





Abstract
Lysine succinylation is a newly identified protein post-translational modification and plays important roles in various biological pathways in both prokaryotes and eukaryotes, but its extent and function in photosynthetic organisms remain largely unknown. Here, we performed the first systematic studies of lysine succinylation in cyanobacteria, which are the only prokaryotes capable of oxygenic photosynthesis and the established model organisms for studying photosynthetic mechanisms. By using mass spectrometry analysis in combination with the enrichment of succinylated peptides from digested cell lysates, we identified 1,704 lysine succinylation sites on 691 proteins in a model cyanobacterium Synechococcus sp. PCC 7002. Bioinformatic analysis revealed that a large proportion of the succinylation sites were present on proteins in photosynthesis and metabolism. Among all identified succinylated proteins involved in photosynthesis, the PSII manganese-stabilizing protein (PsbO) was found to be succinylated on Lys99 and Lys234. Functional studies of PsbO were performed by site-directed mutagenesis, and mutants mimicking either constitutively succinylated (K99E and K234E) or non-succinylated states (K99R and K234R) were constructed. The succinylation-mimicking K234E mutant exhibited a decreased oxygen evolution rate of the PSII center and the efficiency of energy transfer during the photosynthetic reaction. Molecular dynamics simulations suggested a mechanism that may allow succinylation to influence the efficiency of photosynthesis by altering the conformation of PsbO, thereby hindering the interaction between PsbO and the PSII core. Our findings suggest that reversible succinylation may be an important regulatory mechanism during photosynthesis in Synechococcus, as well as in other photosynthetic organisms.
Introduction
Protein lysine succinylation is a recently identified post-translational modification (PTM), in which a succinyl group (-CO-CH2-CH2-CO-) is added to a lysine residue on a protein moiety (Zhang et al. 2011). Succinylation is increasingly recognized as having important roles in various biological pathways in both prokaryotes and eukaryotes (Zhang et al. 2011, Colak et al. 2013, Weinert et al. 2013). Comprehensive so-called ‘succinylome’ studies have been performed to identify the proteins that contain this modification in bacteria (Colak et al. 2013, Weinert et al. 2013, Kosono et al. 2015, Pan et al. 2015, Yang et al. 2015, Mizuno et al. 2016, Okanishi et al. 2017), a protozoan parasite (Hou et al. 2014), fungus (Weinert et al. 2013, Zheng et al. 2016, Xie et al. 2017), mammalian cells (Weinert et al. 2013, Chen 2016, Song et al. 2017), mouse mitochondria (Park et al. 2013, Rardin et al. 2013, Weinert et al. 2013, Boylston et al. 2015) and plants (He et al. 2016, Jin and Wu 2016, Shen et al. 2016, Zhen et al. 2016). These succinylome studies have demonstrated that lysine succinylation is a widespread PTM in diverse model organisms, which changes dynamically in response to cellular physiology and stress (Colak et al. 2013, Park et al. 2013, Hirschey and Zhao 2015). In particular, lysine succinylation is abundant in mitochondrial proteins, and it is enriched in metabolic processes (Park et al. 2013, Rardin et al. 2013). Accumulating evidence suggests that lysine succinylation may be a mechanism involved in the regulation of metabolism and cellular physiology in organisms ranging from bacteria to mammals (Newman et al. 2012, You et al. 2012, Colak et al. 2013, Park et al. 2013, Rardin et al. 2013).
Cyanobacteria (blue-green algae) are a large group of prokaryotes capable of oxygenic photosynthesis, and they play crucial roles in global CO2 assimilation, O2 production and N2 fixation (Nozzi et al. 2013). Cyanobacteria are ancient life forms and evolutionary precursors of plant plastids (Chellamuthu et al. 2013). The photosynthetic activity of cyanobacteria is estimated to comprise more than half of the total primary production on Earth (Johnson et al. 2006). Photosynthesis and carbon metabolism are closely connected processes in photosynthetic organisms (Padmasree et al. 2002, Noctor et al. 2004, Tan et al. 2011), and cyanobacteria have developed intricate mechanisms to balance the supply of electrons from photosynthetic processes with the demands of cellular metabolism (Singh et al. 2008, Wegener et al. 2010, Tan et al. 2011). However, many aspects of the mechanisms that regulate photosynthesis and metabolic processes remain poorly understood. Lysine succinylation has been reported to be a functional PTM, which has the potential to regulate metabolism and co-ordinate different metabolic pathways in diverse model organisms (Newman et al. 2012, You et al. 2012, Colak et al. 2013, Park et al. 2013, Rardin et al. 2013); therefore, we hypothesized that lysine succinylation may play important regulatory roles in multiple metabolic processes in cyanobacteria, such as photosynthesis and carbon metabolism. Knowledge of the succinylated proteins in cyanobacteria is essential for understanding their function, but no succinylated proteins have been reported previously in cyanobacteria to the best of our knowledge.
To obtain a comprehensive understanding of succinylation events in cyanobacteria, we performed a systematic study of the identities and functional roles of the succinylated proteins in cyanobacteria, where we used Synechococcus sp. PCC 7002 (hereafter Synechococcus) as a model. We analyzed the succinylome of Synechococcus using peptide affinity enrichment prior to high accuracy mass spectrometry (MS) analysis. In total, we identified 1,704 unique lysine succinylation sites on 691 proteins in Synechococcus. These results comprise the most extensive data set regarding succinylation that is available for any photosynthetic organism at present, as well as providing insights into the range of functions regulated by succinylation in Synechococcus. The succinylated proteins identified in this study were involved in various biological processes, and they were particularly enriched in photosynthesis and metabolic processes, which are both intimately linked to the cellular energy status. The functional significance of lysine succinylation sites in PSII manganese-stabilizing protein (PsbO) was assessed by site-specific mutagenesis and biochemical studies. Overall, our findings provide novel insights into the range of functions regulated by succinylation, as well as suggesting that reversible succinylation may be an important and previously unknown regulatory mechanism involved in Synechococcus photosynthesis, and other photosynthetic organisms.
Results
Lysine succinylation in photosynthetic organisms
To investigate whether lysine succinylation occurs in photosynthetic organisms, we performed Western blot analysis using anti-succinyl lysine antibody with whole-cell lysates from Synechococcus, Synechococcus elongatus PCC 7942, Synechocystis sp. PCC 6803, Chlorella sp. NJ-18, Chlamydomonas reinhardtii, Arabidopsis thaliana, Spinacia oleracea and Oryza sativa. As shown in Fig. 1A, multiple bands were detected and lysine succinylation occurred on many proteins, suggesting that succinylation is a widespread and frequent PTM in diverse photosynthetic organisms.
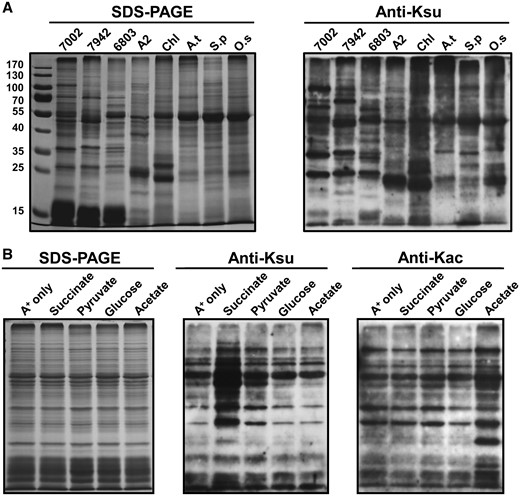
Analysis of lysine succinylation. (A) Profiling of lysine succinylation in photosynthetic organisms. Lane 1, Synechococcus; 2, Synechococcus elongatus PCC 7942; 3, Synechocystis sp. PCC 6803; 4, Chlorella sp. NJ-18; 5, Chlamydomonas reinhardtii; 6, Arabidopsis thaliana; 7, Spinacia oleracea; 8, Oryza sativa. (B) Anti-lysine acetylation/succinylation immunoblots showing that different carbon sources altered acetylation/succinylation profiles in Synechococcus.
