





Abstract
This first-in-human phase I dose-escalation study evaluated the safety, pharmacokinetics, and efficacy of tinengotinib (TT-00420), a multi-kinase inhibitor targeting fibroblast growth factor receptors 1-3 (FGFRs 1-3), Janus kinase 1/2, vascular endothelial growth factor receptors, and Aurora A/B, in patients with advanced solid tumors.
Patients received tinengotinib orally daily in 28-day cycles. Dose escalation was guided by Bayesian modeling using escalation with overdose control. The primary objective was to assess dose-limiting toxicities (DLTs), maximum tolerated dose (MTD), and dose recommended for dose expansion (DRDE). Secondary objectives included pharmacokinetics and efficacy.
Forty-eight patients were enrolled (dose escalation, n = 40; dose expansion, n = 8). MTD was not reached; DRDE was 12 mg daily. DLTs were palmar-plantar erythrodysesthesia syndrome (8 mg, n = 1) and hypertension (15 mg, n = 2). The most common treatment-related adverse event was hypertension (50.0%). In 43 response-evaluable patients, 13 (30.2%) achieved partial response (PR; n = 7) or stable disease (SD) ≥ 24 weeks (n = 6), including 4/11 (36.4%) with FGFR2 mutations/fusions and cholangiocarcinoma (PR n = 3; SD ≥ 24 weeks n = 1), 3/3 (100.0%) with hormone receptor (HR)-positive/HER2-negative breast cancer (PR n = 2; SD ≥ 24 weeks n = 1), 2/5 (40.0%) with triple-negative breast cancer (TNBC; PR n = 1; SD ≥ 24 weeks n = 1), and 1/1 (100.0%) with castrate-resistant prostate cancer (CRPC; PR). Four of 12 patients (33.3%; HR-positive/HER2-negative breast cancer, TNBC, prostate cancer, and cholangiocarcinoma) treated at DRDE had PRs. Tinengotinib’s half-life was 28-34 hours.
Tinengotinib was well tolerated with favorable pharmacokinetic characteristics. Preliminary findings indicated potential clinical benefit in FGFR inhibitor-refractory cholangiocarcinoma, HER2-negative breast cancer (including TNBC), and CRPC. Continued evaluation of tinengotinib is warranted in phase II trials.
This was the first clinical study of tinengotinib (TT-00420), a multi-kinase inhibitor targeting fibroblast growth factor receptors 1-3 (FGFRs 1-3), Janus kinase [JAK] 1/2, vascular endothelial growth factor receptors, and Aurora A/B. Tinengotinib was given to 48 patients with advanced solid tumors and was well tolerated, with favorable pharmacokinetics. Forty-one patients (85.4%) had drug-related side effects, the most common being hypertension (50.0%), diarrhea (33.3%), palmar-plantar erythrodysesthesia syndrome (29.2%), stomatitis (29.2%), nausea (22.9%), and vomiting (20.8%). Preliminary efficacy was reported in patients with FGFR inhibitor-refractory cholangiocarcinoma, hormone-receptor-positive/HER2-negative breast cancer, triple-negative breast cancer, and castrate-resistant prostate cancer. These findings suggest that tinengotinib may be effective for patients with different solid tumors.
Introduction
Receptor tyrosine kinase signaling is frequently dysregulated in human cancers and contributes to many of the hallmark features of tumor biology, including proliferation, angiogenesis, and immune response evasion.1 Selective targeting of these molecular drivers with small-molecule tyrosine kinase inhibitors can produce meaningful clinical benefit, although the effects of many are limited by acquired drug resistance.2 Strategies to overcome resistance include combination therapy with 2 agents directed against the original target kinase and the predominant cause of resistance,1,2 or single-agent therapy with a kinase inhibitor that simultaneously targets multiple tyrosine kinases.
Tinengotinib (TT-00420) is a small-molecule, spectrum-selective, multiple kinase inhibitor that inhibits kinases involved with mitosis (Aurora A/B), angiogenesis (vascular endothelial growth factor receptors [VEGFRs], fibroblast growth factor receptors [FGFRs] 1, 2, and 3), and tumor cell proliferation and immune activity (Janus kinase [JAK] 1/2; colony-stimulating factor 1 receptor) at low nanomolar concentrations.3 In preclinical models, tinengotinib inhibited tumor growth in cell-line-derived and patient-derived xenograft (PDX) models of triple-negative breast cancer (TNBC),3 cholangiocarcinoma, bladder cancer, hepatocellular carcinoma, and thyroid cancer.4 Tinengotinib also demonstrated good oral bioavailability and dose-dependent exposure following oral administration in rats and dogs, with mechanism-related but manageable toxicities.5
This first-in-human study (ClinicalTrials.gov NCT03654547) was conducted to characterize the safety, tolerability, pharmacokinetics, and preliminary efficacy of single-agent tinengotinib in patients with advanced solid tumors.
Patients and Methods
Study Design
This 2-part, open-label, phase I study was conducted at the MD Anderson Cancer Center in Texas, USA and the Cancer Hospital Chinese Academy of Medical Sciences in Beijing, China. The initial dose-escalation phase was designed to determine the maximum tolerated dose (MTD), dose-limiting toxicities (DLTs), and dose recommended for dose expansion (DRDE) for tinengotinib. This was followed by a dose-expansion phase, the primary objective of which was to confirm the safety and tolerability of tinengotinib at the DRDE. Secondary objectives were to characterize the pharmacokinetics and preliminary efficacy of tinengotinib.
Dose escalation was guided by an adaptive Bayesian logistic regression model using the Escalation with Overdose Control principle.6,7 Study data (including DLTs, grade 2 adverse events during cycle 1, and pharmacokinetic data) were reviewed by the sponsor and trial investigators at each dose level and used to guide dose-escalation decisions. MTD was based on the Bayesian model, but with consideration given to available safety and tolerability data. DRDE was based on an evaluation of all available data (safety, clinical activity, and pharmacokinetics).
The study was conducted in accordance with the International Conference on Harmonisation Harmonised Tripartite Guidelines for Good Clinical Practice, with applicable local regulations, and the Declaration of Helsinki. The protocol was reviewed and approved by independent ethics committees at each study site. All patients were required to provide written informed consent prior to enrollment.
Patient Selection
Eligible patients had histologically or cytologically confirmed locally advanced or metastatic solid tumors. For the dose-expansion phase, preferred indications were cholangiocarcinoma harboring FGFR2 alterations (all patients with cholangiocarcinoma were considered eligible, regardless of FGFR2 status), advanced HER2-negative breast cancer (ie, hormone receptor [HR]-positive/HER2-negative or TNBC defined according to American Society of Clinical Oncology/College of American Pathologists guidelines8), and selected advanced solid tumors including urothelial carcinoma, gallbladder cancer, gastric cancer, and small cell lung cancer. Patients were 18-75 years of age with adequate organ function, an Eastern Cooperative Oncology Group performance status ≤1, and at least one measurable lesion defined by Response Evaluation Criteria in Solid Tumors (RECIST), version 1.1.9 Detailed eligibility criteria are outlined in Supplementary Methods.
Drug Treatment
Tinengotinib was administered orally (capsule formulation) once daily on a 28-day cycle until documented progression, unacceptable toxicity, or withdrawal of consent. Provisional tinengotinib dose levels during the dose-escalation phase were 1, 3, 5, 8, 10, 12, 15, 18, and 20 mg. After determination of the DRDE, the dose-expansion phase was opened to further characterize safety and pharmacokinetics, and to assess preliminary efficacy.
Assessments
Molecular screening of tumor samples prior to study entry was not performed and there was no confirmatory testing after study entry. With the exception of an exploratory biomarker analysis in the dose-expansion phase (described later), biomarker data were collected retrospectively from medical records when available.
Routine safety assessments of adverse events, laboratory tests (hematology, coagulation, biochemistry, and urinalysis), vital signs, and 12-lead electrocardiograms were performed. Adverse events were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE), version 5.0. A DLT was defined as an adverse event or abnormal laboratory value assessed as unrelated to disease, disease progression, intercurrent illness, or concomitant medications, that occurred ≤28 days following the first dose of tinengotinib, and that met any of the protocol-defined criteria for DLT (Supplementary Table S1).
Tumor measurements were obtained at baseline and every 2 cycles (8 weeks) using computerized tomography or magnetic resonance imaging until disease progression or study withdrawal. Tumor responses were determined by local investigator assessment per RECIST, version 1.1.9 Confirmed responses were those that were confirmed within 4-8 weeks of the initial response.
Pharmacokinetic Assessments
Blood samples for the pharmacokinetic evaluation of tinengotinib were collected on pharmacokinetic lead-in day 1 (single dose) and day 28 of cycle 1 (steady state). Single-dose and steady-state pharmacokinetic parameters (area under the concentration-time curve from 0 to 24 hours [AUC0-24], maximum concentration [Cmax], time to Cmax [Tmax], and elimination half-life [t1/2]) were estimated when feasible. Plasma concentrations of tinengotinib were measured by validated liquid chromatography-tandem mass spectrometry assay (limit of quantitation, 0.05 ng/mL).
