





Abstract
Self-incompatibility (SI) is an important plant reproduction mechanism that facilitates the maintenance of genetic diversity within species. Three plant families, the Solanaceae, Rosaceae and Plantaginaceae, share an S-RNase-based gametophytic SI (GSI) system that involves a single S-RNase as the pistil S determinant and several F-box genes as pollen S determinants that act via non-self-recognition. Previous evidence has suggested a specific self-recognition mechanism in Prunus (Rosaceae), raising questions about the generality of the S-RNase-based GSI system. We investigated the evolution of the pollen S determinant by comparing the sequences of the Prunus S haplotype-specific F-box gene ( SFB ) with those of its orthologs in other angiosperm genomes. Our results indicate that the Prunus SFB does not cluster with the pollen S of other plants and diverged early after the establishment of the Eudicots. Our results further indicate multiple F-box gene duplication events, specifically in the Rosaceae family, and suggest that the Prunus SFB gene originated in a recent Prunus -specific gene duplication event. Transcriptomic and evolutionary analyses of the Prunus S paralogs are consistent with the establishment of a Prunus -specific SI system, and the possibility of subfunctionalization differentiating the newly generated SFB from the original pollen S determinant.
Introduction
Self-incompatibility (SI) is a genetically controlled reproductive mechanism defined as ‘the inability of a fertile hermaphrodite seed-plant to produce zygotes after self-pollination’ ( de Nettancourt 2001 ). SI contributes to the diversification and differentiation of species by maintaining genetic diversity ( de Nettancourt 2001 ). SI is widely distributed within angiosperms and is present in approximately 60% of angiosperm species, in at least 19 orders, 71 families and 250 genera ( Allen and Hiscock 2008 ). Three plant families, the Solanaceae, the Rosaceae and the Plantaginaceae, use the S-RNase-based gametophytic SI (GSI) system, characterized by a pollen–pistil recognition mechanism that is based on an S-RNase as the single pistil S determinant, and one or multiple F-box proteins as pollen S determinants ( McCubbin and Kao 2000 ). The pollen S determinant is called the S locus F-box ( SLF ) in the Solanaceae and the Plantaginaceae, S locus F-box brothers ( SFBB ) in the subtribe Malinae (Rosaceae) and S haplotype-specific F-box ( SFB ) in the genus Prunus (Rosaceae) ( Sassa et al. 2010 , Tao and Iezzoni 2010 , Meng et al. 2011 , Matsumoto and Tao 2016 ). Although it is unclear whether these pollen S F-box genes are monophyletic ( Wang et al. 2004 , Matsumoto et al. 2008 ), phylogenetic analyses of S-RNase and related sequences have shown that the S-RNase-based GSI system has a single evolutionary origin, that dates back to approximately 120 million years ago ( Igic and Kohn 2001 , Steinbachs and Holsinger 2002 , Vieira et al. 2008 , Nowak et al. 2011 ).
Recognition mechanisms between pistil S and pollen S determinants have been extensively studied. It is generally accepted that the S-RNase exerts its cytotoxicity by arresting pollen tube growth, if it remains active in pollen cytoplasm. Although the mechanism whereby S-RNase exerts its cytotoxicity remains to be clarified, a recent study indicated the possibility that microtubules in the pollen tube play a crucial role in S-RNase transport and initiation of the SI reaction in apple ( Meng et al. 2014 ). The pollen S F-box proteins are thought to target these cytotoxic S-RNases for degradation via the ubiquitin–proteasome and via pathways shared between the Solanaceae, the Plantaginaceae and the subtribe Malinae. In Petunia within the Solanaceae, multiple SLF genes, clustered as tandem repeats generated by lineage-specific duplications, can collaboratively recognize and target non-self S-RNases for degradation, but SLF proteins from each haplotype are unable to target their own S-RNases for degradation ( Kubo et al. 2010 , Williams et al. 2014 , Kubo et al. 2015 ). It has been suggested ( Sassa et al. 2007 , Kakui et al. 2011 , Okada et al. 2011 ) that similar pathways involving the collaborative recognition and degradation of non-self S-RNases by multiple F-box proteins are also present in the subtribe Malinae.
In contrast to the models presented above, SFB , the pollen S F-box protein from Prunus , is thought to function fundamentally differently, via self- instead of non-self-recognition, possibly via the following model: a putative general inhibitor (GI), functionally homologous to the SLF/SFBB factors and expressed in the pollen, targets all S-RNases for degradation or inactivation. SFB encodes a new function, not present in the other systems, and interacts specifically with self-S-RNases to protect them from inactivation and allow them to exert their function, resulting in self-cytotoxicity ( Ushijima et al. 2004 , Tao and Iezzoni 2010 , Matsumoto and Tao 2016 ). This hypothesis is consistent with our current observations of the Prunus SI system. For example, in several instances, the loss of the Prunus SFB function resulted in self-compatibility ( Ushijima et al. 2003 , Yamane et al. 2003 , Sonneveld et al. 2005 , Hauck et al. 2006 , Tsukamoto et al. 2006 , Tao et al. 2007 , Tao and Matsumoto 2012 ). In contrast, loss of function of the SFBB / SLF genes would be expected to result in loss of both self- and non-self-compatibility resulting from the lost ability to degrade the S-RNases. Consistently, loss of function of one of the SFBB genes in Pyrus pyrifolia (subtribe Malinae) was not shown to result in self-compatibility but rather in a change in the specificity of non-self-S-RNase recognition ( Kakui et al. 2011 ). This Prunus -specific SI mechanism may reflect the different lineage of the Prunus SFB from the lineage to which the SLF/SFBB genes belong ( Ushijima et al. 2003 , Wang et al. 2004 , Matsumoto et al. 2008 , Aguiar et al. 2015 ). However, evolutionary paths for the establishment of the current mechanism and the function of the Prunus SFB remain to be elucidated.
Recent studies of the evolution of plant F-box genes suggest that they have experienced frequent gene duplications and rapid diversification ( Gagne et al. 2002 , Jain et al. 2007 , Yang et al. 2008 ), even within a restricted lineage ( Xu et al. 2009 ). This evolutionary pattern is suggested to have played an important role in the acquisition of functional diversification in the superfamily of F-box genes ( Xu et al. 2009 ). The fate of duplicated genes is expected to fit one or more of the following theoretical evolutionary models: pseudogenization (loss of function) ( Moore and Purugganan, 2005 ), subfunctionalization (partitioning of the original function between the duplicated genes) ( Cusack and Wolfe 2007 ), neofunctionalization (functional diversification) ( Blanc and Wolfe 2004 ) and/or ‘escape from adaptive conflict’ (EAC) ( Conant and Wolfe 2008 ). Genes undergoing pseudogenization, neofunctionalization and EAC are expected to experience neutral and/or positive selection [non-synonymous (Ka)/synonymous (or silent; Ks) ≥1]; whereas those under subfunctionalization are expected to experience purifying selection (Ka/Ks < 1) ( Flagel and Wendel 2009 , Roulin et al. 2013 ).
In this study, we attempted to retrace the functional history of the S locus-linked F-box genes by tracking their evolutionary and duplication patterns in the genome. Our results suggest the existence of Prunus -specific duplications (PSDs) and evolution of the S locus-linked F-box genes. We discuss the implications of these findings with respect to our understanding of the functional mechanism of the Prunus -specific GSI.
Results
Duplication and evolutionary pattern of the Prunus SFB/SLFL -like F-box genes
Gene collection and phylogenetic relationship. Prunus species carry two kinds of F-box genes at and close to the S locus: a single SFB gene and at least three S locus F-box genes with low allelic sequence polymorphism, the SLF-like genes ( SLFL1–SLFL3 ) ( Ushijima et al. 2003 , Entani et al. 2003 , Matsumoto et al. 2008 ). The function of the SLFL genes remains to be determined, although they show high sequence similarity to SLF/SFBB genes ( Ushijima et al. 2004 , Matsumoto et al. 2012 ). To obtain a broad phylogenetic context for the F-box genes across angiosperms, comprehensive searches for genes containing an F-box motif and showing significant homology to the Prunus SFB / SLFL genes were performed by BLAST and Gene Ontology (GO) analysis. This searching was performed by scanning Pfam databases ( Finn et al. 2014 ) for the F-box signature and its associated domains, in three genomes from the Rosaceae: peach ( Prunus persica , Prunoideae), diploid wild strawberry ( Fragaria vesca , Rosoideae) and apple ( Malus × domestica, Malinae), as well as 28 other angiosperm species. Along with the F-box genes previously identified as potential pollen S determinants from the Solanaceae, the Plantaginaceae and the subtribe Malinae, a total of 408 genes (all with one gene per locus) were identified ( Supplementary Table S1 ). These included one Arabidopsis gene (AT3G06240) located in one of the major F-box clades, and the FBA family, which has been reported to be orthologous to S locus-linked F-box family genes ( Xu et al. 2009 ).
Bayesian Markov chain Monte Carlo (MCMC) phylogenetic analyses of SLF genes from Petunia and Antirrhinum , SFBB genes from the subtribe Malinae and 29 SFB / SLFL -like F-box genes identified from the peach genome (all with one gene per locus) revealed three main clades: clades S, A and B, each with significant support ( Fig. 1 ), and strongly supporting monophyly (clade S, Bayesian posterior probability > 0.95) for SLF , SFBB , the three Prunus SLFL genes and five other peach SLFL homologs (ppa023668m, ppa024694m, ppa019333m, ppa016207m and ppa016317m). The Prunus pollen SFB gene did not cluster closely with the SLF / SFBB / SLFL clade, but was nested in clade A, suggesting that it was derived from another F-box family ( Fig. 1 ; Supplementary Fig. S1 ). Similar results were obtained after phylogenetic analysis including all sequences obtained from the 31 angiosperm species, suggesting that the divergence between clades S and A occurred early in the diversification of eudicots ( Fig. 2 A shows the tree topology; Supplementary Fig. S2 focuses on the divergence of clades S and A). Our results suggest that a single SFB / SLFL -like F-box gene was present in the most recent common ancestor (MRCA) of angiosperms (monocots and eudicots), according to the Angiosperm Phylogeny Group III (APGIII) classification ( Bremer et al. 2009 ). In contrast, our results suggest the presence of some SFB / SLFL -like F-box genes in the MRCA of eudicots, one of which corresponds to the divergence of clade S. Although the tree topology ( Supplementary Fig. S2A ) did not provide significant support for the definition of some clades, our results suggest that SFB and SLF / SFBB / SLFL were derived from different genes already present in the MRCA of the eudicots [ Fig. 2 A; Supplementary Fig. S2B : maximum likelihood (ML) analysis bootstrap > 77% for clades S and A]. Furthermore, in the MRCA of the eudicots, we could define only one crown node for clade S. Overall, although homologs from some plant species are missing, the divergence pattern of the clade S genes was consistent with the APGIII classification ( Supplementary Fig. S2C ). Taken together, these results suggest that the functions of SLF , SFBB and SLFL in the SI system are derived from a common original function.
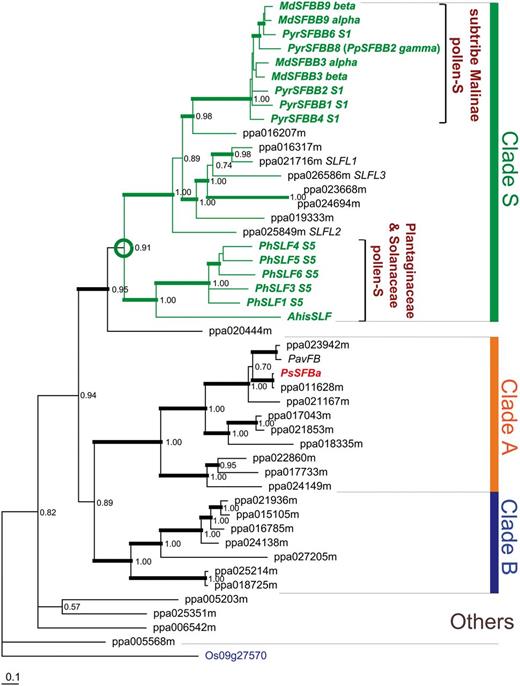
Phylogenetic tree of SFB / SLFL -like F-box genes from the Prunus persica genome. Bayesian MCMC phylogenetic tree with a GTR + I + G + SS model (see the Materials amd Methods) for SFB/SLFL -like F-box genes from P. persica (prefixes of ‘ppa’) along with pollen S determinants from Antirrhinum , Petunia and the subtribe Malinae as well as a SFB/SLFL -like F-box gene from Oryza sativa (LOC_Os09g27570) used as an outgroup gene (gene name in blue). Maximum likelihood (ML) analysis generated a tree with almost the same topology ( Supplementary Fig. S2 ). The pollen S determinants from Antirrhinum , Petunia and the subtribe Malinae and the clade containing them (clade S) are highlighted in green. SFB , the Prunus pollen S , is colored in red. Three large clades (A, B and S) are defined by this topology. The position of the original pollen S in the putative MRCA of the species with S-RNase-based GSI (the Solanaceae, the Plantaginaceae and the Rosaceae) is shown with a green circle. For the disrupted SFB gene, ppa011628m* ( SFB 2 ; AB252416) ( Tao et al. 2007 ) and the truncated ppa023942m from the P. persica genome database, intact alleles from P. salicina ( SFB a ) and P. avium ( PavFB ; JQ322648) were used. Thick branches indicate clades with strong statistical support (Bayesian posterior probability > 0.95).

Phylogenetic trees of SFB / SLFL -like F-box genes from angiosperm genomes. (A) A conceptual evolutionary topology of SFB / SLFL -like F-box genes from 31 angiosperm genomes, along with two single orthologs from Physcomitrella patens and Selaginella moellendorffii , used as outgroups, based on protein sequences and NJ analyses. The pollen S determinants (SFBBs) from Antirrhinum , Petunia (SLFs) and the subtribe Malinae are shown in purple, pink and light blue squares, respectively, while the Prunus pollen S , SFB, is indicated by a thick red triangle. The clade containing the pollen S determinants from Antirrhinum , Petunia and the subtribe Malinae is colored in green. The clade that contains RSD is colored in orange. In this clade, the Prunus -specific duplication (PSD) branches are shown in red. Here, clades other than clades A, B and S ( Fig. 1 ) were temporarily defined as O, and O-2. The putative genes in the MRCA of the angiosperms, the eudicots and the species with S-RNase-based GSI (the Solanaceae, the Plantaginaceae and the Rosaceae) are shown in blue, red and green circles, respectively. The putative genes in the MRCA of the eudicot were defined as the roots of the subclades including both asterid and rosid species. (B) After reconstruction of the tree topology with all genes, Bayesian MCMC phylogenetic analysis was used to re-examine the topology of Rosaceae-specific subclades α and ε using only the components present in each subclade. The closest orthologs in the Cucumis genome were used as outgroups (in blue). The SFBB branches and the PSD branches are shown in green with a single asterisk and in red with a double asterisk, respectively. The putative ancestors of paralogs from the Rosaceae are defined as crown nodes from branches with posterior probability of >0.95 that include at least two species from Fragaria , Malus and Prunus , and are indicated by orange circles. The truncated ppa011628m and ppa023942m sequences were replaced with intact alleles from P. salicina ( SFB a ) and P. avium ( PavFB ). ‘ppa’, ‘MDP’ and ‘mrna’ represent P. persica , Malus × domestica and Fragaria vesca , respectively. Thick branches indicate clades with strong statistical support (Bayesian posterior probability > 0.95).
Our analysis also suggested drastic duplications of the F-box genes in the Rosaceae, in clades S and A ( Fig. 2 B). We defined two subclades, α and ε, corresponding to subclades S and A, respectively ( Fig. 2 B). Note that, although some Fragaria species are thought to use an SI mechanism based on RNase and F-box genes (Bošković et al. 2010), little is known about the underlying loci. Furthermore, the Fragaria reference genomic sequence we used was obtained from the self-compatible F. vesca. The topology of subclade α, which includes the SLFL / SFBB genes , is statistically well supported and suggests that the SFBB genes in the subtribe Malinae were established by lineage-specific gene duplication and that they do not share orthologs in Prunus and Fragaria ( Fig. 2 B). Such lineage-specific gene duplications were also observed for other genes in the Rosaceae family in subclades α and ε ( Fig. 2 B), most of which are organized in tandem in the Rosaceae genome. Subclade ε, which includes the Prunus SFB and encompasses most of clade A, contained multiple paralogs in the Rosaceae. This finding suggests multiple Rosaceae-specific duplication (RSD) events prior to the diversification of Fragaria , Malus and Prunus ( Fig. 2 B; the branches for RSD are shown in orange in Fig. 2 A). It further indicates that SFB was derived from PSD events ( Fig. 2 B; red branches in Fig. 2 A).
Genomic organization and synteny. Analysis of the genomic organization of the F-box genes also indicated their evolutionary history. Prunus SFB / SLFL -like genes often occurred in tandem, whereas interchromosomal duplication events were rare ( Supplementary Fig. S3 ). Previous comprehensive exploitation of F-box genes, in Arabidopsis and Oryza genomes, suggested that 36% and 43%, respectively, of this gene family had been generated from tandem duplications ( Yang et al. 2008 ). This characteristics is thought to be conserved widely in plant genomes ( Gagne et al. 2002 , Jain et al. 2007 , Yang et al. 2008 , Xu et al. 2009 ), which was consistent with our results. Consistently, most F-box genes in clade S, including the SLFL genes ( Figs. 1 , 2 , shown in green), were located in tandem at the end of chromosome 6 (linkage group 6; LG6) in an approximatley 193 kb region of the peach genome that also contains the S locus ( Supplementary Fig. S3 ). However, the F-box genes from the RSD clade ( Fig. 2 , shown in orange), including those in the PSD clade ( Fig. 2 , shown in red), are also organized in tandem but located on chromosome 3 (LG3), except for ppa018335m (LG1), ppa024149m (LG2) and the SFB gene at the S locus on LG6 ( Supplementary Fig. S3 ). In the PSD clade, the SFB gene showed significant sequence conservation with ppa021167m in the approximately 350 bp region upstream of the start codons ( Supplementary Fig. S4 ), whereas we could not detect any significant colinearity in the surrounding regions of SFB and SLFL genes ( Supplementary Fig. S4D ).
Estimated relative time of divergence . We calculated the pairwise nucleotide diversity for the 4-fold synonymous third-codon transversion position (4DTv) between the SFB / SLFL -like F-box genes to estimate the times of their divergence from one another. The average pairwise 4DTv values between the SLFL genes from Prunus and the SFBB genes from the subtribe Malinae or between the SLFL and SLF genes from Petunia were significantly lower than those between the SLFL genes and the SFB from Prunus ( Fig. 3 A). In fact, the 4DTv values between the SLFL genes and the Prunus SFB genes were very similar to the 4DTv values between SLFL genes and the single closest F-box gene from monocots, which was used as the outgroup gene in the phylogenetic analysis. These results are consistent with the topology we obtained ( Fig. 2 A) and suggested that the SFB (clade A) and SLFL (clade S) genes diverged shortly after the establishment of the eudicots. The average 4DTv values between the SFBB genes in the subtribe Malinae and any SLFL genes in Prunus are higher than those among the closest gene sets in Prunus and Malus ( Fig. 3 A). This finding indicates that the SFBB and SLFL genes diverged from each other before the divergence of Prunus and Malus. Although tandem duplication of the SLFL and SFBB genes ( Sassa et al. 2007 , Kakui et al. 2011 , Okada et al. 2011 ) appears to have similar origins, pairwise 4DTv values between them suggest that the tandem duplication that gave rise to the Prunus SLFL genes occurred earlier than those that gave rise to the SFBB genes ( Fig. 3 A, B). Furthermore, most of the pairwise 4DTv values between the SLFL genes were significantly higher than those between the closest gene sets in Prunus and the other two rosaceous species ( Fig. 3 B), indicating that the tandem duplication that gave rise to the SLFL genes occurred before the diversification of the three rosaceous species. Our results suggest that the tandemly repeated SFBB genes are lineage specific and were established after the divergence of Prunus and the subtribe Malinae. The pairwise 4DTv values in clade A indicated that most of the gene duplication events in this clade occurred after the divergence between the Rosales and Cucurbitales and that the gene duplication events that generated SFB in the PSD clade post-dated the divergence of Prunus from the other rosaceous species ( Fig. 3 C; the asterisk indicates the putative timing of the duplication event that generated SFB ).
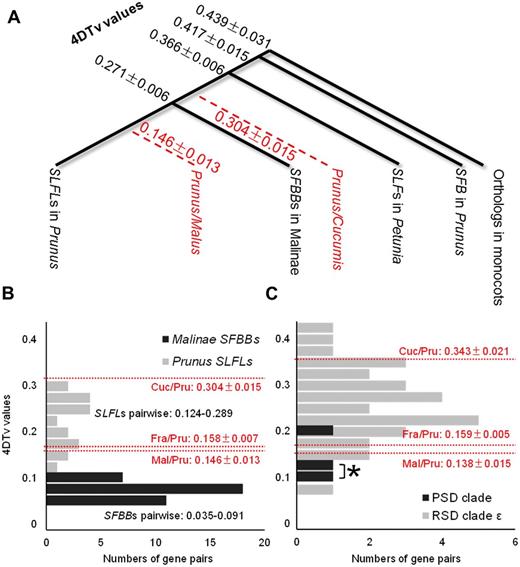
Estimation of time of gene divergence. (A) 4DTv values for the eight Prunus SLFL genes located in clade S ( Fig. 1 ). The average values of the 4DTv distance between two gene groups are shown in black. The values for pairs of species are shown in red and are calculated using the average values of the closest gene sets between the two species (or genera) across all SFB / SLFL -like genes. All 4DTv values are given with the SE. (B) Pairwise 4DTv values between tandemly duplicated genes in SFBB and SLFL clusters. Prunus SLFL genes are represented by six genes located in or flanking the Prunus S locus ( Supplementary Fig. S3 ), except for SFB. The distances between two species in this gene family are indicated by dotted red lines. SEs are indicated. (C) Pairwise 4DTv values for genes within RSD (clade ε) and the subsequent PSD, corresponding to the orange and red branches in Fig. 2 , respectively. 4DTv values for pairs of two species in this gene family are indicated by dotted red lines. *Pairwise values for SFB and the other two genes in the PSD clade. SEs are indicated.
Taken together, our results support a model for the establishment of a Prunus -specific SI system using SFB , as illustrated in Fig. 4 . In this model, the original pollen S clade and clade A, which includes the Prunus pollen S ( SFB ), diverged early in the history of the eudicots. The pollen S function may have been maintained since the divergence between the SLF genes and SFBB / SLFL gene ancestors, until the divergence between the SFBB genes and their orthologs in Prunus. The SFBB genes were generated by lineage-specific duplications after the divergence between the SFBB and SLFL genes. SFB , in contrast, may have been generated by a Prunus -specific F-box gene duplication event on the ancestral LG3. A later interchromosomal duplication or a transposition event could have resulted in the relocation of SFB to the distal part of LG6 with the SLFL genes and the S-RNase ( Fig. 4 ).
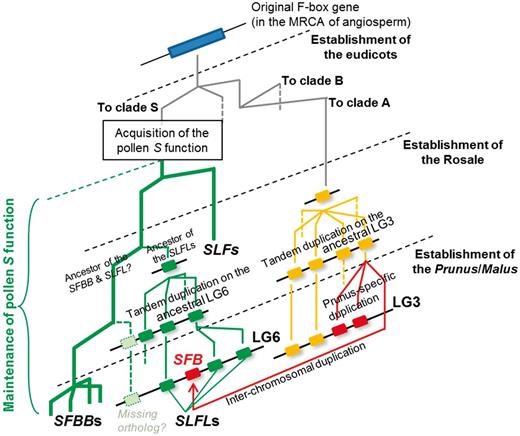
Model for the establishment and organization of the SFB / SLFL -like genes in each genus or family. After the acquisition of the pollen S function, the common function of the pollen S genes must have been maintained at least until the establishment of the original pollen S in the MRCA of Prunus and the subtribe Malinae, unless the mechanism underlying the single S-RNase GSI system is different within the species shown here, or the evolution of the pollen S gene is reversible. Maintenance of the pollen S function is shown by thick green branches in the SLF / SFBB / SLFL clade (clade S). Because pollen S factors must be present in the S haploblock on LG6, where the single S-RNase gene is located, the ancestral SFB could not have been a pollen S factor until the Prunus -specific interchromosomal gene duplication event took place, after the establishment of Prunus .
Expression of SFB/SLFL-like F-box and SI-associated genes in Prunus. Mature pollen grains from three cultivars in two Prunus species, P. avium and P. mume , were subjected to mRNA-Seq analyses. In total, we detected 8,780 of 28,689 mRNA transcript sequences from the P. persica genome [from the Genome Database for Rosaceae (GDR) Prunus_persica_v1.0] with significant expression in pollen of one or more of the three cultivars [>2.0 reads per kilobase of exon per million mapped reads (RPKM)]. We identified genes showing significantly different expression levels among the pollen samples using DESeq, and found that, among the SFB / SLFL -like F-box genes, only ppa026586m [(SLFL3, false discovery rate (FDR) = 0.0002] and ppa017043m (FDR = 0.0024) showed significant differences in pollen expression between P. mume and P. avium. Overall, the expression patterns of SFB / SLFL -like genes were associated with the phylogenetic clades to which they belonged ( Fig. 5 A). Genes in clade S generally showed high expression levels in pollen grains, although there was considerable variation between genes. For example, two of the clade S genes, ppa016207m and ppa016317m, showed no significant expression in pollen. Three genes in the PSD clade possibly generated by PSD events were expressed at similar levels. Most of the genes from clades other than the S and PSD clades showed very low expression levels in pollen. Approximately 90 F-box genes with an F-box GO annotation (in Phytozome v9.1; http://www.phytozome.net/ ) were significantly expressed (RPKM > 2.0) in pollen grains ( Supplementary Fig. S5 ). We performed a more detailed expression analyses of the nine SFB / SLFL -like genes that showed substantial expression in pollen grains ( Fig. 5 A) by evaluating the expression levels in various plant organs of P. avium (cv. Satonishiki) and P. mume (cv. Nanko). All showed the strongest expression in pollen grains ( Fig. 5 B).
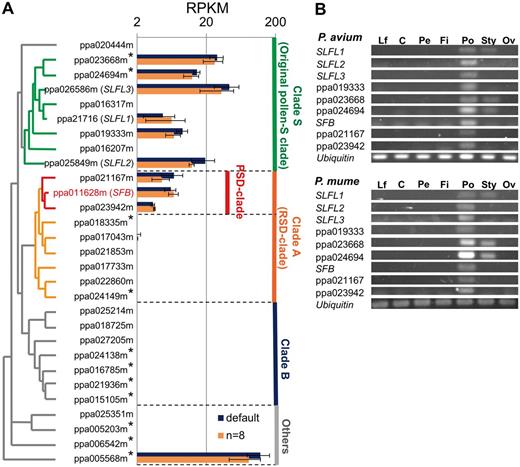
Expression profiles of SFB/SLFL -like genes in Prunus. (A) Comparison of gene expression levels of SFB / SLFL -like genes in Prunus pollen by Illumina sequencing. Expression analysis was performed with tissue from P. avium (cv. Satonishiki) and two P. mume cultivars (Nanko and Kairyo-Uchida-Ume). Average expression levels across the three Prunus accessions are expressed as RPKM with the SE. Read mapping was performed twice, using the default parameters or allowing 8 bp of mismatches per read ( n = 8) (see the Materials and Methods). RPKM values above the expression threshold of 2 are indicated. Genes not located on either LG3 or LG6 are marked by asterisks. (B) PCR analyses of the expression pattern of the nine SFB / SLFL -genes that are located in clades S or A and that showed substantial expression in pollen, using the expression of a ubiquitin gene (ppa005507m) as the reference. Expression was assessed in leaves (Lf), calyx (C), petals (Pe), filaments (Fi), pollen grains (Po), style (Sty) and ovaries (Ov). Although significant expression of a few genes was detected in styles, P. avium and P. mume showed mostly pollen-specific (or pollen-predominant) expression of all genes assayed.
Selective pressure on the evolution of the SFB/SLFL genes
Based on the results of our evolutionary and expression analyses, we tested four genes located in or close to the S locus and expressed in pollen grains, SLFL1–SLFL3 and SFB , for signs of selective pressure. Codon-based models of sequence evolution showed that ratios of Ka (nonsynonymous substitutions) to Ks (synonymous substitutions) substitutions in the three Prunus SLFL genes (Ks = 0.036–0.178, Ka = 0.010–0.061, Ka/Ks = 0.223–0.343) were lower than those of the Prunus S-RNase (Ks = 0.252, Ka = 0.144, Ka/Ks = 0.571). Similar results were found for pollen S from Petunia (Ka/Ks = 0.312 ± 0.030 on average for the six SLF genes and 0.485 for the S-RNase ) and Pyrus (Ka/Ks = 0.335 ± 0.069 on average for the five full-length SFBB genes shown in Fig. 1 , and 0.878 for the S-RNase ). In contrast, in Prunus , the pollen SFB gene showed a Ka/Ks value (0.438) closer to that of the Prunus S-RNase genes (Ka/Ks = 0.571), as discussed previously ( Newbigin et al. 2008 ). Next, we used a sliding window analysis to assess the trend of selective pressure along the lengths of the coding regions. No common Ka/Ks transition pattern could be detected in the SLFL / SFBB / SLF genes ( Fig. 6 ). In contrast, consistent differences between SLFL / SFBB / SLF and the Prunus SFB gene were observed in Ka/Ks values for the C-terminal region, especially for the 930–1,050 bp region (Ka/Ks < 0.5 in SLFL / SFBB / SLF genes), consistent with purifying selection in SLFL / SFBB / SLF in this region. Importantly, the C-terminal region of the SFB gene, which corresponds to the hypervariable regions HVa, and HVb ( Ikeda et al. 2004 ), showed significantly higher (0.9–1.5) Ka/Ks values than the rest of the sequence, suggesting that positive selection affected this region ( Fig. 6 ; Supplementary Figs. S6, S7 ).
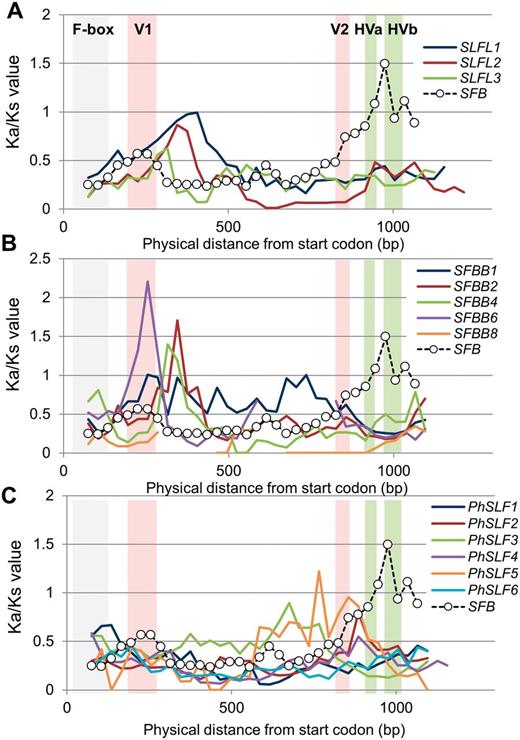
Selective pressure measurements (Ka/Ks) of SFB / SLFL -like genes in Prunus , Pyrus and Petunia. Window-average Ka/Ks values for 150 bp sliding windows with a 30 bp step size. Regions with no significant Ks are not plotted. The F-box region, two variable regions (V1 and V2) and two hypervariable regions (HVa and HVb) of the SFB gene ( Ikeda et al. 2004 ) are indicated by gray, red and green bands, respectively. SFB data are represented by a dotted line and white circle for each plot. Mean pairwise Ka/Ks values between alleles from (A) Prunus SFB and three SLFL genes located on the S haploblock in Prunus , (B) Prunus SFB and five full-length SFBB genes from Pyrus , (C) Prunus SFB and six full-length SLF genes from Petunia .
Next, we focused on PSD that generated SFB , ppa021167m and ppa023942m. Putative ancestral sequences were reconstructed ( Supplementary Text S1 ) as indicated in the Materials and Methods. The full-length average Ka/Ks values showed no clear differences among the branches ( Fig. 7 A, Ka/Ks = 0.382–0.596). However, sliding-window analysis uncovered distinct segmental Ka/Ks transition patterns in the path to the establishment of the SFB alleles from the ancestral gene. From the Rosaceae SFB origin (RSO, black circle in Fig. 7 A) to the inferred sequence of the original Prunus SFB (SFB-ori, red circle in Fig. 7 A), the C-terminal region showed significantly low (<0.3) Ka/Ks values, indicating strong purifying selection on this region until the establishment of the SFB ( Fig. 7 B). In contrast, the Ka/Ks values in this region increased abruptly (Ka/Ks > 1.0) after the divergence of SFB from the other members of its lineage, suggesting a release from purifying selective pressure ( Fig. 7 B). Ka/Ks values between PSD and the two SFB sisters (ppa021167m and ppa023942m), appeared to be low throughout the gene and showed similar transition patterns, indicating weak purifying selection in the coding region for the C-terminal region (Ka/Ks = approximately 0.3–0.6) ( Fig. 7 C). To assess this inference further, we applied a codon-based method using PAML and detected signs of significant positive selection specific to the transition from the PSD events to the SFB alleles ( P < 1.5e −11 ), especially in the C-terminal region (313 and 375 amino acid residues, posterior probability > 0.999 for Ka/Ks > 1). Collectively, our results suggest that SFB experienced positive selection specifically on the C-terminal region, which corresponds to the hypervariable regions (HVa and HVb). This trend is not visible in the paths of establishment of the original SFB or that of the other two sister genes generated by the PSD event.

Variation in selective pressure during the establishment of SFB. (A) Ka/Ks values for the PSD clade genes are shown on each branch of the evolutionary tree. The topology was obtained from ML analysis. Ka/Ks values are shown for the gene diversification path from the Rosaceae SFB origin (RSO, black outlined circle) to PSD (blue outlined circle), and to three major branches for the establishment of the original SFB (SFB-ori, red outlined circle) and two other SFB -like F-box genes (ppa023942m and ppa021167m) from the PSD. Ka/Ks values corresponding to the establishment of the present SFB alleles are given as the average of pairwise Ka/Ks values of SFB alleles (0.438 ± 0.013) and as the average of the Ka/Ks values from the original SFB (single asterisk; 0.498 ± 0.042). **RSO is a part of RSD in clade A. ***PSD is the same as that defined as described above ( Fig. 2 ). (B) Ka/Ks values are shown as window-average values in 150 bp windows with a 30 bp step size. Ka/Ks values from SFB-ori to the present SFB alleles, from the PSD to SFB-ori, from RSO to PSD and from RSO to SFB-ori are shown in thick blue, red, green and light blue, respectively, with transition of pairwise Ka/Ks values in the current SFB alleles. They correspond to branches of the same color in (A). After the establishment of the original SFB, the C-terminal regions corresponding to the HVa and HVb region were released from strong purifying selection. (C) Comparison of Ka/Ks window-average values from PSD to ppa023942m, ppa021167m and the present SFB allele is shown in orange, purple and red, corresponding to the branches with the same colors in (A), except for the SFB alleles. Here, we used PavFB sequences for ppa023942m. Only the path of gene establishment to the current SFB alleles shows relaxation of purifying selection on the HVa and HVb regions, after the PSD.
Discussion
It has been hypothesized that the Prunus SFB gene is phylogenetically distant from the pollen S determinants of other plant species with S-RNase-based SI systems ( Ushijima et al. 2004 , Wang et al. 2004 , Matsumoto et al. 2008 , Aguiar et al. 2015 ). Our genome-wide analysis of the SFB / SLFL -like F-box genes supports this hypothesis and provides novel clues to the evolution of SFB in the Prunus genome. We propose that the divergence between the ancestors of the Prunus pollen SFB and the other pollen S F-box genes occurred early in the establishment of eudicots and that SFB was generated from PSD. These findings suggest that the Prunus species came to use the SFB gene as a pollen S factor after the Prunus -specific F-box duplications, which probably occurred around the time of the Prunus divergence from its common ancestor with Malus or Fragaria. This inference may raise a question to be answered: ‘why did the Prunus SLFL genes, which belong to clade S along with pollen S factors from Petunia and Malinae, not retain the pollen S determinant function, even though they are still physically close to the S-RNase ?’. A clue to the answer might be obtained from gene expression analysis and selective pressures on SLFL genes. Given that SLFL genes are expressed mainly in the pollen ( Fig. 5 ) and they retain low Ka/Ks values (or purifying selection) over their gene sequences ( Fig. 6 ), they may still participate in the SI reaction. Matsumoto et al. (2012) showed that SFB, FB (represented by ppa023942m in this study) and SLFLs interact with a Skp1-like1 homolog in P. avium ( PavSSK1 ) that is proposed to be a component of the SCF complex involved in the polyubiquitination of proteins targeted for degradation. It is thus possible that the Prunus SLFLs are still involved in the degradation of S-RNase proteins in a process similar to that by which SLF / SFBB genes are involved in the degradation of non-self-S-RNase ( Kubo et al. 2010 , Kakui et al. 2011 , Williams et al. 2014 , Kubo et al. 2015 ). In this case, we could define the SLFL genes as strong candidate for the function of GIs that putatively targets all S-RNase for degradation ( Tao and Iezzoni 2012 , Matsumoto and Tao 2016 ).
This evolutionary scenario would suggest the possibility that SFB has taken over the function of discrimination between self- and non-self-S-RNases from the original pollen S factors, which are nested within the S clade S. Although ‘self-recognition’ and ‘non-self-recognition’ are complementary and indicate the same function per se, if we consider protein–protein interactions at the molecular level, they are distinct. Gene duplication often results in relaxed purifying selection on an ancestral gene. In the Prunus SI system, the original ancestral Prunus pollen S must have carried the function to recognize non-self-S-RNases for degradation, which is the function collaboratively carried out by the current pollen S proteins of Petunia. After the establishment of SFB , genes in the SLF / SFBB / SLFL clade (clade S) in Prunus might have escaped from purifying selection on the function to distinguish self- and non-self-S-RNases. This could have resulted in the acquisition of the GI function, i.e. to degrade all S-RNases ( Tao and Iezzoni 2012 , Matsumoto and Tao 2016 ). Instead, the SFB gene, which was newly duplicated in the Prunus S locus, currently acts for self-recognition or discrimination of self- and non-self-S-RNase ( Fig. 8 ). This hypothesis, however, does not fully explain the evolutionary path leading to the current SI system in Prunus and the switch from a system based on ‘non-self-recognition’ in Petunia and the subtribe Malinae to one based on ‘self-recognition’ for the Prunus SFB. Evidence that SFB experienced positive selective pressure in the C-terminal region after PSD may be consistent with the neofunctionalization or EAC models. This could also be related to why the newly generated proto- SFB was fixed in the population, although, at this point, what selective pressure drives the acquisition of the current SFB functions remains unclear.
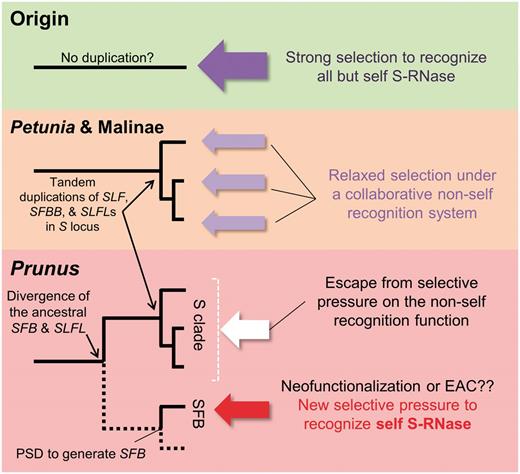
Model for the various types of selective pressures that acted on the duplicated pollen S F-box genes. Dotted lines in the schematic phylogeny of Prunus indicate genes not located within the S locus. In the original or incipient states of the S locus, a putative single-copy F-box gene was under purifying selections to recognize all but self-S-RNase. In the cases of Petunia and the subtribe Malinae, the function of non-self recognition is performed collaboratively by all gene copies, such that S-RNase types are apportioned to each tandemly duplicated SLF / SFBB gene for degradation. Thus, the purifying selection on the function of non-self recognition is weaker than on a single-copy SLF gene. In Prunus , the SLFL genes (genes in the clade S) escape from selective pressure on the function of non-self recognition (white arrow). Instead, the SFB gene, generated by PSD, has taken over the self-recognition function (red arrow).
The concept of relaxation from purifying selection after gene duplication might also be consistent with the non-self recognition function present in Petunia and the subtribe Malinae ( Kubo et al. 2010 , Kakui et al. 2011 , Williams et al. 2014 , Kubo et al. 2015 ). There may have been strong selective pressure on the ancestral F-box protein at the S locus to target all but self-S-RNase for degradation. Tandem duplications of the ancestral genes of the SLF / SFBB genes may have led to functional redundancy, resulting in relaxed purifying selection, and accumulation of loss-of-function mutations that were complemented by other copies, such that each targeted a subset of S-RNase for degradation. Eventually, the S locus F-box proteins collaboratively target all but self-S-RNases for degradation, a process known as the ‘collaborative non-self recognition model’ ( Kubo et al. 2010 , Kubo et al. 2015 ) ( Fig. 8 ). Recent studies also have suggested that duplicated genes most often experience diversification of expression patterns rather than protein functional diversification ( Adams 2007 , Buggs et al. 2010 , Liu and Adams 2010 , Roulin et al. 2013 ). Our study indicated no substantial changes in expression patterns between the duplicated F-box genes belonging to clade S and the PSD clade in Prunus , whereas functional divergence of SLFL and SFB was apparent.
Overall, our results are consistent with a model in which F-box genes evolved in a sudden radiation, accumulating a wide variety of structures and specific motifs among each subfamily, each contributing to changes in function ( Xu et al. 2009 ). In particular, the C-terminal region of the F-box gene harbors important motifs for target protein recognition ( Dharmasiri et al. 2005 , Thomas 2006 , Xu et al. 2009 ) and may contribute to rapid adaptive evolution by providing variability in protein–protein interactions ( Dharmasiri et al. 2005 , Xu et al. 2009 ), although little is known about the detailed functions of each motif in the C-terminal sequences. This situation would be consistent with the hypothesis that the Prunus -specific F-box duplications and the resulting establishment of hypervariable regions in the proto- SFB C-terminal sequences enabled self-recognition and created a SI mechanism distinct from the one found in the other species with S-RNase-based SI systems. Thus, the SFB-mediated self-recognition SI system in Prunus is a good example of rapid functional divergence after lineage-specific duplication of F-box genes.
Materials and Methods
Gene collection
The putative full-length sequences and locations of 411 genes with significant homology to SFB/SLFL genes were identified from the genomes of 31 angiosperms and two species outside the angiosperms, Selaginella and Physcomitrella , using BLASTP in Phytozome (JGI release version 9.1, http://www.phytozome.net/ ). We selected genes with significant homology (BLASTP e −19 cut-offs for all eudicots, and the highest hit with an e −10 cut-off in BLASTP for the others) to SFB ( SFB -S4; AB111521) or SLFL2 ( SLFL2 -S4; AB280954) from P. avium , according to the previous comprehensive identification of F-box genes ( Yang et al. 2008 ). These e-value cut-offs had previously been used to identify F-box genes from transcripts and protein sequences from a variety of plant species ( Childs et al. 2007 , Yang et al. 2008 ). Protein sequences with significant BLASTP hits were subjected to GO analysis by scanning the Pfam database for signature F-box domains (PF00646) and F-box-associated domains (PF04300, PF07734, PF07735 and PF08268), as described previously ( Xu et al. 2009 ). Some of the genes selected by BLASTP with the F-box motif were not associated with the F-box annotation in Phytozome but were included in the construction of the phylogeny and subsequent analyses.
Construction of evolutionary topology
Nucleotide sequence alignment was performed using MAFFT version 7 ( Katoh and Standley 2013 ). Custom Perl and Python scripts were used to adjust the nucleotide alignments to correspond to the amino acid alignments, also constructed using MAFFT. Raw alignments were adjusted manually, using SeaView version 4 ( Gouy et al. 2010 ), and then used for assignment of the best available evolutionary model in jModeltest 2.1 ( Darriba et al. 2012 ), using unequal base frequencies, a proportion of invariable sites and rate variation among sites and number of categories (‐i -f -g 4 -s 11, parameters in jModeltest), based on both the Akaike information criterion (AIC) and the Bayesian information criterion (BIC).
Nucleotide alignments were used to construct phylogenies using the full-length SFB / SLFL -like genes from the Prunus genome along with two Rosaceae-specific subclades α and ε. In jModeltest, the general-time reversible (GTR) + gamma (G) + proportion of invariable sites (I) model was selected for the full-length SFB / SLFL -like genes from the Prunus genome; whereas TIM1 (TIM = transversion model) and TPM3uf + I + G (TPM, three-parameter model; uf, unequal base frequencies) were adopted for the analysis of the two Rosaceae-specific subclades α and ε, respectively. The TPM model is a submodel of GTR, and the substitution pattern of TIM (numbers of substitutions=4) is not implemented in MrBayes 3.1.2 ( Ronquist and Huelsenbeck 2003 ). Thus, in this study, the GTR with a site-specific (SS) model, which is considered a codon model, was used to construct the evolutionary tree using the +I and/or +G options. The optimal tree and estimation of branch support were determined using a Bayesian MCMC approach using MrBayes or an ML approach using MEGA 5.05 ( Tamura et al. 2011 ). MCMC analyses used flat priors and were run for 3,000,000–10,000,000 generations and four Markov chains (using the default heating values), and sampled every 1,000 generations. We inferred that the chains converged on a stable set of parameters by calculating the potential scale reduction factor using MrBayes. ML phylogenetic analysis was performed according to the GTR model with invariant sites and gamma-distributed rates (three categories), with NNI as the tree-searching heuristic and 1,000 replications. All (remaining) sites, including missing and gap data, were used for the construction of the phylogeny.
For the alignment of amino acid sequences, MAFFT with the L-INS-i model and SeaView version 4 were used as described above. Phylogenetic reconstructions were generated from the amino acid alignments of the full length of SFB / SLFL -like genes from 31 angiosperm genomes. In all alignments, unnecessary long gap sequences disturbing proper alignment construction and genes showing significant homology but apparently different structure from other SLFL / SFB -like genes were removed. ML and Neighbor–Joining (NJ) approaches were applied to the resulting alignment file for phylogenetic analysis, using MEGA with 1,000 bootstrap replications. Under ML, the WAG model with invariant sites and gamma-distributed rates (three categories) was used, with NNI as the tree-searching heuristic. All (remaining) sites including missing and gap data were used for construction of a phylogeny. Under NJ, the Poisson matrix, with gamma-distributed rates (alpha parameter 3), was used, with pairwise deletion for the treatment of missing data.
Genomic information and definition of synteny
Information about the structure and physical location of the F-box genes in the Prunus genome was based on the Prunus persica genome (from the GDR, Prunus_persica_v1.0, http://www.rosaceae.org/projects/peach_genome/v1.0/genes ). Local synteny analysis around the SFB/SLFL-like genes was performed with the Mulan tool ( http://mulan.dcode.org/ ) in NCBI.
Estimation of the timing of gene divergence
We aligned the protein sequences of each pair of homologs using MAFFT and the L-INS-i algorithm, and converted the protein alignments to nucleotide alignments. We calculated the nucleotide diversity for the 4DTv between gene pairs using custom Python scripts. Here 4-fold synonymous sites are third codons of amino acid residues G, A, T, P, V, R, S and L. 4DTv values were calculated for F-box gene pairs retaining at least eight 4-fold synonymous sites. For calculation of 4DTv values from SLFL genes to a specific gene group (i.e. SLF genes in Petunia ), we averaged the 4DTv values of each pairwise combination of the genes between the two groups. For estimation of 4DTv values between two species (or genera), we averaged values of the putatively closest SFB / SLFL -like F-box gene sets, which show the smallest values in 4DTv, between the two taxa.
Gene expression profiling
Mature pollen grains of cv. Satonishiki ( P. avium ), and cvs. Nanko and Kairyo-Uchida-Ume ( P. mume ) were collected from anthers sampled from flower buds at the balloon stage of development in the experimental orchard of Kyoto University. Total RNA was extracted using cold-phenol extraction, as previously reported ( Ushijima et al. 2003 ). mRNA isolation and construction of RNA-Seq libraries were performed by TAKARA (Tokyo). All procedures for sample preparation were conducted using TruSeq RNA Sample Prep Kits (Illumina). Sequencing was performed using the TruSeq SBS Kit v3-HS on the Illumina HiSeq 2000 (Illumina) as paired-end reads of 100 bp (PE100).
One-fifth of a sequencing lane was dedicated to each of the three cultivars, and 29,812,682, 35,162,899 and 32,728,911 informative reads were obtained for cvs. Satonishiki, Kairyo-Uchida-Ume and Nanko, respectively. All bioinformatic and statistical analyses were performed on local servers at the UC Davis Genome Center. Raw reads without adaptor sequences were subjected to trimming procedures by using custom Python scripts ( http://comailab.genomecenter.ucdavis.edu/index.php/Barcoded_data_preparation_tools ). The reads were trimmed at the first point where nucleotide sequences showed a Phred quality score ≤20 over mean sliding windows of 5 bp. The paired-end reads of which at least either end showed length <35 bp were removed. Reads were then mapped to the 28,689 predicted mRNA transcript sequences including untranslated regions in the P. persica genome database (from the GDR, Prunus_persica_v1.0) using the Burrows–Wheeler Aligner (BWA) tools ( Li and Durbin 2009 ), either using default parameters or specifying a maximum of an 8 bp mismatch allowance per read. The number of reads mapping to each gene were calculated from the alignment file produced by the Sequence Alignment/Map (SAM) tool ( Li et al. 2009 ). Generally, the expression levels were normalized per RPKM. To define the pollen expression levels of all F-box genes from the Prunus genome, we used a series of ontology analyses from Phytozome. Searching for the term ‘F-box’ in the pfam, panther, KOG and GO databases generated 140 ontologies corresponding to 355 genes.
Gene expression statistics
The statistical tests for expression differences among the pollen samples were performed using the Bioconductor package DESeq in R ( Anders and Huber 2010 ) coupled with TbT normalization ( Kadota et al. 2012 ) for detection of the FDR, according to the DESeq analysis manual ( http://www-huber.embl.de/users/anders/DESeq/ ). To test for expression differences between P. mume and P. avium , we used the data from the two P. mume cultivars as two biological replicates and compared them with the expression levels in the P. avium sample. To compare the expression levels (RPKM) of F-box genes from Prunus , we used the values from all three cultivars as triple biological replicates.
Identification of selective pressure patterns on the F-box genes
For SFB and the three SLFL genes from the Prunus S haplogroup ( Ushijima et al. 2003 ), full-length allele sequences were obtained from the NCBI or Phytozome databases or by sequencing of PCR products, mainly in P. avium and P. mume cultivars that showed different allele types. All primers were designed outside the open reading frame region in each locus, using sequences from the peach genome database (GDR, Prunus_persica_v.1.0) ( Supplementary Table S2 ). PCR products were sequenced after cloning into the pGEM-T vector. Amino acid sequence alignments were constructed with MAFFT and converted from amino acid to nucleotide alignments. Aligned nucleotide sequences were used to determine putative ancestral sequences with MEGA. Informative single nucleotide polymorphisms (SNPs) in alleles were analyzed by DnaSP 5.1 ( Librado and Rozas 2009 ) and used to calculate an indicator of selective pressure (ω; Ka/Ks ratio). To calculate the selective pressure in Petunia ( SLF genes) and the subtribe Malinae ( SFBB genes), sequences from databases (NCBI, http://www.ncbi.nlm.nih.gov/ ; and DDBJ, http://www.ddbj.nig.ac.jp/index-e.html ) ( Supplementary Table S3 ) were aligned and analyzed by DnaSP 5.1, as described. Window-average Ka/Ks values were calculated from the start codon (ATG) in a 150 bp window with a 30 bp step size in DnaSP 5.1, until the walking window reached the stop codon.
The codon-based detection of branch-specific and SS positive selection was performed using PAML ( Yang 1997 ). The aligned nucleotide sequences and the topology constructed by MEGA v5.05 were analyzed using the branch site model. The statistical significance of positive selection on the foreground branches was evaluated using the likelihood ratio test of the null hypothesis that Ka/Ks = 1. SS positive selection was assessed by Bayes Empirical Bayes analysis.
Funding
This work was supported by the Japan Society for the Promotion of Science [Grants-in Aid for Scientific Research (A) Nos. 20248004, 24248007 and 15H02431 to R.T.]
Abbreviations
- EAC
escape from adaptive conflict
- 4DTv
4-fold synonymous third codon transversion position
- FDR
false discovery rate
- GDR
Genome Database for Rosaceae
- GI
general inhibitor
- GO
Gene Ontology
- GSI
gametophytic self-incompatibility
- LG
linkage group
- MCMC
Markov chain Monte Carlo
- ML
maximum likelihood
- MRCA
most recent common ancestor
- NJ
Neighbor–Joining
- PSD
Prunus -specific duplication
- RPKM
reads per kilobase of exon per million mapped reads
- RSD
Rosaceae-specific duplication
- SFB
S haplotype-specific F-box gene
- SFBB
S locus F-box brothers
- SI
self-incompatibility
- SLF
S locus F-box
- SLFL
S locus F-box genes with low allelic sequence polymorphism
- SS
site-specidic
Acknowledgments
We thank Meric Lieberman, Kathie J. Ngo and Luca Comai, Genome Center, University of California Davis (Davis, CA, USA) for bioinformatics support.
Disclosures
The authors have no conflicts of interest to declare.
References
Author notes
Illumina sequence data generated in this study has been submitted to the DDBJ Sequence Read Archive (DRA) database (BioProject ID: DRP000624, Submission ID: DRA000593).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}