To gain functional insights into how succinylation might regulate cellular functions, we assessed the relative abundance of lysine succinylation in whole-cell extracts from Synechococcus under different culture conditions, including succinate, acetate, glucose and pyruvate. We found that Synechococcus exhibited different succinylation or acetylation levels in media that contained different carbon sources (Fig. 1B). Synechococcus exhibited different growth rates (Fig. 2A) and doubling times (Fig. 2B) in media that contained different carbon sources. As expected, Synechococcus exhibited different growth rates in media containing different concentrations of succinate (Fig. 2A, B), and the succinylation levels increased as the concentration of succinate increased (Fig. 2C). In contrast, the protein acetylation levels remained unchanged in Synechococcus under increased concentrations of succinate (Fig. 2C). Synechococcus can tolerate extreme high light intensities (Nomura et al. 2006b), so we determined whether different light intensities might affect the succinylation levels of Synechococcus proteins in vivo. As shown in Fig. 2D, the succinylation levels of Synechococcus proteins were modulated in response to changes in light intensity, suggesting that lysine succinylation may be a mechanism during the response to high light stress in Synechococcus.
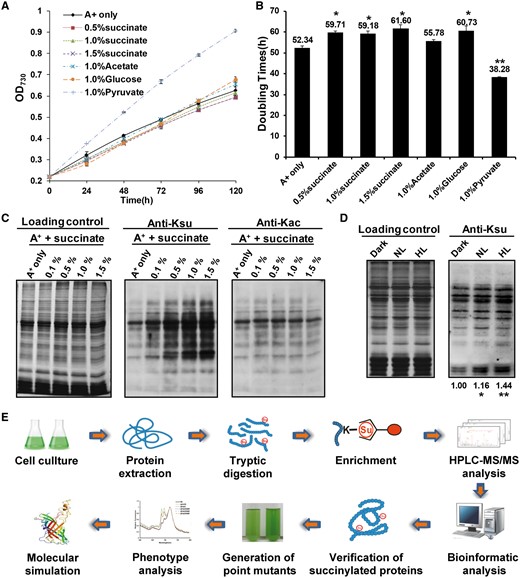
Growth and lysine acetylation/succinylation profiles of Synechococcus. (A) Growth curves of Synechococcus in culture media containing different carbon sources. Cells were grown in A+ medium or A+ medium supplemented with 1.0% (w/v) succinate, 1.0% (w/v) pyruvate, 1.0% (w/v) glucose or 1.0% (w/v) acetate. Growth curves are representative of at least three independent experiments. (B) Doubling times of Synechococcus grown in different carbon sources. (C) Anti-lysine acetylation/succinylation immunoblots showing that different concentrations of succinate altered acetylation/succinylation profiles of Synechococcus. (D) Profiling of lysine succinylation of Synechococcus under dark (0 μmol m–2 s–1), normal light (NL, 250 μmol m–2 s–1) and high light (HL, 2,000 μmol m-2 s-1) conditions. (E) Flowchart showing the experimental procedure for the succinylation proteomics study.
Lysine succinylation proteome in Synechococcus
To identify the lysine succinylation sites in Synechococcus, we used a proteomic approach to detect succinylated proteins and their succinylation sites in Synechococcus proteins (Fig. 2E). In total, we identified 1,703 unique modified peptides encompassing 1,704 succinylation sites (class I) from 691 proteins with a false discovery rate (FDR) <1% for modified peptides. Detailed information about all of the succinylated peptides identified is provided in Supplementary Table S1. The average site localization score for modified peptides was 101.3031 (Supplementary Fig. S1A), and the overall absolute peptide mass accuracy was 0.0651 p.p.m. (SD 1.3841 p.p.m.) (Supplementary Fig. S1B), which confirmed the high accuracy of the modified peptide data obtained by MS. MS raw data files are publicly available through PeptideAtlas (www.peptideatlas.org) (data set ID PASS00670). Annotated peptide spectra for all succinylated peptides are deposited in Peptideatlas and supplied as Supplementary Data.
Functional annotation of the Synechococcus succinylome
To understand the possible roles of the lysine succinylation pathway, we performed functional annotation using Gene Ontology (GO) (Supplementary Table S2A; Fig. S1C). The classification results obtained for biological process showed that most of the succinylated proteins were involved in metabolic process (43%) and cellular process (35%). The major cellular localization of succinylated proteins included cell (42%), macromolecular complex (25%), structural membrane (19%) and organelle (14%). Within the molecular functions cluster, the largest group of succinylated proteins was related to catalytic activity (45%), followed by binding (39%) and structural molecule activity (7%).
Similar to the GO classification results, the GO enrichment analysis showed that succinylated proteins were mostly enriched in translation (P = 3.2E-22) and photosynthesis (P = 1.54E-6) (Supplementary Table S2B; Fig. S2A). In agreement with this, the GO enrichment analysis for cellular component demonstrated that photosynthetic components were mostly enriched in our data, including thylakoid (P =1.68E-06), thylakoid membrane (P =2.52E-04), phycobilisome (P = 4.25E-04) and photosynthetic membrane (P = 8.93E-04). Thus, the molecular functions attributed to succinylated proteins included structural constituent of ribosome (P = 4.86E-20), structural molecule activity (P =8.26E-19) and rRNA binding (P = 3.25E-13). These results suggest that succinylation may play regulatory roles in photosynthesis. Furthermore, enrichment analysis of the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway showed that a large proportion of the succinylated proteins identified in this study were also mapped to photosynthetic pathways (Supplementary Fig. S2A). Therefore, the KEGG pathway, GO annotation and enrichment analyses suggested that succinylation is associated with the photosynthesis process in Synechococcus.
Sequence recognition motifs and structural preference of the Synechococcus succinylome
To determine whether common sequence preferences were present in the succinylated proteins, we evaluated the flanking sequences of the lysine succinylation sites identified in the present study. We tested the 12 residues flanking the succinylation sites for the over-representation of specific amino acids relative to their expression in the proteome background distribution and an intensity map was generated, as described previously (Yang et al. 2013, Liu et al. 2014, Yang et al. 2015) (Supplementary Fig. S2B). A significant preference was observed for arginine at the +5 position, whereas non-polar hydrophobic residues (isoleucine and methionine) were found most commonly at the –1 position, and lysine occurred frequently at the –5 and +5 position.
We also investigated the structural features of lysine succinylation sites in proteins using the NetSurfP algorithm (Petersen et al. 2009), and all the lysine residues were categorized based on the structured regions of proteins (α-helix, β-strand) and non-structured regions of proteins (coil region). We found that 58.5% of the identified succinylated lysines tended to be present in the structured regions of the identified proteins. Meanwhile, there were still a large proportion of non-succinylated lysines on the identified proteins, and 55.7% of these lysines were localized in the structured regions (Supplementary Fig. S2C). In agreement with previous reports (Weinert et al. 2013, Yang et al. 2015), the succinylated lysine residues were found more frequently in structured regions, in contrast to the non-succinylated lysines. Therefore, we speculated that lysine succinylation may affect the surface properties of target proteins, leading to the potential changes of structure or function as in previous findings (Yang et al. 2015).
Evolutionary conservation of succinylated proteins
To explore succinylome conservation in Synechococcus, we performed a systematic analysis with all the identified succinylated proteins and all the proteomes as previously described (Liu et al. 2014). The percentages of Synechococcus homologs of non-succinylated proteins and succinylated proteins are presented in Supplementary Fig. S2D. Interestingly, the succinylated proteins of Synechococcus showed markedly higher conservation, ranging from archaea to eukarya, particularly in bacteria, than the non-acetylated proteins.
We further aligned the best scoring homologous proteins from the following six previously reported bacterial succinylomes: Escherichia coli (Weinert et al. 2013), Mycobacterium tuberculosis (Yang et al. 2015), Corynebacterium glutamicum (Mizuno et al. 2016), Geobacillus kaustophilus (Okanishi et al. 2017), Bacillus subtilis (Kosono et al. 2015) and Vibrio parahaemolyticus (Pan et al. 2015). In total, 322 homologous succinylated proteins in Synechococcus were overlapped in the succinylome of the six bacteria (Supplementary Table S3A), and it is noteworthy that most of these orthologs are involved in central metabolism (Supplementary Table S3B). Together, these results suggest that succinylation is conserved in different species and may play an important role in metabolic processes.