Statistical Considerations
Dose-escalation sample size was determined by dose levels and emerging toxicities. Per protocol, a minimum of 21 patients was required for enrollment during dose escalation and 6 evaluable patients had to be treated at the dose level declared as the DRDE. Descriptive statistics were used for safety, pharmacokinetic, and efficacy data as outlined in the protocol and as specified in the statistical analysis plan. Pharmacokinetic parameters were estimated using Phoenix WinNonlin version 7.0 or higher (Certara USA, Inc., Princeton, NJ). All other statistical analyses were performed with SAS software version 9.4 (SAS Institute Inc., Cary, NC). The cutoff date for all analyses was September 9, 2022.
Exploratory Biomarker and Pharmacodynamic Assessments
Biomarker Analysis
An exploratory biomarker analysis was performed in patients enrolled in the dose-expansion cohort only. When feasible, archival tumor tissue (baseline), fresh tumor biopsy, and/or blood samples were collected at baseline, day 1/cycle 3, and at disease progression. For patients with cholangiocarcinoma, assays included next-generation sequencing for FGFR2 alterations in tumor or plasma, and measurement of plasma FGF-23 levels, increases in which represent FGFR inhibition.
Subcutaneous Xenograft Models
To validate its effectiveness in FGFR2 fusion-driven cholangiocarcinoma in vivo, tinengotinib was administered orally at 15 mg/kg in the CC6204 FGFR2-BICC1 gene fusion PDX model. Tumor fragments of a PDX model of intrahepatic cholangiocarcinoma with the FGFR2-BICC1 fusion were implanted subcutaneously in female BALB/c nude mice. Test animals were dosed as indicated and relative tumor volume and tumor growth inhibition (TGI) were calculated. Studies were conducted at Crown Bioscience International (San Diego, CA) according to a protocol approved by the Institutional Animal Care and Use Committee as previously described.3
ELISA Assay for Cellular phosphorylated FGFR2
Ex vivo pharmacodynamic analysis of phosphorylated FGFR2α inhibition was previously established as a reliable biomarker for predicting clinical efficacy in the FIGHT-101 trial.10 We therefore assessed FGFR2 phosphorylation in the KATO III cell line following treatment with tinengotinib. Cells were cultured in healthy donor plasma spiked with serial dilutions of tinengotinib. Cellular phosphorylated FGFR2 was determined using the commercial enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems) according to the manufacturer’s instructions. Pemigatinib was included as a positive control10 to indicate if the tinengotinib analysis was reliable.
Results
Patient Characteristics
Between January 8, 2019 and May 13, 2021, 48 patients were enrolled and received tinengotinib (dose escalation, n = 40; dose expansion, n = 8; DRDE, n = 20), of whom 43 (DRDE, n = 18) were evaluable for tumor response (Supplementary Fig. S1). Patient characteristics at baseline are presented in Table 1, and by dose level in Supplementary Table S2. Patients were predominantly White (68.8%) with a median age of 56.8 years (range, 25-79). The most common primary tumor types were cholangiocarcinoma (n = 14; 29.2%) and HER2-negative breast cancer (n = 9; 18.8%), including 6 patients with TNBC and 3 patients with HR-positive/HER2-negative breast cancer. Overall, 70.8% of patients had received 3 or more prior systemic anticancer therapies.
Baseline patient demographics and characteristics (full analysis set).
| Dose escalation | Dose expansion (12 mg) | Total | |
|---|---|---|---|
| (n = 40) | (n = 8) | (n = 48) | |
| Median (range) age, years | 57.4 (28‒79) | 44.6 (25‒73) | 56.8 (25‒79) |
| <60, n (%) | 23 (57.5) | 6 (75.0) | 29 (60.4) |
| ≥60, n (%) | 17 (42.5) | 2 (25.0) | 19 (39.6) |
| Sex, n (%) | |||
| Male | 16 (40.0) | 6 (75.0) | 22 (45.8) |
| Female | 24 (60.0) | 2 (25.0) | 26 (54.2) |
| Race, n (%) | |||
| White | 26 (65.0) | 7 (87.5) | 33 (68.8) |
| Black or African American | 6 (15.0) | 0 | 6 (12.5) |
| Asian | 6 (15.0) | 0 | 6 (12.5) |
| Other | 2 (5.0) | 1 (12.5) | 3 (6.3) |
| ECOG performance status, n (%) | |||
| 0 | 8 (20.0) | 1 (12.5) | 9 (18.8) |
| 1 | 32 (80.0) | 7 (87.5) | 39 (81.3) |
| Primary tumor site, n (%) | |||
| Liver | 7 (17.5) | 3 (37.5) | 10 (20.8) |
| Breast | 8 (20.0) | 1 (12.5) | 9 (18.8) |
| Colon | 1 (2.5) | 1 (12.5) | 2 (4.2) |
| Lung | 2 (5.0) | 0 | 2 (4.2) |
| Esophagus | 2 (5.0) | 0 | 2 (4.2) |
| Ovary | 2 (5.0) | 0 | 2 (4.2) |
| Salivary gland | 2 (5.0) | 0 | 2 (4.2) |
| Soft-tissue sarcoma | 2 (5.0) | 0 | 2 (4.2) |
| Other | 14 (35.0) | 3 (37.5) | 17 (35.4) |
| Prior therapies, n (%) | 40 (100) | 8 (100) | 48 (100) |
| Radiation | 22 (55.0) | 2 (25.0) | 24 (50.0) |
| Surgery | 40 (100) | 8 (100) | 48 (100) |
| Anticancer medication | 39 (97.5) | 8 (100) | 47 (97.9) |
| Chemotherapy | 37 (92.5) | 8 (100) | 45 (93.8) |
| Hormonal therapy | 5 (12.5) | 1 (12.5) | 6 (12.5) |
| Immunotherapy | 17 (42.5) | 1 (12.5) | 18 (37.5) |
| Targeted therapy | 13 (32.5) | 2 (25.0) | 15 (31.3) |
| Other | 2 (5.0) | 0 | 2 (4.2) |
| Lines of therapy, n (%) | |||
| 0 | 1 (2.5) | 0 | 1 (2.1) |
| 1 | 6 (15.0) | 4 (50.0) | 10 (20.8) |
| 2 | 2 (5.0) | 1 (12.5) | 3 (6.3) |
| 3 | 13 (32.5) | 0 | 13 (27.1) |
| ≥4 | 18 (45.0) | 3 (37.5) | 21 (43.8) |
| Dose escalation | Dose expansion (12 mg) | Total | |
|---|---|---|---|
| (n = 40) | (n = 8) | (n = 48) | |
| Median (range) age, years | 57.4 (28‒79) | 44.6 (25‒73) | 56.8 (25‒79) |
| <60, n (%) | 23 (57.5) | 6 (75.0) | 29 (60.4) |
| ≥60, n (%) | 17 (42.5) | 2 (25.0) | 19 (39.6) |
| Sex, n (%) | |||
| Male | 16 (40.0) | 6 (75.0) | 22 (45.8) |
| Female | 24 (60.0) | 2 (25.0) | 26 (54.2) |
| Race, n (%) | |||
| White | 26 (65.0) | 7 (87.5) | 33 (68.8) |
| Black or African American | 6 (15.0) | 0 | 6 (12.5) |
| Asian | 6 (15.0) | 0 | 6 (12.5) |
| Other | 2 (5.0) | 1 (12.5) | 3 (6.3) |
| ECOG performance status, n (%) | |||
| 0 | 8 (20.0) | 1 (12.5) | 9 (18.8) |
| 1 | 32 (80.0) | 7 (87.5) | 39 (81.3) |
| Primary tumor site, n (%) | |||
| Liver | 7 (17.5) | 3 (37.5) | 10 (20.8) |
| Breast | 8 (20.0) | 1 (12.5) | 9 (18.8) |
| Colon | 1 (2.5) | 1 (12.5) | 2 (4.2) |
| Lung | 2 (5.0) | 0 | 2 (4.2) |
| Esophagus | 2 (5.0) | 0 | 2 (4.2) |
| Ovary | 2 (5.0) | 0 | 2 (4.2) |
| Salivary gland | 2 (5.0) | 0 | 2 (4.2) |
| Soft-tissue sarcoma | 2 (5.0) | 0 | 2 (4.2) |
| Other | 14 (35.0) | 3 (37.5) | 17 (35.4) |
| Prior therapies, n (%) | 40 (100) | 8 (100) | 48 (100) |
| Radiation | 22 (55.0) | 2 (25.0) | 24 (50.0) |
| Surgery | 40 (100) | 8 (100) | 48 (100) |
| Anticancer medication | 39 (97.5) | 8 (100) | 47 (97.9) |
| Chemotherapy | 37 (92.5) | 8 (100) | 45 (93.8) |
| Hormonal therapy | 5 (12.5) | 1 (12.5) | 6 (12.5) |
| Immunotherapy | 17 (42.5) | 1 (12.5) | 18 (37.5) |
| Targeted therapy | 13 (32.5) | 2 (25.0) | 15 (31.3) |
| Other | 2 (5.0) | 0 | 2 (4.2) |
| Lines of therapy, n (%) | |||
| 0 | 1 (2.5) | 0 | 1 (2.1) |
| 1 | 6 (15.0) | 4 (50.0) | 10 (20.8) |
| 2 | 2 (5.0) | 1 (12.5) | 3 (6.3) |
| 3 | 13 (32.5) | 0 | 13 (27.1) |
| ≥4 | 18 (45.0) | 3 (37.5) | 21 (43.8) |
Abbreviation: ECOG, Eastern Cooperative Oncology Group.
Baseline patient demographics and characteristics (full analysis set).
| Dose escalation | Dose expansion (12 mg) | Total | |
|---|---|---|---|
| (n = 40) | (n = 8) | (n = 48) | |
| Median (range) age, years | 57.4 (28‒79) | 44.6 (25‒73) | 56.8 (25‒79) |
| <60, n (%) | 23 (57.5) | 6 (75.0) | 29 (60.4) |
| ≥60, n (%) | 17 (42.5) | 2 (25.0) | 19 (39.6) |
| Sex, n (%) | |||
| Male | 16 (40.0) | 6 (75.0) | 22 (45.8) |
| Female | 24 (60.0) | 2 (25.0) | 26 (54.2) |
| Race, n (%) | |||
| White | 26 (65.0) | 7 (87.5) | 33 (68.8) |
| Black or African American | 6 (15.0) | 0 | 6 (12.5) |
| Asian | 6 (15.0) | 0 | 6 (12.5) |
| Other | 2 (5.0) | 1 (12.5) | 3 (6.3) |
| ECOG performance status, n (%) | |||
| 0 | 8 (20.0) | 1 (12.5) | 9 (18.8) |
| 1 | 32 (80.0) | 7 (87.5) | 39 (81.3) |
| Primary tumor site, n (%) | |||
| Liver | 7 (17.5) | 3 (37.5) | 10 (20.8) |
| Breast | 8 (20.0) | 1 (12.5) | 9 (18.8) |
| Colon | 1 (2.5) | 1 (12.5) | 2 (4.2) |
| Lung | 2 (5.0) | 0 | 2 (4.2) |
| Esophagus | 2 (5.0) | 0 | 2 (4.2) |
| Ovary | 2 (5.0) | 0 | 2 (4.2) |
| Salivary gland | 2 (5.0) | 0 | 2 (4.2) |
| Soft-tissue sarcoma | 2 (5.0) | 0 | 2 (4.2) |
| Other | 14 (35.0) | 3 (37.5) | 17 (35.4) |
| Prior therapies, n (%) | 40 (100) | 8 (100) | 48 (100) |
| Radiation | 22 (55.0) | 2 (25.0) | 24 (50.0) |
| Surgery | 40 (100) | 8 (100) | 48 (100) |
| Anticancer medication | 39 (97.5) | 8 (100) | 47 (97.9) |
| Chemotherapy | 37 (92.5) | 8 (100) | 45 (93.8) |
| Hormonal therapy | 5 (12.5) | 1 (12.5) | 6 (12.5) |
| Immunotherapy | 17 (42.5) | 1 (12.5) | 18 (37.5) |
| Targeted therapy | 13 (32.5) | 2 (25.0) | 15 (31.3) |
| Other | 2 (5.0) | 0 | 2 (4.2) |
| Lines of therapy, n (%) | |||
| 0 | 1 (2.5) | 0 | 1 (2.1) |
| 1 | 6 (15.0) | 4 (50.0) | 10 (20.8) |
| 2 | 2 (5.0) | 1 (12.5) | 3 (6.3) |
| 3 | 13 (32.5) | 0 | 13 (27.1) |
| ≥4 | 18 (45.0) | 3 (37.5) | 21 (43.8) |
| Dose escalation | Dose expansion (12 mg) | Total | |
|---|---|---|---|
| (n = 40) | (n = 8) | (n = 48) | |
| Median (range) age, years | 57.4 (28‒79) | 44.6 (25‒73) | 56.8 (25‒79) |
| <60, n (%) | 23 (57.5) | 6 (75.0) | 29 (60.4) |
| ≥60, n (%) | 17 (42.5) | 2 (25.0) | 19 (39.6) |
| Sex, n (%) | |||
| Male | 16 (40.0) | 6 (75.0) | 22 (45.8) |
| Female | 24 (60.0) | 2 (25.0) | 26 (54.2) |
| Race, n (%) | |||
| White | 26 (65.0) | 7 (87.5) | 33 (68.8) |
| Black or African American | 6 (15.0) | 0 | 6 (12.5) |
| Asian | 6 (15.0) | 0 | 6 (12.5) |
| Other | 2 (5.0) | 1 (12.5) | 3 (6.3) |
| ECOG performance status, n (%) | |||
| 0 | 8 (20.0) | 1 (12.5) | 9 (18.8) |
| 1 | 32 (80.0) | 7 (87.5) | 39 (81.3) |
| Primary tumor site, n (%) | |||
| Liver | 7 (17.5) | 3 (37.5) | 10 (20.8) |
| Breast | 8 (20.0) | 1 (12.5) | 9 (18.8) |
| Colon | 1 (2.5) | 1 (12.5) | 2 (4.2) |
| Lung | 2 (5.0) | 0 | 2 (4.2) |
| Esophagus | 2 (5.0) | 0 | 2 (4.2) |
| Ovary | 2 (5.0) | 0 | 2 (4.2) |
| Salivary gland | 2 (5.0) | 0 | 2 (4.2) |
| Soft-tissue sarcoma | 2 (5.0) | 0 | 2 (4.2) |
| Other | 14 (35.0) | 3 (37.5) | 17 (35.4) |
| Prior therapies, n (%) | 40 (100) | 8 (100) | 48 (100) |
| Radiation | 22 (55.0) | 2 (25.0) | 24 (50.0) |
| Surgery | 40 (100) | 8 (100) | 48 (100) |
| Anticancer medication | 39 (97.5) | 8 (100) | 47 (97.9) |
| Chemotherapy | 37 (92.5) | 8 (100) | 45 (93.8) |
| Hormonal therapy | 5 (12.5) | 1 (12.5) | 6 (12.5) |
| Immunotherapy | 17 (42.5) | 1 (12.5) | 18 (37.5) |
| Targeted therapy | 13 (32.5) | 2 (25.0) | 15 (31.3) |
| Other | 2 (5.0) | 0 | 2 (4.2) |
| Lines of therapy, n (%) | |||
| 0 | 1 (2.5) | 0 | 1 (2.1) |
| 1 | 6 (15.0) | 4 (50.0) | 10 (20.8) |
| 2 | 2 (5.0) | 1 (12.5) | 3 (6.3) |
| 3 | 13 (32.5) | 0 | 13 (27.1) |
| ≥4 | 18 (45.0) | 3 (37.5) | 21 (43.8) |
Abbreviation: ECOG, Eastern Cooperative Oncology Group.
Although biomarker data collection was not mandatory and FGFR mutation status did not need to be determined before inclusion in the study, 10 of the 14 patients with cholangiocarcinoma had historical next-generation sequencing data. These were FGFR2-KCNH7 fusion, FGFR2 N549K mutation; FGFR2-AHCYL1.F17A2 fusion; FGFR2 C382R mutation; FGFR2 F17S6, N549H, and N549K mutations; FGFR2 fusion; FGFR2 rearrangement, and TP53 and BAP1 mutations; FGFR2-DNAJC12 fusion; FGFR2 D101V fusion; FGFR2-TACC2.F17T11 fusion; and FGFR2-CCDC6.F17C2 fusion.
Dose Escalation
During dose escalation, sequential patient cohorts received tinengotinib 1 mg (n = 1), 3 mg (n = 1), 5 mg (n = 4), 8 mg (n = 10), 10 mg (n = 6), 12 mg (n = 12), and 15 mg (n = 6). Three DLTs were observed at the 8 mg dose (grade 3 palmar-plantar erythrodysesthesia syndrome, n = 1) and at the 15 mg dose (grade 3 hypertension, n = 2; Supplementary Table S3). There were no other DLTs at the 12 mg dose during the dose-expansion phase. The MTD for tinengotinib was not reached based on the Bayesian model; however, based on the evaluation of safety, pharmacokinetic, and efficacy data, a DRDE of 12 mg once daily was selected for patients with advanced solid tumors.
Safety and Tolerability
The median duration of treatment with tinengotinib across all dose levels was 2.9 months (range, 0.5‒14.8). Among the 48 patients who received tinengotinib and were evaluable for safety, 41 patients (85.4%) had study drug-related treatment-emergent adverse events (TEAEs; Table 2; Supplementary Table S4). The most common drug-related TEAEs were hypertension (50.0%), diarrhea (33.3%), palmar-plantar erythrodysesthesia syndrome (29.2%), stomatitis (29.2%), nausea (22.9%), and vomiting (20.8%). Twenty-one patients (43.8%) had drug-related grade 3 TEAEs, including hypertension (27.1%), stomatitis (4.2%), diarrhea (2.1%), palmar-plantar erythrodysesthesia syndrome (2.1%), and nausea (2.1%). No drug-related grade 4 or 5 TEAEs were reported. Hypertension was manageable by dose modifications and/or concomitant antihypertensive medication; no patients permanently discontinued study treatment because of drug-related hypertension. An exposure-response model predicted that the risk of grade ≥3 hypertension was uniform and consistent (<25%) for all doses (Supplementary Fig. S2). Seven patients (14.6%) had drug-related serious adverse events (hypertension, n = 1; nausea and hypertension, n = 1; anemia and gastrointestinal hemorrhage, n = 1; mucosal inflammation, n = 1; gastrointestinal infection, n = 1; cerebrovascular accident, n = 1; palmar-plantar erythrodysesthesia syndrome, n = 1). Dose modifications (dose adjustment or temporary interruption) were required because of drug-related TEAEs in 25 patients (52.1%), most commonly hypertension (22.9%). Drug-related TEAEs leading to permanent discontinuation of tinengotinib were documented in 6 patients (6.3%): 1 case of grade 3 palmar-plantar erythrodysesthesia syndrome (8 mg); 2 cases of grade 3 hypertension, and 1 case each of grade 2 nausea, grade 2 vomiting, grade 3 cerebrovascular accident, and grade 2 palmar-plantar erythrodysesthesia syndrome (10 mg); and grade 1 retinal vein occlusion (12 mg).
Summary of study drug-related TEAEs (safety set).
| Dose escalation (n = 40) | Dose expansion (12 mg, n = 8) | Total (n = 48) | ||||
|---|---|---|---|---|---|---|
| Any grade | Grade ≥3 | Any grade | Grade ≥3 | Any grade | Grade ≥3 | |
| Study drug-related TEAE | 33 (82.5) | 16 (40.0) | 8 (100) | 5 (62.5) | 41 (85.4) | 21 (43.8) |
| Study drug-related serious TEAE | 5 (12.5) | 0 | 2 (25.0) | 0 | 7 (14.6) | 0 |
| Common study drug-related TEAEa | ||||||
| Hypertension | 19 (47.5) | 9 (22.5) | 5 (62.5) | 4 (50.0) | 24 (50.0) | 13 (27.1) |
| Diarrhea | 15 (37.5) | 1 (2.5) | 1 (12.5) | 0 | 16 (33.3) | 1 (2.1) |
| Palmar-plantar erythrodysesthesia syndrome | 10 (25.0) | 1 (2.5) | 4 (50.0) | 0 | 14 (29.2) | 1 (2.1) |
| Stomatitis | 9 (22.5) | 2 (5.0) | 5 (62.5) | 0 | 14 (29.2) | 2 (4.2) |
| Nausea | 11 (27.5) | 1 (2.5) | 0 | 0 | 11 (22.9) | 1 (2.1) |
| Vomiting | 10 (25.0) | 0 | 0 | 0 | 10 (20.8) | 0 |
| Decreased appetite | 5 (12.5) | 0 | 2 (25.0) | 0 | 7 (14.6) | 0 |
| Blood thyroid-stimulating hormone increased | 4 (10.0) | 0 | 3 (37.5) | 0 | 7 (14.6) | 0 |
| Proteinuria | 6 (15.0) | 0 | 0 | 0 | 6 (12.5) | 0 |
| Alanine aminotransferase increased | 4 (10.0) | 0 | 1 (12.5) | 0 | 5 (10.4) | 0 |
| Aspartate aminotransferase increased | 4 (10.0) | 0 | 1 (12.5) | 0 | 5 (10.4) | 0 |
| Dry mouth | 5 (12.5) | 0 | 0 | 0 | 5 (10.4) | 0 |
| Myalgia | 5 (12.5) | 0 | 0 | 0 | 5 (10.4) | 0 |
| Dose escalation (n = 40) | Dose expansion (12 mg, n = 8) | Total (n = 48) | ||||
|---|---|---|---|---|---|---|
| Any grade | Grade ≥3 | Any grade | Grade ≥3 | Any grade | Grade ≥3 | |
| Study drug-related TEAE | 33 (82.5) | 16 (40.0) | 8 (100) | 5 (62.5) | 41 (85.4) | 21 (43.8) |
| Study drug-related serious TEAE | 5 (12.5) | 0 | 2 (25.0) | 0 | 7 (14.6) | 0 |
| Common study drug-related TEAEa | ||||||
| Hypertension | 19 (47.5) | 9 (22.5) | 5 (62.5) | 4 (50.0) | 24 (50.0) | 13 (27.1) |
| Diarrhea | 15 (37.5) | 1 (2.5) | 1 (12.5) | 0 | 16 (33.3) | 1 (2.1) |
| Palmar-plantar erythrodysesthesia syndrome | 10 (25.0) | 1 (2.5) | 4 (50.0) | 0 | 14 (29.2) | 1 (2.1) |
| Stomatitis | 9 (22.5) | 2 (5.0) | 5 (62.5) | 0 | 14 (29.2) | 2 (4.2) |
| Nausea | 11 (27.5) | 1 (2.5) | 0 | 0 | 11 (22.9) | 1 (2.1) |
| Vomiting | 10 (25.0) | 0 | 0 | 0 | 10 (20.8) | 0 |
| Decreased appetite | 5 (12.5) | 0 | 2 (25.0) | 0 | 7 (14.6) | 0 |
| Blood thyroid-stimulating hormone increased | 4 (10.0) | 0 | 3 (37.5) | 0 | 7 (14.6) | 0 |
| Proteinuria | 6 (15.0) | 0 | 0 | 0 | 6 (12.5) | 0 |
| Alanine aminotransferase increased | 4 (10.0) | 0 | 1 (12.5) | 0 | 5 (10.4) | 0 |
| Aspartate aminotransferase increased | 4 (10.0) | 0 | 1 (12.5) | 0 | 5 (10.4) | 0 |
| Dry mouth | 5 (12.5) | 0 | 0 | 0 | 5 (10.4) | 0 |
| Myalgia | 5 (12.5) | 0 | 0 | 0 | 5 (10.4) | 0 |
Data are presented as No. (%).
Abbreviation: TEAE, treatment-emergent adverse event.
aDefined as any grade study drug-related TEAE occurring in ≥10% of the total study population. No grade 4 or 5 study drug-related TEAEs were reported.
Summary of study drug-related TEAEs (safety set).
| Dose escalation (n = 40) | Dose expansion (12 mg, n = 8) | Total (n = 48) | ||||
|---|---|---|---|---|---|---|
| Any grade | Grade ≥3 | Any grade | Grade ≥3 | Any grade | Grade ≥3 | |
| Study drug-related TEAE | 33 (82.5) | 16 (40.0) | 8 (100) | 5 (62.5) | 41 (85.4) | 21 (43.8) |
| Study drug-related serious TEAE | 5 (12.5) | 0 | 2 (25.0) | 0 | 7 (14.6) | 0 |
| Common study drug-related TEAEa | ||||||
| Hypertension | 19 (47.5) | 9 (22.5) | 5 (62.5) | 4 (50.0) | 24 (50.0) | 13 (27.1) |
| Diarrhea | 15 (37.5) | 1 (2.5) | 1 (12.5) | 0 | 16 (33.3) | 1 (2.1) |
| Palmar-plantar erythrodysesthesia syndrome | 10 (25.0) | 1 (2.5) | 4 (50.0) | 0 | 14 (29.2) | 1 (2.1) |
| Stomatitis | 9 (22.5) | 2 (5.0) | 5 (62.5) | 0 | 14 (29.2) | 2 (4.2) |
| Nausea | 11 (27.5) | 1 (2.5) | 0 | 0 | 11 (22.9) | 1 (2.1) |
| Vomiting | 10 (25.0) | 0 | 0 | 0 | 10 (20.8) | 0 |
| Decreased appetite | 5 (12.5) | 0 | 2 (25.0) | 0 | 7 (14.6) | 0 |
| Blood thyroid-stimulating hormone increased | 4 (10.0) | 0 | 3 (37.5) | 0 | 7 (14.6) | 0 |
| Proteinuria | 6 (15.0) | 0 | 0 | 0 | 6 (12.5) | 0 |
| Alanine aminotransferase increased | 4 (10.0) | 0 | 1 (12.5) | 0 | 5 (10.4) | 0 |
| Aspartate aminotransferase increased | 4 (10.0) | 0 | 1 (12.5) | 0 | 5 (10.4) | 0 |
| Dry mouth | 5 (12.5) | 0 | 0 | 0 | 5 (10.4) | 0 |
| Myalgia | 5 (12.5) | 0 | 0 | 0 | 5 (10.4) | 0 |
| Dose escalation (n = 40) | Dose expansion (12 mg, n = 8) | Total (n = 48) | ||||
|---|---|---|---|---|---|---|
| Any grade | Grade ≥3 | Any grade | Grade ≥3 | Any grade | Grade ≥3 | |
| Study drug-related TEAE | 33 (82.5) | 16 (40.0) | 8 (100) | 5 (62.5) | 41 (85.4) | 21 (43.8) |
| Study drug-related serious TEAE | 5 (12.5) | 0 | 2 (25.0) | 0 | 7 (14.6) | 0 |
| Common study drug-related TEAEa | ||||||
| Hypertension | 19 (47.5) | 9 (22.5) | 5 (62.5) | 4 (50.0) | 24 (50.0) | 13 (27.1) |
| Diarrhea | 15 (37.5) | 1 (2.5) | 1 (12.5) | 0 | 16 (33.3) | 1 (2.1) |
| Palmar-plantar erythrodysesthesia syndrome | 10 (25.0) | 1 (2.5) | 4 (50.0) | 0 | 14 (29.2) | 1 (2.1) |
| Stomatitis | 9 (22.5) | 2 (5.0) | 5 (62.5) | 0 | 14 (29.2) | 2 (4.2) |
| Nausea | 11 (27.5) | 1 (2.5) | 0 | 0 | 11 (22.9) | 1 (2.1) |
| Vomiting | 10 (25.0) | 0 | 0 | 0 | 10 (20.8) | 0 |
| Decreased appetite | 5 (12.5) | 0 | 2 (25.0) | 0 | 7 (14.6) | 0 |
| Blood thyroid-stimulating hormone increased | 4 (10.0) | 0 | 3 (37.5) | 0 | 7 (14.6) | 0 |
| Proteinuria | 6 (15.0) | 0 | 0 | 0 | 6 (12.5) | 0 |
| Alanine aminotransferase increased | 4 (10.0) | 0 | 1 (12.5) | 0 | 5 (10.4) | 0 |
| Aspartate aminotransferase increased | 4 (10.0) | 0 | 1 (12.5) | 0 | 5 (10.4) | 0 |
| Dry mouth | 5 (12.5) | 0 | 0 | 0 | 5 (10.4) | 0 |
| Myalgia | 5 (12.5) | 0 | 0 | 0 | 5 (10.4) | 0 |
Data are presented as No. (%).
Abbreviation: TEAE, treatment-emergent adverse event.
aDefined as any grade study drug-related TEAE occurring in ≥10% of the total study population. No grade 4 or 5 study drug-related TEAEs were reported.
Antitumor Activity
Forty-three patients received at least one dose of study drug and had at least one post-baseline scan (dose escalation, n = 36; dose expansion, n = 7). As summarized in Fig. 1, partial responses (PRs) were observed in 7 patients (16.3%), including 3 patients with cholangiocarcinoma (8, 10, and 12 mg doses), 2 patients with HR-positive/HER2-negative breast cancer (10 and 12 mg doses), 1 patient with TNBC (12 mg dose), and 1 patient with castration-resistant prostate cancer (CRPC; 12 mg dose). Stable disease (SD) was noted in 23 patients (53.3%), including 6 patients (14.0%) with SD lasting ≥24 weeks (Table 3). At the DRDE of 12 mg once daily, a PR was observed in 4 of 18 (22%) evaluable patients (Table 3).
Patients with SD for ≥ 24 weeks or PR by RECIST v1.1.
| Patient characteristics | Tinengotinib | Best response by RECIST version 1.1 | |||||
|---|---|---|---|---|---|---|---|
| Cancer type | FGFR alterationsa | Prior systemic therapies, n | Previous targeted therapies | Dose, mg/day | Duration of treatment, weeks | ||
| 1001-25 | Cholangiocarcinoma | FGFR2 N549K, FGFR2-KCNH7 fusion | 3 | Derazantinib | 8 | 38.3 | PR |
| 1001-37 | Cholangiocarcinoma | FGFR2 C382R | 3 | Derazantinib | 10 | 32.8 | PR |
| 1001-41b | HR+/HER2− breast cancer | FGFR2 rearrangement | 13 | Palbociclib; SYK/FLT3 inhibitord; PIM kinase inhibitord | 12 | 12.1 | PR |
| 2001-50 | HR+/HER2− breast cancer | None | 6 | None | 10 | 25 | PR |
| 1001-74b | TNBC | None | 8 | None | 12 | 16.1 | PR |
| 1001-64b | Prostate adenocarcinoma | FGFR2 amplification | 13 | SUMO-activating enzyme inhibitord | 12 | 21.9 | PR |
| 1001-79b | Cholangiocarcinoma | FGFR2-CCDC6.F17C2c | 4 | Pemigatinib | 12 | 28 | PR |
| 1001-34 | Cholangiocarcinoma | FGFR2-AHCYL1.F17A2 | 4 | Infigratinib | 10 | 33.9 | SD |
| 1001-15 | HR+/HER2− breast cancer | None | 7 | NO synthase inhibitord | 8 | 26.1 | SD |
| 2001-24 | TNBC | None | 3 | None | 5 | 24.6 | SD |
| 1001-66b | Rectum adenocarcinoma | FGFR1 amplification | 5 | Pemigatinib | 12 | 26 | SD |
| 1001-67b | Epithelioid mesothelioma | None | 3 | Bevacizumab | 12 | 50.4 | SD |
| 1001-05 | Salivary gland adenocarcinoma | None | 3 | Ceritinib; abemaciclib | 5 | 36.4 | SD |
| Patient characteristics | Tinengotinib | Best response by RECIST version 1.1 | |||||
|---|---|---|---|---|---|---|---|
| Cancer type | FGFR alterationsa | Prior systemic therapies, n | Previous targeted therapies | Dose, mg/day | Duration of treatment, weeks | ||
| 1001-25 | Cholangiocarcinoma | FGFR2 N549K, FGFR2-KCNH7 fusion | 3 | Derazantinib | 8 | 38.3 | PR |
| 1001-37 | Cholangiocarcinoma | FGFR2 C382R | 3 | Derazantinib | 10 | 32.8 | PR |
| 1001-41b | HR+/HER2− breast cancer | FGFR2 rearrangement | 13 | Palbociclib; SYK/FLT3 inhibitord; PIM kinase inhibitord | 12 | 12.1 | PR |
| 2001-50 | HR+/HER2− breast cancer | None | 6 | None | 10 | 25 | PR |
| 1001-74b | TNBC | None | 8 | None | 12 | 16.1 | PR |
| 1001-64b | Prostate adenocarcinoma | FGFR2 amplification | 13 | SUMO-activating enzyme inhibitord | 12 | 21.9 | PR |
| 1001-79b | Cholangiocarcinoma | FGFR2-CCDC6.F17C2c | 4 | Pemigatinib | 12 | 28 | PR |
| 1001-34 | Cholangiocarcinoma | FGFR2-AHCYL1.F17A2 | 4 | Infigratinib | 10 | 33.9 | SD |
| 1001-15 | HR+/HER2− breast cancer | None | 7 | NO synthase inhibitord | 8 | 26.1 | SD |
| 2001-24 | TNBC | None | 3 | None | 5 | 24.6 | SD |
| 1001-66b | Rectum adenocarcinoma | FGFR1 amplification | 5 | Pemigatinib | 12 | 26 | SD |
| 1001-67b | Epithelioid mesothelioma | None | 3 | Bevacizumab | 12 | 50.4 | SD |
| 1001-05 | Salivary gland adenocarcinoma | None | 3 | Ceritinib; abemaciclib | 5 | 36.4 | SD |
Abbreviations: DRDE, dose recommended for dose expansion; FGFR, fibroblast growth factor receptor; FLT3, FMS-like tyrosine kinase 3; HER2−, human epidermal growth factor 2-negative; HR+, hormone receptor-positive; NO, nitric oxide; PIM, proviral integration site of Moloney murine leukemia virus; PR, partial response; RECIST, Response Evaluation Criteria in Solid Tumors; SD, stable disease; SUMO, small ubiquitin-like modifier; SYK, spleen tyrosine kinase; TNBC, triple-negative breast cancer.
aIndicates historical FGFR mutational status obtained from medical records review.
bIndicates patient treated at DRDE.
cIndicates FGFR mutational status obtained from next-generation sequencing performed during study screening and prior to study entry.
dInvestigational agent under study.
Patients with SD for ≥ 24 weeks or PR by RECIST v1.1.
| Patient characteristics | Tinengotinib | Best response by RECIST version 1.1 | |||||
|---|---|---|---|---|---|---|---|
| Cancer type | FGFR alterationsa | Prior systemic therapies, n | Previous targeted therapies | Dose, mg/day | Duration of treatment, weeks | ||
| 1001-25 | Cholangiocarcinoma | FGFR2 N549K, FGFR2-KCNH7 fusion | 3 | Derazantinib | 8 | 38.3 | PR |
| 1001-37 | Cholangiocarcinoma | FGFR2 C382R | 3 | Derazantinib | 10 | 32.8 | PR |
| 1001-41b | HR+/HER2− breast cancer | FGFR2 rearrangement | 13 | Palbociclib; SYK/FLT3 inhibitord; PIM kinase inhibitord | 12 | 12.1 | PR |
| 2001-50 | HR+/HER2− breast cancer | None | 6 | None | 10 | 25 | PR |
| 1001-74b | TNBC | None | 8 | None | 12 | 16.1 | PR |
| 1001-64b | Prostate adenocarcinoma | FGFR2 amplification | 13 | SUMO-activating enzyme inhibitord | 12 | 21.9 | PR |
| 1001-79b | Cholangiocarcinoma | FGFR2-CCDC6.F17C2c | 4 | Pemigatinib | 12 | 28 | PR |
| 1001-34 | Cholangiocarcinoma | FGFR2-AHCYL1.F17A2 | 4 | Infigratinib | 10 | 33.9 | SD |
| 1001-15 | HR+/HER2− breast cancer | None | 7 | NO synthase inhibitord | 8 | 26.1 | SD |
| 2001-24 | TNBC | None | 3 | None | 5 | 24.6 | SD |
| 1001-66b | Rectum adenocarcinoma | FGFR1 amplification | 5 | Pemigatinib | 12 | 26 | SD |
| 1001-67b | Epithelioid mesothelioma | None | 3 | Bevacizumab | 12 | 50.4 | SD |
| 1001-05 | Salivary gland adenocarcinoma | None | 3 | Ceritinib; abemaciclib | 5 | 36.4 | SD |
| Patient characteristics | Tinengotinib | Best response by RECIST version 1.1 | |||||
|---|---|---|---|---|---|---|---|
| Cancer type | FGFR alterationsa | Prior systemic therapies, n | Previous targeted therapies | Dose, mg/day | Duration of treatment, weeks | ||
| 1001-25 | Cholangiocarcinoma | FGFR2 N549K, FGFR2-KCNH7 fusion | 3 | Derazantinib | 8 | 38.3 | PR |
| 1001-37 | Cholangiocarcinoma | FGFR2 C382R | 3 | Derazantinib | 10 | 32.8 | PR |
| 1001-41b | HR+/HER2− breast cancer | FGFR2 rearrangement | 13 | Palbociclib; SYK/FLT3 inhibitord; PIM kinase inhibitord | 12 | 12.1 | PR |
| 2001-50 | HR+/HER2− breast cancer | None | 6 | None | 10 | 25 | PR |
| 1001-74b | TNBC | None | 8 | None | 12 | 16.1 | PR |
| 1001-64b | Prostate adenocarcinoma | FGFR2 amplification | 13 | SUMO-activating enzyme inhibitord | 12 | 21.9 | PR |
| 1001-79b | Cholangiocarcinoma | FGFR2-CCDC6.F17C2c | 4 | Pemigatinib | 12 | 28 | PR |
| 1001-34 | Cholangiocarcinoma | FGFR2-AHCYL1.F17A2 | 4 | Infigratinib | 10 | 33.9 | SD |
| 1001-15 | HR+/HER2− breast cancer | None | 7 | NO synthase inhibitord | 8 | 26.1 | SD |
| 2001-24 | TNBC | None | 3 | None | 5 | 24.6 | SD |
| 1001-66b | Rectum adenocarcinoma | FGFR1 amplification | 5 | Pemigatinib | 12 | 26 | SD |
| 1001-67b | Epithelioid mesothelioma | None | 3 | Bevacizumab | 12 | 50.4 | SD |
| 1001-05 | Salivary gland adenocarcinoma | None | 3 | Ceritinib; abemaciclib | 5 | 36.4 | SD |
Abbreviations: DRDE, dose recommended for dose expansion; FGFR, fibroblast growth factor receptor; FLT3, FMS-like tyrosine kinase 3; HER2−, human epidermal growth factor 2-negative; HR+, hormone receptor-positive; NO, nitric oxide; PIM, proviral integration site of Moloney murine leukemia virus; PR, partial response; RECIST, Response Evaluation Criteria in Solid Tumors; SD, stable disease; SUMO, small ubiquitin-like modifier; SYK, spleen tyrosine kinase; TNBC, triple-negative breast cancer.
aIndicates historical FGFR mutational status obtained from medical records review.
bIndicates patient treated at DRDE.
cIndicates FGFR mutational status obtained from next-generation sequencing performed during study screening and prior to study entry.
dInvestigational agent under study.
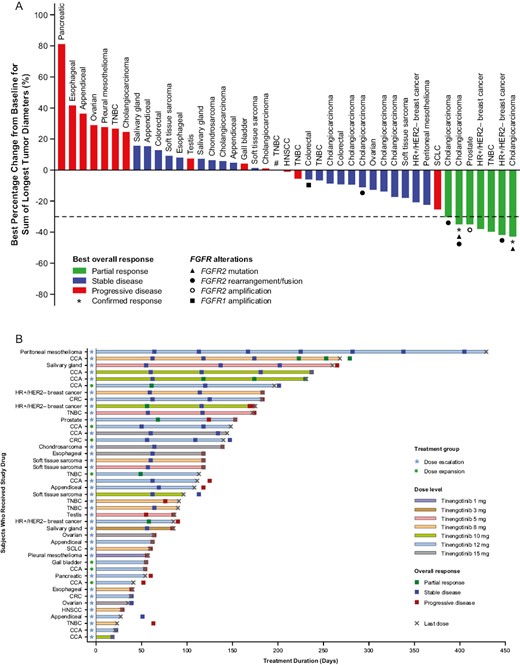
(A) Waterfall plot depicting best overall RECIST responsea (efficacy analysis set); (B) Swimmer plot showing duration of therapy until last dose or data-cut of September 9, 2022 (efficacy analysis set). aForty-three of 48 patients had at least one post-baseline scan and were evaluable for response. Dotted line shows 30% decrease in tumor size by RECIST version 1.1. #Patient had no target lesions at baseline and their best overall response was SD. Abbreviations: CCA, cholangiocarcinoma; CRC, colorectal; HER2−, human epidermal growth factor 2-negative; HNSCC, head and neck squamous cell carcinoma; HR+, hormone receptor-positive; RECIST, Response Evaluation Criteria in Solid Tumors; SCLC, small cell lung cancer; TNBC, triple-negative breast cancer.
Among the 11 efficacy-evaluable patients with cholangiocarcinoma, PRs were noted in 3 patients and SD lasting ≥24 weeks was observed in one other. All 4 patients had FGFR2 alterations [FGFR2 fusions (n = 2), FGFR2 fusion and FGFR2 mutation (n = 1), and FGFR2 mutation (n = 1)] documented either historically from medical records (n = 3) or prospectively by next-generation sequencing at baseline (n = 1; see Table 3 for details). All 4 patients had been previously treated with one or more FGFR tyrosine kinase inhibitor. Further PRs were noted in 2 of 3 efficacy-evaluable patients with HR-positive/HER2-negative breast cancer, with SD lasting ≥24 weeks in one other; PRs were noted in 1 of 5 patients with TNBC and one patient with CRPC; an additional SD lasting ≥24 weeks was observed in a patient with TNBC.
Pharmacokinetics
Preliminary clinical pharmacokinetics were analyzed in 40 evaluable patients who received at least one dose of tinengotinib and had at least one post-baseline pharmacokinetic sample collected (Supplementary Table S5). Mean tinengotinib plasma concentrations (linear and semi-logarithmic forms) on day 28 of cycle 1 are shown in Fig. 2. Following oral administration, Tmax was attained after ~2-5 hours on average. Mean t1/2 was measurable for doses of 8 mg and above and ranged between 28 and 34 hours. Consistent with the observed t1/2 and daily dosing regimen, mean accumulation ratios for AUC0-24 ranged from 2.5 to 2.8 for all doses. Exposure was observed to increase in a less-than-proportional manner, and mean AUC0-24 values for both single doses and at steady state for the 15 mg dose were approximately twice those observed for the 5 mg dose (1.98 and 1.91, respectively).
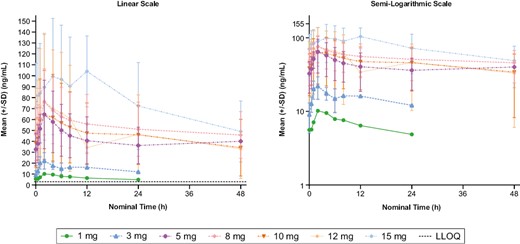
Mean (±SD) plasma concentration-time plots of multiple-dose tinengotinib on cycle 1 day 28 by dose (dose-escalation phase). In lower-dose cohorts (1-5 mg), the multiple-dose plasma concentrations were measured over 24 hours; in other dose cohorts, concentrations were measured over 48 hours. Abbreviations: LLOQ, lower limit of quantification; SD, standard deviation.
Exploratory Biomarker and Pharmacodynamic Analyses
Prospective biomarker data were available for one patient with a PR who had cholangiocarcinoma harboring an FGFR2 fusion. Between baseline and cycle 3 day 1, the patient had a 4.8-fold increase in plasma FGF-23 (a marker of FGFR inhibition) levels, which was accompanied by 30% reduction in target tumor size (Supplementary Fig. S3). Additionally, tinengotinib displayed robust antitumor activity and achieved 80% TGI in the CC6204 PDX FGFR2-BICC1 gene fusion model (Supplementary Fig. S4), suggesting its potential clinical application in patients with FGFR2 fusion-positive cholangiocarcinoma.
The median inhibitory concentration (IC50) for pemigatinib, the positive control in the KATO III cell line model, was 4.4 nM, consistent with published data.11 As shown in Supplementary Table S6, the IC50, IC70, and IC85 values for tinengotinib were 16.5 nM (6.51 ng/mL), 37.85 nM (14.95 ng/mL), and 90.28 nM (35.65 ng/mL), respectively, in the same assay. The steady-state Cmin of 5 mg of tinengotinib was 32.7 ng/mL and the steady-state Cmin of 12 mg/day of tinengotinib was 46.0 ng/mL as shown in Supplementary Table S5.
Discussion
This first-in-human phase I study evaluated tinengotinib, a novel multi-target tyrosine kinase inhibitor targeting FGFR 1-3, JAK 1 and 2, VEGFRs, and Aurora A and B, in patients with advanced solid tumors. The DRDE was established at 12 mg orally once daily on a 28-day cycle. The most common drug-related adverse events were vascular (hypertension), gastrointestinal (diarrhea, stomatitis, nausea, vomiting), and dermatologic (palmar-plantar erythrodysesthesia syndrome), a safety profile that is consistent with other multi-kinase inhibitors that target FGFRs and VEGFRs.12,13 Most drug-related adverse events associated with tinengotinib were mild-to-moderate in severity, and manageable with dose modifications or standard therapies.
Hypertension, a well-documented on-target effect of VEGF inhibition,14 was the most frequently reported adverse event with tinengotinib. An exposure-response model suggested that the risk of grade ≥3 hypertension was uniform and consistent across all doses (Supplementary Fig. S2). Treatment-emergent hypertension was effectively managed with dose modifications and/or antihypertensive agents in this study; however, a more proactive approach has been adopted in ongoing and future clinical studies to minimize unnecessary dose interruptions. This includes the exclusion of patients with uncontrolled hypertension at screening (defined as systolic blood pressure ≥150 mmHg and/or diastolic blood pressure ≥90 mmHg), and detailed guidelines for the management of treatment-emergent hypertension (ie, daily monitoring and reporting of blood pressure, and guidelines for tinengotinib dose modifications and the use of antihypertensive agents by CTCAE grade).
To date, a number of Aurora kinase inhibitors have been evaluated in clinical trials.15 The most commonly observed adverse events in those trials were febrile neutropenia and stomatitis/mucosal inflammation, which were considered to be mechanism-based toxicities for this class of inhibitors.15,16 As a potent pan-Aurora inhibitor, tinengotinib resulted in myelosuppression including a reduction in neutrophils, monocytes, and lymphocytes in preclinical toxicity studies (in-house source data). Interestingly, although stomatitis was reported in 9 (28.1%) of the patients in the phase II study (NCT04919642)17 and 14 (29.2%) in this study, hematologic toxicity was uncommon in both studies. This might be due to reduced bone marrow distribution of the drug in human subjects, but further investigation is needed to determine the reason for this observation.
Preclinical pharmacokinetic/pharmacodynamic (PK/PD) models indicated that the lowest active dose of tinengotinib was 5 mg/kg, which resulted in 61.5% TGI and an AUC of 707 h·ng/mL.3 In the present study, steady-state exposure (AUC) at a dose of 5 mg/day was 1053 h·ng/mL, and the exposure of AUC at a dose of 12 mg/day was 1374.4 h·ng/mL, which exceeded the minimal efficacious AUC of 707 h·ng/mL defined in preclinical PK/PD models.3 The IC70 and IC85 values and Cmin at 5 mg/day and 12 mg/day suggest that sufficient FGFR2 inhibition (>70%) can be maintained if tinengotinib is dosed at ≥5 mg once daily or FGFR2 inhibition >85% can be maintained if tinengotinib is dosed at ≥12 mg once daily. This dose level was also accompanied by disease control in some patients, suggesting that the minimum dose associated with clinical benefit is 5 mg/day. Although exposure to tinengotinib increased in a less-than-proportional manner with dose, the efficacious range could be defined from 5 mg/day to 12 mg/day. Two ongoing trials of tinengotinib (Clinicaltrials.gov NCT04742959 and NCT04919642) include a pharmacokinetic run-in study, intensive pharmacokinetic sample collection to determine how the body metabolizes tinengotinib, and additional sample collection to generate pooled pharmacokinetic parameters of tinengotinib in a larger population.
Tinengotinib showed encouraging preliminary antitumor activity in heavily pretreated patients with a variety of advanced solid tumors including cholangiocarcinoma, HR-positive/HER2-negative breast cancer, TNBC, and CRPC. A notable observation was the strong efficacy signal with tinengotinib in patients with cholangiocarcinoma despite those patients having had 3-5 prior lines of standard therapy. All 4 patients who achieved a PR or SD ≥24 weeks with tinengotinib had FGFR2 genomic alterations (rearrangements, fusions, or mutations) at baseline and had been previously treated with an FGFR inhibitor. It has been reported that a spectrum of FGFR alterations occurs in cholangiocarcinoma through amplification, activating mutations, or fusions via gene rearrangements.18,19 The development of FGFR inhibitors has led to approvals of pemigatinib, infigratinib, and futibatinib for patients with FGFR2 fusion or rearranged cholangiocarcinoma after failure of first-line chemotherapy based on objective response rates of 36%, 23%, and 42%, respectively.20-22 Although FGFR inhibitors have clinical efficacy in cholangiocarcinoma harboring FGFR2 fusions, many patients ultimately develop disease progression after 7-9 months20,23 as a result of secondary mutations in the FGFR2 kinase domain. Treatment of FGFR inhibitor-refractory cholangiocarcinoma represents an area of unmet need.19,24 It is notable that tinengotinib, as a multi-target tyrosine kinase inhibitor, demonstrated inhibitory activity against FGFR kinases in vitro3 as well as in patients harboring FGFR2 kinase domain (N549K) or transmembrane domain (C382R) mutations in the present study. Supportive biomarker data were also documented in one patient with cholangiocarcinoma after tinengotinib therapy that showed a 4.8-fold increase in FGF-23 (a marker of FGFR inhibition) plasma levels accompanied by 30% reduction in target tumor size at cycle 3 day 1. Based on these encouraging findings, tinengotinib has received a fast-track designation from the US Food and Drug Administration for further development in cholangiocarcinoma. A phase II study is underway to better understand the underlying mechanisms of response and resistance to tinengotinib in patients with FGFR-altered or FGFR-wild-type cholangiocarcinoma (Clinicaltrials.gov, NCT04919642). That study will address the limitations of this study, in which biomarker data regarding FGFR status were collected retrospectively from historical records in most patients, and includes biomarker profiling at baseline, during treatment, and upon response/progression. That study will investigate tinengotinib monotherapy in patients with cholangiocarcinoma with FGFR2 fusions for whom prior FGFR inhibitor therapy was unsuccessful, or those who responded to previous FGFR inhibitor(s), or with other FGFR alterations, or wild-type FGFR.
The efficacy signal in patients with TNBC is also encouraging and consistent with preclinical studies that identified tinengotinib as a potential candidate for this type of tumor.3 TNBC encompasses multiple subtypes characterized by distinct genetic drivers, including Aurora A and B kinase in the basal-like 1 subtype, and FGFR, PDGFR, and VEGF signaling pathways in the mesenchymal and mesenchymal stem-like subtypes.25 In a preclinical study, exposure to tinengotinib inhibited proliferation across all subtypes of TNBC in vitro and in vivo, but did not inhibit breast cancer luminal cell lines in vitro.3 Inhibition of Aurora A and B kinase activity was identified as the predominant mechanism of action of tinengotinib in TNBC, with inhibition of other pathways (VEGFR, JAK-STAT, CSF1R, and epidermal growth factor receptor) further contributing to its activity.3 FGF/FGFR signaling also plays a pivotal role in prostate cancer and supports FGF- or FGFR-targeted therapeutic strategies for the treatment of prostate cancer.26 Based on the PR observed in a patient with CRPC in this study, a phase Ib/II study of tinengotinib, which includes patients with TNBC and CRPC, has recently been initiated (ClinicalTrials.gov: NCT04742959).
The broad spectrum of activity of tinengotinib, in particular, dual inhibition of JAK-STAT and FGFR signaling and blockade of lineage plasticity, may be a factor in the responders as efficacy assessments observed in patients with HR-positive/HER2-negative breast cancer, TNBC, and metastatic CRPC (mCRPC), among others, who did not have FGFR alterations at study entry. As previously reported, dual inhibition of FGFR and JAK might benefit patients with mCRPC via lineage reprogramming.27 Detailed molecular studies aiming to illustrate pharmacological effects subsequent to JAK-STAT/FGFR inhibition are being conducted in models of mCRPC, and a clinical proof-of-concept study of tinengotinib plus hormonal therapies to treat mCRPC is being planned. Although the underlying molecular mechanism for tinengotinib activity in patients with breast cancer is unclear, there is growing recognition that lineage plasticity plays a critical role in relapse and drug resistance.28,29 In TNBC, tumors that are induced to acquire vulnerability by Aurora inhibition may be subsequently targeted and killed by inhibition of additional signal pathways. Thus, targeting a number of cancer hallmarks including proliferation, angiogenesis, epithelial-mesenchymal transition, and immune oncology through the inhibition of several prominent oncogenic pathways may be mechanisms for the activity of tinengotinib in patients with FGFR-unaltered breast and prostate cancers. Further studies are needed to confirm these hypotheses.
Some limitations of the present study warrant consideration. These include the limited collection of PK/PD analyses. The ongoing phase Ib/II trial includes one arm specifically designed to evaluate the pharmacokinetics of tinengotinib at a range of doses and using several dosing schedules, with a pharmacokinetics run-in period. Another limitation is the fact that correlative biomarker studies of markers associated with response were not conducted because of the explorative nature of the study. Biomarker analysis of patients with non-FGFR alterations would be of particular interest and may further shed light on other mechanisms involved in response to tinengotinib in our patient population. The phase Ib/II study will also evaluate patient biomarker status, including FGFR mutation status.
Conclusion
In conclusion, tinengotinib was safe and well-tolerated. Based on the totality of safety, efficacy, and PK/PD modeling, the DRDE of 12 mg was established, which exceeded threshold pharmacokinetic levels for efficacy. The preliminary responses observed in patients with cholangiocarcinoma harboring FGFR2 fusions and/or mutations, HER2-negative breast cancer including TNBC, and CRPC form the basis for further investigations of tinengotinib. Phase II trials have been initiated to evaluate the safety and efficacy of tinengotinib as a single agent or as a combination, and to explore biomarkers for response.
Supplementary Material
Supplementary material is available at The Oncologist online.
Acknowledgments
We acknowledge the investigative center study staff, the study patients, and their families. Support for the development of this article, including medical writing and editorial assistance, under the authors’ guidance, was provided by Shu Xiao, PharmD, PhD, funded by CRC Oncology, and Miller Medical Communications, Ltd., funded by CRC Oncology. The statistical analysis was provided by Pharmaron (formerly CR Medicon).
Funding
This study was sponsored by TransThera Sciences (Nanjing), Inc.
Conflict of Interest
Sarina A. Piha-Paul reports research support/grant funding (inst) from AbbVie, Inc., ABM Therapeutics, Inc., Acepodia, Inc, Alkermes, Aminex Therapeutics, Amphivena Therapeutics, Inc., BioMarin Pharmaceutical, Inc., Boehringer Ingelheim, Bristol Myers Squib, Cerulean Pharma, Inc., Chugai Pharmaceutical Co., Ltd, Curis, Inc., Cyclacel Pharmaceuticals, Daiichi Sankyo, Eli Lilly, ENB Therapeutics, Five Prime Therapeutics, F-Star Beta Limited, F-Star Therapeutics, Gene Quantum, Genmab A/S, GlaxoSmithKline, Helix BioPharma Corp., HiberCell, Inc., Incyte Corp., Jacobio Pharmaceuticals Co., Ltd., Lytix Biopharma AS, Medimmune, LLC., Medivation, Inc., Merck Sharp and Dohme Corp., Novartis Pharmaceuticals, Pieris Pharmaceuticals, Inc., Pfizer, Principia Biopharma, Inc., Puma Biotechnology, Inc., Rapt Therapeutics, Inc., Seattle Genetics, Silverback Therapeutics, Taiho Oncology, Tesaro, Inc., TransThera Bio, NCI/NIH, P30CA016672—Core Grant (CCSG Shared Resources). She acts as a consultant for CRC Oncology. Binghe Xu reports participating in speakers’ bureau for AstraZeneca, Eisai, Pfizer, Roche and receiving research funding from Jiangsu Hengrui Medicine (Inst). Ecaterina E. Dumbrava reports research or grant funding from Bayer HealthCare Pharmaceuticals Inc, Immunocore LTD, Amgen, Aileron Therapeutics, Compugen Ltd, TRACON Pharmaceuticals Inc, Unum Therapeutics, Gilead Immunomedics, BOLT Therapeutics, Aprea Therapeutics, Bellicum Pharmaceuticals, PMV Pharma, Triumvira, Seagen Inc, Mereo BioPharma 5 Inc, Sanofi, Rain Oncology, Astex Therapeutics, Sotio, Poseida, Mersana Therapeutics, Genentech, Boehringer Ingelheim; advisory board participation for BOLT Therapeutics, Mersana Therapeutics, Orum Therapeutics, Summit Therapeutics. She is an unpaid consultant for Catamaran Bio, speaker for PMV Pharma and has received travel, accommodations, expenses funding from ASCO, LFSA Association. Siqing Fu reports research funding from Abbisko (Inst), Anaeropharma (Inst), Arrien Pharmaceuticals (Inst), AstraZeneca (Inst), BeiGene (Inst), BioAtla (Inst), Boehringer Ingelheim (Inst), Hookipa Pharma (Inst), Huya Bioscience International (Inst), IMV (Inst), Innovent Biologics (Inst), Lilly (Inst), Lyvgen Biopharma (Inst), MacroGenics (Inst), Medivir (Inst), NCCN (Inst), NCCN (Inst), Nerviano Medical Sciences (Inst), NeuPharma, Inc. (Inst), NIH/NCI (Inst), Novartis (Inst), NovoCure (Inst), OncoMed (Inst), Parexel International, LLC (Inst), Sellas Life Sciences (Inst), Soricimed (Inst), Taiho Oncology (Inst), Tolero Pharmaceuticals (Inst), and Turnstone Bio (Inst). Daniel D. Karp has no financial relationships to disclose. Funda Meric-Bernstam is employed by the MD Anderson Cancer Center and reports honoraria from Mayo Clinic, Rutgers Cancer Institute of New Jersey, consulting or Advisory Role for Aduro Biotech, Alkermes, Debiopharm Group, eFFECTOR Therapeutics, Genentech, IBM Watson Health, Immunomedics, Inflection Biosciences, Jackson Laboratory for Genomic Medicine, Kolon Life Sciences, Mersana, Origimed, PACT Pharma, Parexel International, Pfizer, Puma Biotechnology, Roche, Samsung Bioepis, Seattle Genetics, Silverback Therapeutics, Spectrum Pharmaceuticals, Tyra Biosciences, Xencor, Zentalis, and Zymeworks; speakers’ bureau for Chugai Pharma, research funding from AbbVie (Inst), Aileron Therapeutics (Inst), AstraZeneca (Inst), Bayer (Inst), Boehringer Ingelheim (I), Calithera Biosciences (Inst), Curis (Inst), CytomX Therapeutics (Inst), Daiichi Sankyo (Inst), Debiopharm Group (Inst), eFFECTOR Therapeutics (Inst), Genentech (Inst), GlaxoSmithKline (Inst), Guardant Health (Inst), Jounce Therapeutics (Inst), Millennium (Inst), Novartis (Inst), Pfizer (Inst), PUMA Biotechnology (Inst), Seattle Genetics (Inst), Taiho Pharmaceutical (Inst), and Zymeworks (Inst); and speakers’ bureau for Chugai Pharma. She reports research funding from AbbVie (Inst), Aileron Therapeutics (Inst), AstraZeneca (Inst), Bayer (Inst), Boehringer Ingelheim (I), Calithera Biosciences (Inst), Curis (Inst), CytomX Therapeutics (Inst), Daiichi Sankyo (Inst), Debiopharm Group (Inst), eFFECTOR Therapeutics (Inst), Genentech (Inst), GlaxoSmithKline (Inst), Guardant Health (Inst), Jounce Therapeutics (Inst), Millennium (Inst), Novartis (Inst), Pfizer (Inst), PUMA Biotechnology (Inst), Seattle Genetics (Inst), Taiho Pharmaceutical (Inst), and Zymeworks (Inst); and travel, accommodation and expenses from Beth Israel Deaconess Medical Center and Taiho Pharmaceutical. David S. Hong has stock and other ownership interests in MolecularMatch, OncoResponse, and Presagia; and reports consulting or advisory role activities for Acuta, Adaptimmune, Alphasights, Amgen, Axiom Biotechnologies, Baxter, Bayer, Boxer Capital, COG, EcoR1 Capital, Genentech, GLG, GroupH, Guidepoint Global, H.C. Wainwright & Co., HCW Precision, Infinity Pharmaceuticals, Janssen, Medscape, Merrimack, Numab, Pfizer, Prime Oncology, Seattle Genetics, ST Cube, Takeda, Tavistock Life Sciences, Trieza Therapeutics, and WebMD. He reports research funding from Abbvie (Inst), Adaptimmune (Inst), Aldai Norte (Inst), Amgen (Inst), AstraZeneca (Inst), Bayer (Inst), BMS (Inst), Daiichi Sankyo (Inst), Eisai (Inst), Erasca, Inc (Inst), Fate Therapeutics (Inst), Genentech (Inst), Genmab (Inst), Ignyta (Inst), Infinity Pharmaceuticals (Inst), