reads were removed by Trimmomatic (ver. 0.39; Bolger et al. 2014) with the following parameters: CROP:100 ILLUMINACLIP:2:30:10 LEADING:20 TRAILING:20 SLIDINGWINDOW:4:15 MINLEN:50. To equalize the total reads of each set of ChIP-seq data, seqtk (ver. 1.3; https://github.com/lh3/seqtk) was used. All files were aligned to the reference A. thaliana genome (TAIR10; https://www.arabidopsis.org/) and processed using a classical ChIP-seq pipeline: Bowtie2 (ver. 2.4.5; Langmead and Salzberg 2012) for mapping and SAMtools (ver. 1.6; Li et al. 2009) for the removal of PCR-based duplicated reads (rmdup). Coverage calculations and normalization with input samples for each aligned ChIP-seq dataset (BAM files) were performed in deepTools (ver. 3.5.1; Ramírez et al. 2014). Coverage data were exported in bigwig file format for further processing. To verify consistency between our ChIP-seq data and published ones (H4K16ac; SRR6364625, H2A.Z; SRR11304836, SRR11304837, H3; SRR2046529, H2A.X; SRR11304834, SRR11304835), Pearson Correlation Coefficient (PCC) genome-wide analysis was carried out by deepTools (multiBigwigSummary and plotCorrelation tool). The reproducibility of each set of ChIP-seq data is shown in Supplementary Fig. S4A–C, S5B.
Comprehensive analysis for genome-wide DSB detection using ChIP-seq input data
ChIP-seq data from the input DNA (0, 4, and 52 h samples) were merged with three biological replicates before genome-wide DSB analysis. Using deepTools (computeMatrix tool), we calculated the total coverages of the upstream and downstream 50 bp from Sbf I RS. Final DSB efficiency was calculated using the following formula:
If the above calculated value was greater than 0, we judged that cleavage had occurred.
Determination of random positions
To generate a negative control set of non-DSB regions, we first extracted 10,000 random positions on the Arabidopsis genome (excluding chloroplasts and mitochondria) using Perl. These random sites were filtered for being at least 10 kb away from the Sbf I RS. Finally, we obtained 1,000 random positions from them (Supplementary Table S3).
Methods for extraction of accessible chromatin regions and classification of DSB loci in the plant genome.
According to Ding et al. (2022), we analyzed publicly available datasets (ATAC-seq; SRR6410823, SRR6410824, DNase-seq; SRR11144416, SRR11144417, FAIRE-seq; SRR11144419, SRR11144420, and MNase-seq; SRR10753559–SRR10753562) obtained from Plant Chromatin Accessibility DataBase (PlantCADB; https://bioinfor.nefu.edu.cn/PlantCADB/), and obtained 105,301 ACR candidates. After integrating these accessible chromatin regions (ACRs) and Sbf I RS information (Supplementary Table S1), all Sbf I RS were classified as 125 ACRs or 235 Non-ACRs in 360 cut sites and as 85 ACRs or 178 non-ACRs in 263 uncut sites. (Supplementary Table S4).
Classification of DSB-dependent alterations for H4K16ac and H2A.Z ChIP-seq data
ChIP-seq data for each sample were analyzed by two biological replicates, and their respective genome-wide profiles were entirely consistent (Supplementary Fig. S5B). Because the ChIP-seq data obtained from the two independently prepared biological replicates showed similar profiles, each sequencing data from the two biological replicates was merged and subjected to ChIP-seq data processing. Using deepTools (computeMatrix tool), we calculated the total coverages of the upstream and downstream 250 bp from Sbf I RS (Supplementary Table S5). The alteration patterns of H4K16ac and H2A.Z distributions after DSB induction were classified as follows:
Three categories; (i) increased, (ii) decreased and (iii) unchanged, were defined as Dchange ≧ 1.05, Dchange ≦ 0.95 and the interval between them, respectively. After the classification of H4K16ac and H2A.Z distribution patterns at 360 DSB loci in Sbf I-7 and WT plants, in accordance with Fig. 5A, DSB-dependent/independent fluctuations were identified (Supplementary Table S6). In addition, when Drepair ≦ 1.0, these histone marks were considered restored after DSB repair; otherwise, they were considered unrestored.
Profiling methods for H4K16ac and H2A.Z ChIP-seq data before and after DSB induction
Averaged profiles and heatmaps of H4K16ac and H2A.Z ChIP-seq data were created by deepTools (plotProfile or plotHeatmap tool). The x-axis shows the genomic position relative to Sbf I RS and the y-axis represents the mean coverage at each window (10 bp). All signals of H4K16ac and H2A.Z were normalized by input signals using deepTools (bamCompare tool). In each box-plot representation, the central line represents the median, box ends represent the first and third quartiles, and whiskers represent the minimum and maximum values without outliers. To test differences in distribution between two populations, statistical hypothesis testing was performed using Wilcoxon rank sum test. Statistical analysis was performed using R 4.1.3 with a significance level of 5% (p < 0.05).
Supplementary Data
Supplementary Data are available at PCP online.
Data Availability
The data underlying this article are available in the article and in its online supplementary material. ChIP-seq data have been deposited to the DNA Data Bank of Japan (DDBJ) database under the accession codes DRA015426 (DRR427324-DRR427350).
Funding
This work was supported by Japan Society for the Promotion of Science (JSPS) KAKENHI (22K06342 to S.S.) and Kyoto Prefectural University Academic Promotion Fund Research Encouragement Program (SK20221024 to K.K.).
Acknowledgments
We thank A.F., K.M., K.S., Y.S., S.M. and H.N. for discussion and encouragement. We also thank Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.
Author Contributions
Conceptualization: K.K., S.S.
Data curation: K.K., S.S.
Formal analysis: K.K., S.S.
Funding acquisition: K.K., S.S., J.O.
Investigation: K.K., M.K., S.S.
Methodology: K.K., M.K., T.H., S.S.
Project administration: S.S.
Supervision: S.S., J.O.
Validation: K.K., S.S.
Writing - original draft: K.K., S.S.
Writing - review & editing: K.K., T.H., S.S., J.O.
Disclosures
The authors have no conflicts of interest to declare.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}