Succinylated proteins involved in metabolism and photosynthesis
Based on a biological interaction network of all the succinylated proteins (Supplementary Table S4A), we evaluated the whole interaction networks of succinylated proteins with the MCODE plugin tool in Cytoscape. Remarkably, three highly interconnected subnetworks were retrieved in Synechococcus which comprised several succinylated proteins involved in photosynthesis and metabolism-related proteins (Supplementary Table S4B; Fig. S3A). These findings suggested that lysine succinylation may play crucial roles in the regulation of photosynthesis and central metabolism.
We also investigated the succinylated proteins by mapping to KEGG pathways (Supplementary Table S5), and we found that a large proportion of the succinylated proteins were involved in photosynthesis. In total, 56 proteins were involved in photosynthesis (including carbon fixation), as shown in Supplementary Fig. S3B. Consistent with previous studies on lysine succinylation (Colak et al. 2013, Weinert et al. 2013, Li et al. 2014, Hirschey and Zhao 2015, Yang et al. 2015), many metabolic enzymes involved in central metabolism were found to be succinylated, such as enzymes involved in glycolysis/gluconeogenesis, the citric acid cycle and fatty acid metabolism (Supplementary Fig. S3B). Overall, our results indicate the involvement of lysine succinylation in the regulation of photosynthesis and cellular metabolism in Synechococcus.
Analysis of PsbO succinylation
PsbO is indispensable in the oxygen evolution reaction and plays critical roles in maintaining the integrity and activity of the Mn cluster in all oxygenic photosynthetic organisms (Suorsa and Aro 2007). Based on our succinylome results, we identified two reliable succinylation sites (Lys99 and Lys234) in PsbO (Supplementary Table S1). The MS/MS spectra of these peptides, which indicate their exact succinylation sites, are shown in Fig. 3A and B. Further analysis revealed the that the lysines at positions 99 (in the loop) and 234 (β-sheet) in PsbO were conserved in Synechococcus orthologs ranging from cyanobacteria to plants, thereby suggesting that these residues may be important for the evolutionarily conserved function of PsbO (Fig. 3C). The crystallographic model showed that PsbO could form a stable β-barrel structure and that succinylation may alter the conformation of PsbO and affect its interaction with PSII complexes due to a change in the lysine residue charge status from +1 to −1 (Fig. 3D). In addition, the succinylation levels of PsbO were found to increase significantly in response to high light intensity, succinate stimulation and nitrogen deficiency conditions (Supplementary Fig. S4), which suggests that lysine succinylation may play a regulatory role in response to different treatments in Synechococcus. Based on this observation, we aimed to determine how the modulation of PsbO succinylation might affect its function in Synechococcus.
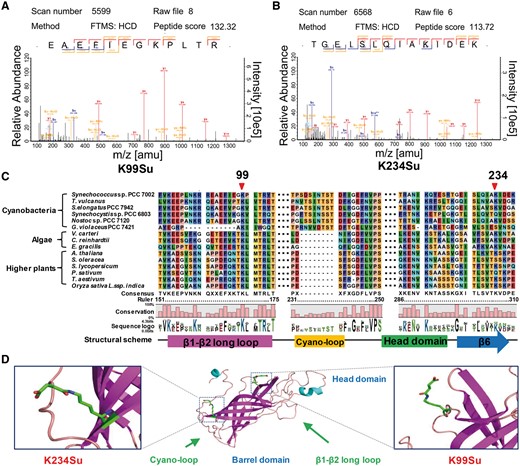
Analysis of PsbO succinylation. (A, B) The MS/MS spectra of two identified succinylated peptides of PsbO. (C) Multiple sequence alignment of PsbO from different species (cyanobacteria, algae and plants). The conserved succinylation sites are marked by the red arrow. (D) Localization of the succinylation sites in PsbO.
Effects of PsbO succinylation on growth and oxygen evolution
To elucidate the roles of succinylation at Lys99 and Lys234 in PsbO, we disrupted the psbO gene by interposon mutagenesis, and site-directed mutations were generated in the Synechococcus psbO gene. First, we constructed a neutral platform at the SYNPCC7002_A1479 locus to express the psbO gene under the control of the cpcAB promoter as previously described (Chen et al. 2017). Next, we mutated the modified residue to arginine (R) to prevent succinylation by using a similar structure (K99R and K234R) and to glutamic acid (E) to mimic a constitutively succinylated lysine (K99E and K234E) (Xie et al. 2012) (Supplementary Fig. S5A). All of the mutations were then verified by both DNA sequencing (Supplementary Fig. S5B) and MS analysis (Supplementary Fig. S5C). Further Western blotting analyses showed that the expression levels of PsbO, the PSII subunit (CP43), PSI subunit (PsaC) and phycobilisome subunit (CpcG) were not affected by site-directed mutagenesis (Fig. 4). Interestingly, the mutations to mimic the constitutive succinylation of PsbO protein led to significantly lower accumulation of D1 protein compared with the results obtained from the wild type. We speculate that PsbO succinylation may regulate the turnover of D1 protein in Synechococcus.
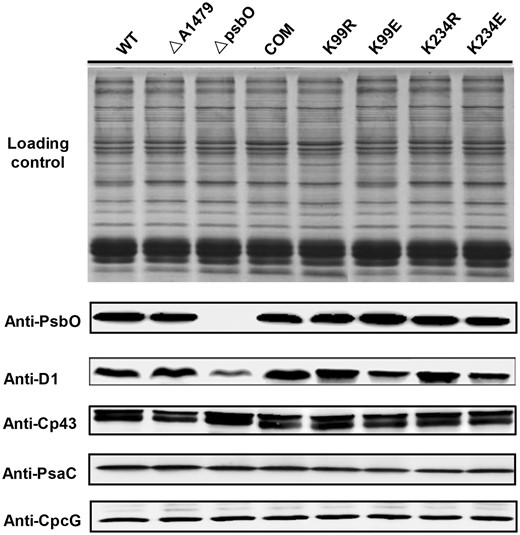
Western blotting analysis of different Synechococcus strains. Western blotting analysis of PsbO, D1, Cp43, PsaC and CpcG from the wild type (WT), ΔA1479 mutant strain, ΔpsbO mutant strain and trans-complemented psbO mutant strains (COM, K99R, K99E, K234R and K234E). Coomassie blue staining of protein lysates from different Synechococcus strains was used for the loading control. PsbO, PSII manganese-stabilizing polypeptide; D1, PSII P680 reaction center D1 protein; CP43, PSII 44 kDa subunit reaction center protein; PsaC, PSI subunit VII; CpcG, phycobilisome rod–core linker polypeptide.
Next, we examined the growth of the wild-type and mutated Synechococcus strains in normal light (NL) and high light (HL) conditions. All of the mutants behaved in a similar manner to the complementary strain when grown in NL conditions (Fig. 5A). However, in HL conditions, the ΔpsbO mutant exhibited a reduced growth rate compared with the wild type and complementary strains (Fig. 5B). Similar to the ΔpsbO mutant, the growth rates of the K234E mutants, which mimicked constitutive succinylation, were significantly slower than those of the wild type and complementary strains (Fig. 5B). In contrast, the growth rates of all the other mutants did not differ from those of their complementary strains in HL conditions (Fig. 5B). We conclude that K234 mutation (K234E) that mimicked the constitutive succinylation of PsbO, may play regulatory roles during the response to HL acclimation in Synechococcus.
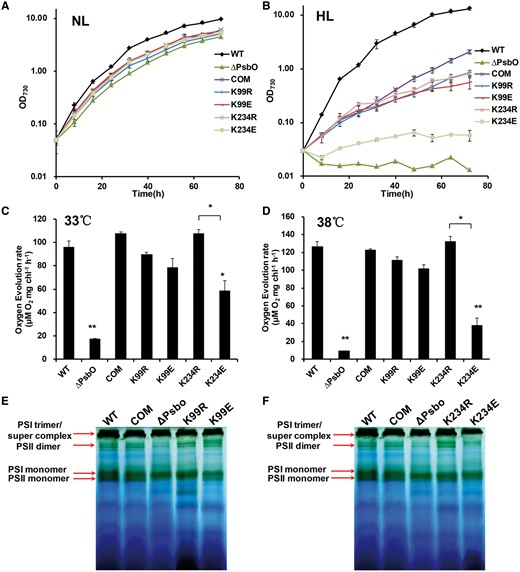
Growth curves, oxygen evolution and BN–PAGE analysis of thylakoid protein complexes for different Synechococcus strains. (A) Growth curves for the wild type, the ΔpsbO mutant strain and trans-complemented psbO mutant strains (COM, K99R, K99E, K234R and K234E). Cells were grown in A+ medium at 38°C under 250 μmol m−2 s−1 and (B) 2,000 μmol m−2 s−1 with aeration by 1.0% (v/v) CO2 in air. Cells were grown in A+ medium at 38°C under 250 μmol m–2 s–1 with aeration by 1% (v/v) CO2 in air. Data are expressed as the mean and SD based on three individual experiments. (C) Oxygen evolution rates for the wild type, ΔpsbO mutant strain and trans-complemented psbO mutant strains (COM, K99R, K99E, K234R and K234E). Oxygen evolution was measured under 500 μmol m–2 s–1 using as an electron acceptor at 33°C and (D) 38°C. Significant differences in oxygen evolution were determined using a two-tailed t-test. (E) BN–PAGE analysis of thylakoid protein complexes isolated from the wild type, the ΔpsbO mutant strain and trans-complemented psbO mutant strains (COM, K99R and K99E). (F) BN–PAGE analysis of thylakoid protein complexes isolated from the wild type, the ΔpsbO mutant strain and trans-complemented psbO mutant strains (COM, K234R and K234E). The PSI trimer, PSII dimer, PSI monomer and PSII monomer of Synechococcus are shown on the left.
PsbO is reported to have an indispensable role in the oxygen evolution reaction (Popelkova and Yocum 2011). Thus, we hypothesized that the succinylation of PsbO might affect the oxygen evolution reaction in Synechococcus. To test our hypothesis, we measured the oxygen evolution rates with the wild type and mutants at different temperatures. Similar to their effects on growth, the K234E mutation led to significant decreases in the oxygen evolution rate, whereas all other mutations had no effect (Fig. 5C, D). These results suggest that K234 mutation (K234E) negatively regulates oxygen evolution in Synechococcus.
To determine further whether PsbO succinylation might affect the assembly/stability of the PSII complex in Synechococcus, the thylakoid membranes of all strains were isolated from the whole-cell extracts (Watanabe et al. 2011, Yang et al. 2014) and further separated by a 5–13% gradient Blue Native (BN)–PAGE as previously described (Watanabe et al. 2011, He and Mi 2016, Yamamoto et al. 2016, Bersanini et al. 2017). Consistent with previous reports (Watanabe et al. 2011, He and Mi 2016, Yamamoto et al. 2016), the major green bands, including PSI trimer, PSII dimer, PSI monomer and PSII monomer, were clearly detected in all strains. As excepted, a remarkable decrease in the amount of PSI and PSII complexes, in both the dimer and monomer form in the ΔPsbO strain, was observed on the gel (Fig. 5E, F), compared with the wild type and complementary strains. The amount of PSI and PSII complexes in K99E and K99R mutants was almost identical in the wild type and complementary strains, suggesting that the mutation at K99 may not affect the assembly of photosynthesis (Fig. 5E). Notably, we detected an evident decrease of the PSII dimer intensity in the K234E mutant, while the K234R mutant exhibited similar bands to the wild type and complementary strains (Fig. 5F). These results suggested that a deficiency in PsbO and a site-specific mutation at K234 (K234E) that mimicked the constitutive succinylation of PsbO may affect the photosynthetic complex assembly/stability in Synechococcus.
Effects of PsbO succinylation on the 77 K fluorescence emission spectra
To investigate energy transfer from the phycobilisome to Chl a in the thylakoid membrane, we obtained low temperature (77 K) fluorescence emission spectra. We detected three major fluorescence emission peaks using whole cells from the wild type and various mutants at 685, 695 and 720 nm, where the excitation wavelength employed was 430 nm, to excite Chl a (Fig. 6A). It is well established that the first two peaks are due principally to PSII, whereas the emission peak at 720 nm is related to PSI (Shen et al. 2002). The fluorescence emission spectrum of the ΔpsbO mutant had larger emission peaks due to PSII at 685 nm (mostly from PsbC/CP43) and 695 nm (mostly from the PsbB/CP47 protein) compared with the wild-type strain. After complementation with the wild-type gene, the emission spectrum produced by the ΔpsbO mutant did not differ from that of the wild type. These results demonstrate that a deficiency in PsbO affects the assembly of the PSII reaction centers. In particular, the K234E mutant had similar fluorescence emission peaks at 685 and 695 nm compared with the ΔpsbO mutant, whereas those produced by all of the other mutants were almost indistinguishable from those of the wild type.
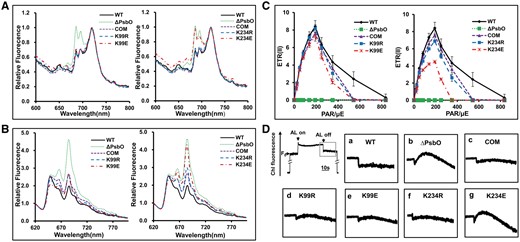
Fluorescence emission spectra for different Synechococcus strains. (A) Fluorescence emission spectra at 77 K for different Synechococcus strains, which were obtained using dark-adapted whole cells excited at 430 nm (Chl excitation). The concentrations of all samples was adjusted to 3 μg ml−1 Chl a. (B) Fluorescence emission spectra at 77 K for different Synechococcus strains, which were obtained using dark-adapted whole cells excited at 600 nm (phycobilisome excitation). The concentrations of all samples were adjusted to 3 μg ml−1 Chl a. (C) Electron transport characteristics of different Synechococcus strains. PSII-mediated electron transport rates (ETRs) of intact Synechococcus cells, which were derived using fluorescence parameters recorded from rapid light curves. (D) Analysis of cyclic electron flow around PSI based on the detection of transient increases in Chl fluorescence. The upper left curve represents a typical trace of Chl fluorescence from the wild type. The inset shows magnified traces from the boxed area. ML, measuring light; SF, saturating flash. a, wild type; b, ΔpsbO mutant; c, complemented strain; d–g, trans-complemented psbO mutants (K99R, K99E, K234R and K234E).
In addition, the excitation of phycobilisomes with 600 nm light also indicated that there were large differences among the wild type and various mutants (Fig. 6B). Compared with the wild type strain, the ΔpsbO and K234E mutants exhibited increased emissions from allophycocyanin at 665 nm as well as the terminal phycobilin emitters at 685 nm (Veerman et al. 2005), whereas all of the other mutants had similar fluorescence emission spectra. We conclude that K234 mutation (K234E) of PsbO affects the assembly of PsbO in the PSII reaction centers, which leads to further decreases in the efficiency of energy transfer from PBS to PSII Chl a.
Effects of PsbO succinylation on electron transport
The modulated source used to obtain pulse amplitude modulated (PAM) fluorescence measurements is constant, so it can be used to measure the modulated fluorescence signal if the background irradiance is sufficiently high to be close to all of the reaction centers (Geider and Osborne 1992). Thus, to investigate whether linear electron transport was affected by the succinylation status of PsbO, we performed room temperature PAM fluorescence studies to determine the electron transport rates through PSII [ETR(II)] in various Synechococcus strains. As shown in Fig. 6C, the ETR(II) for the ΔpsbO mutant was close to zero. However, the K234E mutant exhibited significantly decreased ETR(II)s compared with the wild type and complementary strains, whereas all of the other mutants differed little in terms of their ETR(II)s (Fig. 6C). In addition, the maximal electron transport rates [ETR(II)max] in the K234E mutant were reduced by 34.97% (Fig. 6C). Therefore, K234 mutation (K234E) that mimicked the constitutive succinylation of PsbO decreased the efficiency of electron transport through PSII.
Post-illumination increases in Chl fluorescence are thought to be attributable to the reduction of plastoquinone by NAD(P)H or other reducing substances that accumulate under light, which mainly involves PSI cyclic electron transport (Asada et al. 1993, Mano et al. 1995, Mi et al. 1995). We also investigated the effect of succinylation on the PsbO-mediated cycle electron flow around PSI. As shown in Fig. 6D, the ΔpsbO and K234E mutants exhibited significant increases in Chl fluorescence compared with the wild type and complementary strains, whereas all of the other mutants did not differ in terms of Chl fluorescence. Our results demonstrated that K234 mutation (K234E) of PsbO increased PSI cyclic electron transport, whereas mutation of K99 did not have the same effect.
Succinylation at K234 influences the head domain stability of PsbO
To obtain insights into the potential mechanisms that might allow succinylation to affect the functions of PsbO, we used molecular dynamics (MD) simulations to determine the effects of succinylation at K99 and K234 on the structure of PsbO. We found that succinylation at K99, which is located in the β-strand loop region, had little or no effect on the structure of PsbO in our MD simulations (Fig. 7A). In contrast, K234 is positively charged before succinylation, and it is located in the β-strand adjacent to the large flexible loop of the head domain. The MD simulations showed that succinylation at K234 might change the microenvironment of the β-strand domain, which could subsequently alter the conformation of the head domain of PsbO (Fig. 7B). The K234E mutation, which mimics succinylation with a negative charge, was also predicted to destabilize the head domain of PsbO. The destabilizing effect was observed in the MD simulations when we introduced the K234E mutation, and thus this suggests that the K234E mutation mimics succinylation at the molecular level (Fig. 7B). Moreover, the structures with the K99E and K99R mutations agreed with those with K99su, unmodified K99, respectively (Fig. 7A). Similarly, the structures with the K234R mutation corresponded well with the unmodified PsbO structure, thereby indicating that the mutation of the lysine residue to R could lock PsbO into a constitutively desuccinylated state and maintain the protein’s structure (Fig. 7B). The values of the root mean square deviation (Fig. 7C) trajectories and residue-specific root mean square fluctuations (Fig. 7D) also confirmed the outcomes of the MD simulations for PsbO. Thus, the findings of the MD simulations agreed with the results of the functional studies.
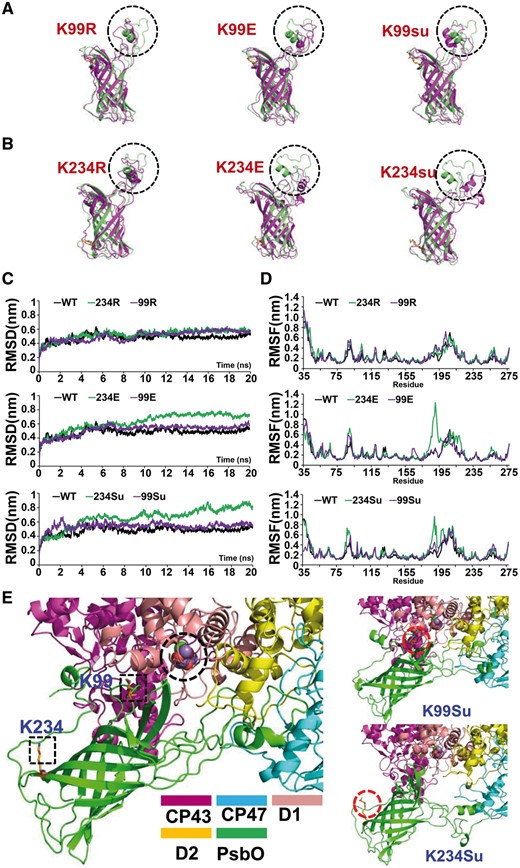
Molecular dynamics simulation of PsbO structure. (A, B) The structures of the simulations of various PsbO systems. The black dotted circles mark the head domain of PsbO. (C) Trajectories of the overall RMSDs of the various PsbO systems. (D) Residue-specific RMSF profiles of the various PsbO systems. (E) Schematic illustration of effects of lysine succinylation on PsbO. Final structures of the simulations of PsbO are shown, and the square section marks the lysine succinylation site of PsbO.
Accumulating evidence indicates that the extended head domain of PsbO is critical for correct interactions with other photosynthetic proteins, such as CP43, CP47, D1 and D2 (De Las Rivas and Barber 2004, Popelkova and Yocum 2011). As shown in Fig. 7E, the destabilization of the head domain by K234 succinylation leads to displacement of the head domain region. Thus, based on the results of MD simulations, we hypothesized that succinylation at K234 may influence the photosynthetic efficiency by partially altering the conformation of PsbO, thereby hindering the interaction between PsbO and the PSII core.
Discussion
To the best of our knowledge, this is the first systematic study of lysine succinylation in cyanobacteria. In this study, we identified 1,704 succinylation sites (class I) on 691 proteins, which are involved in a wide range of pathways and processes in various cellular compartments. A characteristic group comprised photosynthesis-related proteins. In processes such as light harvesting, PSI and PSII, and energy metabolism, we found that representative proteins from the full photosynthetic pathway harbored this PTM. Thus, understanding the physiological functions of modifications of these sites would certainly help to elucidate the regulation of the photosynthesis pathway in cyanobacteria, as well as in other photosynthetic organisms.
In the present study, a large proportion of proteins related to photosynthesis were subjected to succinylation according to MS analysis. Among them, PsbO was found to be succinylated on Lys99 and Lys234. PsbO, a 33 kDa polypeptide located on the lumen side of thylakoid membranes, is the largest extrinsic subunit of PSII. PSII is a large membrane-bound enzyme complex located in the thylakoid membranes of all photosynthetic organisms (Slowik et al. 2011), which catalyzes a series of light-induced electron transfer reactions that split water into protons and molecular oxygen. The latter maintains the oxygenic atmosphere, which is indispensable for sustaining oxygen-based life on the Earth (Slowik et al. 2011). Three inorganic cofactors (4 Mn, 1 Ca2+ and 1 Cl−) and at least three extrinsic proteins (including PsbO) bind to the PSII core proteins, where they together form a module called the oxygen-evolving complex (OEC), which is the catalytic center for water oxidation. PsbO is an intrinsically disordered protein (Lydakis-Simantiris et al. 1999), which relies on its conformational promiscuity in solution to bind to a target or an array of target proteins (Tompa 2012). PsbO provides a template for the organization of its target, the PSII reaction center (Offenbacher et al. 2013), and it is required for stabilization of the inorganic core of the OEC and to achieve the maximum rate for the water oxidation reaction (Yamamoto et al. 1998, Roose et al. 2010, Pigolev et al. 2012). In addition, PsbO plays regulatory roles in the PSII repair cycle (Lundin et al. 2007b, Bricker and Frankel 2011) as well as the dephosphorylation and turnover of the PSII reaction center D1 protein (Komenda and Barber 1995, Lundin et al. 2007a, Lundin et al. 2007b). The X-ray structure of PsbO bound to the PSII center indicates that PsbO is a protein with an elongated shape and two major domains, i.e. head and β-barrel domains (Ferreira et al. 2004). The loop in the head domain is conformationally flexible, where it participates in the interaction with PSII as well as affecting the subsequent functional folding and assembly of PsbO into PSII (Rosenberg et al. 1999, Ferreira et al. 2004, Popelkova et al. 2006, Roose et al. 2010, Popelkova and Yocum 2011, Umena et al. 2011). A previous study demonstrated that mutation of a tryptophan residue (W241F) in spinach PsbO resulted in defective binding to PSII and a reduction in the steady-state oxygen evolution activity (Wyman et al. 2008). Mutations at Arg182, Asp188, Lys190 or Arg192 also reduced binding and significantly affected the functional assembly of PsbO into PSII (Motoki et al. 2002, Popelkova et al. 2006, Popelkova et al. 2009, Roose et al. 2010). Thus, it was concluded that these conserved charged residues in the large flexible loop of PsbO play important roles in PSII binding, and that PsbO flexibility is essential for effective PSII binding and the restoration of high oxygen evolution activity (Offenbacher et al. 2013). However, the precise role of PsbO in photosynthesis remains elusive, and the regulation of PsbO is highly complex (Popelkova and Yocum 2011).
In the present study, we showed that a succinylation mimic (E) mutation of K234 negatively regulated growth, oxygen evolution and the assembly/stability of PSII complex (Fig. 5) by decreasing the photosynthetic activity of PSII (Fig. 6A–C) and increasing PSI cyclic electron transport (Fig. 6D). Further MD simulations demonstrated that succinylation of K234 could alter the conformation of the head domain of PsbO (Fig. 7B) and lead to the migration of the head domain from its docking site on the lumenal surface of PSII (Fig. 7E), which is where the correct interactions between PsbO and other photosynthetic proteins occur, such as CP43, CP47, D1 and D2 (De Las Rivas and Barber 2004, Popelkova and Yocum 2011). K234 is located on the C-terminus of the sixth β-sheet of the PsbO β-barrel domain where it is the only positively charged residue in this β-sheet (Fig. 3D). The β-sheets are the primary structural element in the major PsbO domain (β-barrel), and they are important for PsbO stability (Popelkova and Yocum 2011). Previous research has shown that C-terminal truncations of the last (eighth) β-sheet dramatically reduced or eliminated the ability of PsbO to interact with its binding sites on spinach PSII (Betts et al. 1998, Lydakis-Simantiris et al. 1999), whereas amino acid substitutions (Wyman et al. 2008, Popelkova et al. 2010) and the acetylation of Tyr242 (spinach numbering) (Zhang et al. 2005) in this region also disrupted the binding of PsbO to PSII. Analysis of the structure of PsbO showed that the sixth β-sheet is highly similar to the last (eighth) β-sheet and both β-sheets are essential for the conformational stability of PsbO (Popelkova and Yocum 2011). In addition, the sixth β-sheet is tightly connected to the head domain of PsbO. Thus, succinylation at K234 may change the secondary and tertiary structures of PsbO to perturb the loop in the head domain (Fig. 7B). Succinylation at K234 may disrupt specific intramolecular electrostatic interactions and increase the structural stability of PsbO, thereby interfering with the inherent flexibility required for PsbO binding and the functional assembly of PsbO in PSII.
The roles of lysine succinylation in the regulation of photosynthesis are largely unknown. However, their importance for the control of metabolic pathways in bacteria, yeast and humans, as well as the conservation of the enzymes involved in these pathways in photosynthetic organisms, suggest that lysine succinylation may play an important role in the control of photosynthetic pathways because photosynthesis and metabolism are physically and functionally interconnected processes that are linked to the cellular energy status (Wulff-Zottele et al. 2010, Mettler et al. 2014, Mo et al. 2015). Plants are also likely to possess unique mechanisms related to the control of metabolic and photosynthetic pathways via lysine succinylation because much of the energy status of plants is related to the chloroplast via photosynthesis. Thus, to elucidate the role of lysine succinylation in photosynthesis, it will be essential to acquire in-depth descriptions of the succinylome in photosynthetic organisms and to demonstrate the extent to which succinylation is important for the control of photosynthesis. This will involve several key approaches including: (i) determining how environmental and growth conditions can influence the level of protein succinylation; (ii) determining the effects of succinylation on the functions of proteins; and (iii) identifying the involvement of lysine succinyltransferase and desuccinylase in the control of the succinylation status of proteins involved in photosynthesis, as well as understanding their regulation. In addition, protein succinylation is only one of many types of PTM, which competes with other forms of acylation, such as acetylation, malonylation, crotonylation, propionylation and butyrylation, for the same lysine residues (Lee 2013, Hirschey and Zhao 2015). Further examples of co-operation between different PTMs have been described previously. Thus, it is expected that a complex interplay and cross-talk exists between protein succinylation and other PTMs, which involves both antagonistic and co-operative interactions that ultimately modulate protein functions and photosynthesis. Unraveling the combinatorial and hierarchical patterns of succinylation and other PTMs will be essential for understanding metabolic and photosynthetic pathways.
Overall, our results indicate that lysine succinylation is widespread in cyanobacteria and that it is probably an important mechanism that regulates photosynthesis in both plants and cyanobacteria. The underlying molecular mechanisms will have to be examined thoroughly because they might be crucial for fine tuning the photosynthetic efficiency and carbon fixation in various environmental conditions.
Materials and Methods
Cyanobacterial strains and growth conditions
The wild-type strain of Synechococcus was grown in medium A supplemented with 1 mg ml−1 NaNO3 as the nitrogen source (designated as medium A+) (Ludwig and Bryant 2011). The growth temperature was 38°C with continuous illumination at 250 μmol photons m−2 s−1 and the liquid culture was bubbled with 1% (v/v) CO2 in air. Then cells were grown under several conditions and harvested at different growth stages. A total of nine culture extracts were prepared from Synechococcus cultures grown to exponential phase (OD730nm ∼0.7–0.8) and stationary phase (OD730nm ∼5.0), and exposed to stresses. For iron, phosphate, nitrogen and A5 (trace metal elements) deficiency treatments, cells in the exponential phase of growth were harvested by centrifugation, washed twice with nitrogen deficiency medium A (lacking the specified nutrient) and immediately resuspended in medium A without the specified nutrient or other medium as previously described (Ludwig and Bryant 2012): in Fe-free medium; in phosphate-free medium A+; in medium (lacking nitrate); or in A5-free medium A+. The resuspended cultures were adjusted to an initial OD730nm of 0.4 and subsequently incubated under 38°C with continuous illumination at 250 μmol photons m−2 s−1 and 1% (v/v) CO2 in air when OD730nm reached 0.7–0.8. For HL intensity treatment, cells were grown to exponential phase and immediately illuminated at 2,000 μmol photons m−2 s−1 for 2 h. For high salt concentration treatment, cells were grown to exponential phase and immediately resuspended in medium contained additional 2.5 M NaCl for 2 h. For carbon source utilization studies, cells were resuspended in fresh medium A containing an additional carbon sources, such as 1% pyruvate, 1% glucose, 1% succinate or 1% acetate, respectively. Cultures were harvested, lysed and Western blotted with anti-succinyl/anti-acetyl lysine polyclonal rabbit antibodies.
Preparation of protein lysate, in-solution trypsin digestion and immunoaffinity enrichment
Whole-cell lysates from S. elongatus PCC 7942 (Moronta-Barrios et al. 2012), Synechocystis sp. PCC 6803 (Mo et al. 2015), Chlorella sp. NJ-18 (Lin et al. 2017), Chlamydomonas reinhardtii (Mettler et al. 2014), A. thaliana (Lundin et al. 2007a), Spinacia oleracea (Yamamoto et al. 1998) and Oryza sativa (He et al. 2016) were prepared essentially as described. Synechococcus cultures under different treatments were harvested by centrifugation at 8,000×g for 15 min at 4°C. The cells were washed twice with phosphate-buffered saline (PBS) and resuspended in lysis buffer containing 20 mM Tris–HCl (pH 7.5), 150 mM NaCl, 1% Triton X-100, 50 mM nicotinamide, pH 7.5 and 1× protease inhibitor cocktail (Thermo Fisher Scientific). The mixture was subjected to sonication (2 s on, 2 s off) for about 30 min on ice with an output of 135 W (JY92-IIN, Ningbo Scientz Biotechnology Co., Ltd.). Cellular debris was removed by centrifugation at 6,000×g for 30 min at 4°C, and protein concentrations were measured using a BCA protein assay kit (Beyotimea).
The clarified lysate was precipitated by using 10% trichloroacetic acid and 1% sodium deoxycholate, and then washed twice with ice-cold acetone. The precipitated proteins (1 mg) were redissolved in 50 mM ammonium bicarbonate, then in-solution digested by trypsin as previously described (Colak et al. 2013, Yang et al. 2015). The digested peptides were then separated into 14 fractions by high pH reverse-phase HPLC and dried by vacuum centrifuging.
Succinylated peptides were enriched using agarose-conjugated anti-succinyl lysine polyclonal rabbit antibody (PTM Biolabs Inc.) as previously described (Yang et al. 2015). Briefly, tryptic peptides redissolved in NETN buffer (100 mM NaCl, 1 mM EDTA, 50 mM Tris–HCl, 0.5% NP-40, pH 8.0) were incubated with pre-washed anti-succinyl lysine antibody-conjugated protein A agarose beads at 4°C for 6 h with gentle rotation. After washing three times with NETN buffer, twice with ETN buffer (100 mM NaCl, 1 mM EDTA and 50 mM Tris, pH 8.0) and twice with ddH2O, the bound peptides were eluted by washing three times with 1% trifluoroacetic acid. The eluates were combined and dried in a SpeedVac. The resulting succinylated peptides were loaded onto self-packed C18 STAGE tips to desalt the sample, prior to nano-HPLC-MS/MS analysis.
In-gel trypsin digestion
Protein extracts were resolved on a 12% SDS–polyacrylamide gel (1.5 mm thick, 80 mm wide, 70 mm long), stained with Coomassie blue R250, excised into different slices and diced into approximately 1 mm3 cubes for further in-gel digestion as described (Yang et al. 2013). Briefly, each section was further destained, reduced and alkylated prior to trypsin digestion (dilution 1:100, w/w). After digestion at 37°C overnight, the supernatants were transferred into a new microcentrifuge tube, and the gels were sonicated twice with extraction buffer [67% (v/v) acetonitrile containing 5% (v/v) trifluoroacetic acid]. The peptide extract and the supernatant of the gel slice were combined and then completely dried in a SpeedVac.
LC-MS/MS analysis
The enriched peptides were dissolved in the HPLC buffer A [0.1% (v/v) formic acid in water], and analyzed by online nanoflow LC-MS/MS using an easy nLC-1000 system (Thermo Fisher Scientific) connected to a Q-Exactive (Thermo Fisher Scientific) mass spectrometer. Briefly, samples were injected onto the analytical C18-nanocapillary LC column (C18 resin with 2 μm particle size, 100 Å pore diameter, 150 mm length×50 μm inner diameter, Acclaim PepMap RSLC, Thermo Scientific) and eluted at a flow rate of 300 nl min–1 with a 42 min gradient from 5% solvent B (90% acetonitrile/0.1% formic acid, v/v) to 80% solvent B. The mass spectrometer under Xcalibur 3.0 was operated in a data-dependent mode with an automatic switch between MS and MS/MS acquisition. Full MS spectra from m/z 350 to 1,600 were acquired with a resolution of 70,000 at m/z = 200 in profile mode. Lock mass at m/z 445.12003 was enabled for the full MS scan. The 15 most intense ions in each full MS spectrum were sequentially picked for MS/MS fragmentation and detected at a resolution of 17,500 in an orbitrap analyser. All the tandem mass spectra were produced by higher energy C-trap dissociation (HCD) with a normalized collision energy (NCE) of 28%. The top 15 most intense ions with charge state 2–5 were allowed for fragmentation by HCD NCE of 28% and stepped NCE of 5%. The max ion times of full MS and each MS/MS were 50 and 150 ms, respectively. The dynamic exclusion duration was set to 5 s. The intensity threshold was set to 3.0×104. The electrospray voltage applied was 2.0 kV. Automatic gain control (AGC) was used to prevent overfilling of the ion trap; 5×104 ions were accumulated for generation of MS/MS spectra.
Data analysis
All acquired raw data were processed with the MaxQuant software (version 1.3.0.5) (Cox and Mann 2008). The peak lists were searched against the Synechococcus protein database from cyanobase site (http://genome.microbedb.jp/cyanobase/GCA000019485.1/genes.faa; 3,186 sequences, released 2012) concatenated with a reverse decoy database and protein sequences of common contaminants. Two missed cleavages were allowed for trypsin. The precursor and fragment ion mass tolerances were 10 p.p.m. and 0.02 Da, respectively. Carbamidomethylation (Cys) was set as a fixed modification, whereas oxidation (Met), deamidation (Asn/Gln), succinylation (Lys) and acetylation (protein N-terminus) were set as variable modifications. Minimum peptide length was set at 6. The estimated FDR thresholds for modification site, peptide and protein were specified at maximum 1%. All succinylation sites assigned to the peptide C-terminus were removed and all MS/MS spectra for the identified peptides with succinylation modifications were manually inspected using criteria as previously reported (Chen et al. 2005, Macek et al. 2008), prior to bioinformatics analysis.
Bioinformatics analysis
The identified succinylated proteins were grouped into biological process and molecular function class based on the GO terms by Blast2GO software (Conesa and Gotz 2008). The protein subcellular localization was analyzed with the PSORTb program (Yu et al. 2010). To gain information on the over-representations, GO term and KEGG pathway enrichment were performed using the DAVID bioinformatics resources (Huang et al. 2009a, Huang et al. 2009b). In our data, the corresponding P-value < 0.05 was considered statistically significant. Functional interaction network analysis was performed using interaction data as previously reported (Yang et al. 2013). The MCODE plug-in toolkit was used to identify highly connected clusters, and the interaction network was visualized by Cytoscape (version 3.0.2) (Bader and Hogue 2003, Shannon et al. 2003). Amino acid sequence motifs were analyzed using motif-X (Schwartz and Gygi 2005) and an in-house script as previously reported (Yang et al. 2013). The secondary structure types were determined for positions with succinylated lysine using the NetSurfP tool (Petersen et al. 2009). The evolutionary conservation analysis was performed with two-directional BLASTP, and the best-scoring homologous protein was selected if the corresponding BLAST E values were <1E-5 as previously described (Liu et al. 2014, Mo et al. 2015). Eighteen archaeal, 653 bacterial and 69 eukaryotic protein databases were retrieved from Uniprot (http://www.uniprot.org/uniprot/), NCBI (http://www.ncbi.nlm.nih.gov/) and EnsEMBL (http://www.ensembl.org), respectively.
Immunoprecipitation and Western blotting
Immunoprecipitation experiments were performed using the PsbO primary antibody (Agrisera). The PsbO primary antibody was conjugated to protein G Dynabeads (Life Technologies AS) in PBS by gentle rocking for 4 h at 4ºC. The conjugated beads were washed three times with PBS and incubated with the whole-cell lysates overnight at 4ºC. The beads were then washed three times with PBS to remove the unbound proteins. Bound proteins were boiled in SDS loading buffer for 5 min, then subjected to 12% SDS–PAGE and transferred to a polyvinylidene difluoride (PVDF) membrane. For Western blotting, the membrane was blocked overnight at 4 or 37°C for 2 h in Tris-buffered saline (TBS) buffer (25 mM Tris–HCl, pH 8.0, 150 mM NaCl) containing 5% bovine serum albumin (BSA) and incubated with either the specific PsbO primary antibody or the anti-succinyl lysine antibody (PTM Biolabs, Inc.) (1:2,000, in TBS/5% BSA) overnight at 4°C. After washing three times with TBST buffer (25 mM Tris–HCl, pH 8.0, 150 mM NaCl, 0.1% Tween-20), the membrane was incubated with horseradish peroxidase-conjugated goat anti-rabbit antibody (1:5,000 dilutions) for 1 h at 37°C. The membrane was then washed with TBST buffer and visualized with enhanced chemiluminescence (ECL) immunoblotting detection reagents (Advansta Inc.). The density of each band was determined with a fluorescence scanner (ImageQuant TL, GE Healthcare). Densitometry analysis was performed via the ImageJ suite (http://rsbweb.nih.gov/ij/).
The Western blotting analyses of D1, CP43, PsaC and CpcG proteins were performed as described above using the primary antibodies purchased from Agrisera.
Molecular dynamics simulations
The initial model of the PsbO module was prepared from the available crystal structure (PDB code 3WU2) via structural modeling using the SWISS-MODEL Server (http://swissmodel.expasy.org/). All simulations were based on the initial model, and a total of 13 systems were prepared for simulation using GROMACS 4.6.5 (Van Der Spoel et al. 2005) in conjunction with the GROMOS96(54a7) force field parameters (Stocker and van Gunsteren 2000). Each system was solvated in simple point charge (SPC) water molecules (Price and Brooks 2004) in a triclinic box, with the box edges approximately 1.0 nm from any atom of the protein, and additional Na+ ions were added to neutralize the charge of each system. Energy minimization for the solvated structures was carried out by the steepest descent method until the maximum force was <100 KJ mol–1 nm–1. Next, all the simulations were performed under a constant temperature of 311 K, and the V-rescale algorithm (Bussi et al. 2007) was used with a temperature coupling time constant of 0.1 ps. The pressure was maintained at 1 bar by coupling the system to an isotropic pressure bath using a coupling constant of 2 ps. Van dar Waals and electrostatic interactions used a cut-off at 1.2 nm, and long-range electrostatic interactions were handled using the particle mesh Ewald (PME) method (Essmann et al. 1995) with a fourth-order spline interpolation and a 0.12 nm Fourier grid spacing. All bond lengths were constrained using the LINCS algorithm (Hess et al. 1997). The SETTLE algorithm was used to constrain the geometry of the water molecules (Miyamoto and Kollman 1992). Finally, each system was subjected to 20 ns of MD simulation, and the time step used in the simulation was 2 fs. All analyses were performed using the GROMACS suite of tools. The PyMOL Molecular Graphics System (version 1.7.2, http://www.pymol.org) was employed to present the structural results of this study. For the structural models of D1, D2, CP43 and CP47, each initial model was prepared from the previously determined crystal structure via the modeling server SWISS-MODEL.
Construction of the psbO interruption mutant and point mutants by site-directed mutagenesis
The Synechococcus gene interruption mutant and point mutants were constructed and confirmed by methods previously described (Chen et al. 2017). First, using Synechococcus genomic DNA as a template, the full-length psbO or synPCC7002_A1479 genes were amplified with the primers shown in Supplementary Table S6. The PCR amplicon was inserted into pMD18-T (TAKARA). Then, the plasmids containing the psbO or synPCC7002_A1479 genes (pMDpsbO, pMDA1479) were interrupted at a unique SmaI site within the psbO or synPCC7002_A1479 genes by a kanamycin resistance cassette (derived from the PRL446 plasmid), respectively. Finally, the plasmids (pMDpsbO::Kan and pMDA1479::Kan) containing the interruption were transformed into Synechococcus as previously described (Frigaard et al. 2004) and the transformants were selected on kanamycin (50 μg ml–1). The psbO and synPCC7002_A1479 genes were then inactivated by a homologous recombination strategy. The resulting mutants (ΔpsbO, and ΔA1479) were continuously subcultured until the mutants contained homoplasmic interrupted genes.
For the generation of site-directed mutants, the promoter region for the cpcAB gene was amplified from genomic DNA of Synechocystis sp. PCC 6803 and the fragment was fused to the psbO gene containing the chloramphenicol resistance cassette (Cmr) at the C-terminus, termed the PcpcAB::psbO::Cmrfragment, by fusion PCR using the primers shown in Supplementary Table S6. This fragment was cloned and subjected to site-directed mutagenesis using a Quick Change II site-directed mutagenesis kit (Agilent Technologies Inc.) according to the manufacturer’s instructions. The wild-type strain codon AAG or AAA coding for lysine was replaced by the codon CGC for arginine or GAA for glutamic acid. The wild-type and point mutated psbO gene fragments were digested by HindIII and SacI, and introduced into the unique SmaI site within the synPCC7002_A1479 gene of plasmid pMDA1479. The resulting plasmids were then transformed into the ΔA1479 mutant to generate the site-directed mutants as previously described (Frigaard et al. 2004). For the generation of the psbO-complemented strain, the psbO gene was amplified and cloned into the endogenous plasmid-based pAQ1Ex_PcpcBA expression system to generate plasmid pAQ1Ex_PcpcBA_psbO (Xu et al. 2011). The recombined plasmid was transformed into the ΔpsbO mutant to generate the complemented strain. All the transformants were selected on appropriate antibiotics as required at 50 μg ml–1 kanamycin and 25 μg ml–1 chloromycetin. The point mutations in the transformed cells were further confirmed by DNA sequencing and MS analysis.
Measurement of the photosynthetic oxygen evolution rate
Photosynthetic oxygen evolution rates of various Synechococcus strains were determined on cell cultures using an oxygen electrode system (Hansatech Instruments Ltd.) maintained at 33 or 38°C by a circulating water bath. Cells in exponential growth phase were harvested, washed once with fresh A+ medium and adjusted to an equal final cell concentration (OD730nm ∼1.0) in fresh A+ medium. The rate of oxygen evolution was measured at a saturating light intensity of 800 μE m–2 s–1 in the presence of 10 mM NaHCO3 according to a previously described method (Nomura et al. 2006a).
Isolation of crude thylakoid membranes and BN–PAGE analysis
The logarithmic phase cells (OD730nm ∼1.0) were harvested and washed twice with fresh A+ medium. The thylakoid membranes were isolated from the whole-cell extracts as previously described with minor modification (Watanabe et al. 2011, Yang et al. 2014). Briefly, the cells were resuspended in a buffer containing 50 mM HEPES-NaOH (pH 7.0), 5 mM MgCl2, 5 mM CaCl2 and 25% glycerol (v/v). The mixture was then subjected to sonication (3 s on, 3 s off) for about 60 min on ice with an output of 135 W (JY92-IIN, Ningbo Scientz Biotechnology Co., Ltd.). Cellular debris was removed by centrifugation at 1,800×g for 10 min at 4°C. The thylakoid membranes were subsequently separated by ultracentrifugation at 20,000×g for 30 min at 4°C.
BN–PAGE was performed as previously described with minor modifications (Watanabe et al. 2011, He and Mi 2016, Yamamoto et al. 2016, Bersanini et al. 2017). The thylakoid membranes were washed with washing buffer (330 mM sorbitol, 50 mM Bis-Tris–HCl, pH 7.0) and resuspended in a buffer containing 10 mM MgCl2, 20% glycerol (v/v), 0.1 U of RNase-free DNase RQ1, 25 mM Bis-Tris–HCl (pH 7.0) to a final concentration of 0.5 mg ml−1 Chl. The mixtures were incubated on ice for 10 min, and an equal volume of 4% (w/v) n-dodecyl-β-D-maltopyranoside (DM) was added. Thylakoids were then solubilized on ice for 40 min and centrifuged at 18,000×g at 4°C for 15 min. The supernatant was supplemented with 1/10 vol. of sample buffer [100 mM Bis-Tris–HCl (pH 7.0), 0.5 M ε-amino-n-caproic acid, 75% (w/v) sucrose and 5% (w/v) Serva Blue G]. Solubilized membranes were then applied to a gel with a 5–12% (w/v) gradient of acrylamide in the separation gel. A 9 μg aliquot of Chl a of samples was loaded in each lane. Electrophoresis was performed at 4°C for 5.5 h by gradually increasing the voltage from 50 to 200 V.
Measurements of fluorescence
Low temperature (77 K) fluorescence emission spectra were recorded with a single-beam fluorometer (Photon Technology International) as described (Shen and Bryant 1995). Briefly, cells in the exponential growth phase were harvested, washed once with fresh A+ medium and resuspended in fresh A+ medium. Glycerol was added to a final concentration of 60% (v/v). Cells were adjusted to a concentration of OD730 nm of approximately 0.5 and quickly frozen in liquid nitrogen. Excitation was provided at 430 nm for Chl and 600 nm for phycobilisomes, and the fluorescence emission was measured at every 1 nm between 600 and 800 nm.
Chl fluorescence was measured using the pulse-amplitude fluorimeter model Dual-PAM-100 (Heinz Walz Gmbh) as described (Genty et al. 1989, Shikanai et al. 1998, Nomura et al. 2006a). Briefly, cells in exponential growth phase were harvested, washed once with fresh A+ medium and adjusted to an equal final cell concentration (OD730nm ∼2.0) in fresh A+ medium. The dark-adapted samples were incubated for 15 min at room temperature and dispensed into a quartz glass cuvette containing a micro magnetic stirrer. The cuvette was then inserted into the PAM fluorometer and the rapid light curves (RLCs) were generated by applying stepwise-increased (logarithmic increment) actinic light intensities ranging from 0 to 850 mmol photons m–2 s–1. All data were recorded and used to generate the RLCs by Dual-PAM software. The steady-state fluorescence yields were determined by the RLCs, and each actinic light incubation lasted for 30 s before a saturation pulse was applied to determine the maximal fluorescence yields. The electron transport rates (ETRs) were calculated with the steady-state and maximal Chl fluorescence by Dual-PAM software. The transient increase in Chl fluorescence after turning off the actinic light was monitored as described (Shikanai et al. 1998).
Supplementary Data
Supplementary data are available at PCP online.
Funding
This work was supported by the National Natural Science Foundation of China [grant No. 31570829]; the National Key Research and Development Program (2017YFA0503700); the Chinese Academy of Sciences [grant QYZDY-SSW-SMC004]; and the Strategic Priority Research Program of the Chinese Academy of Sciences [grant No. XDB14030202).
Disclosures
The authors have no conflicts of interest to declare.
References
Abbreviations
- BN–PAGE
Blue Native PAGE
- ETR(II)
electron transport rate through PSII
- FDR
false discovery rate
- GO
Gene Ontology
- HL
high light
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- MD
molecular dynamics
- MS/MS
tandem mass spectrometry
- PAM
pulse amplitude modulated
- PBS
phosphate-buffered saline
- PsbO
PSII manganese-stabilizing protein
- PTM
post-translational modification
Author notes
Xin Liu and Mingkun Yang authors contributed equally to this work

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}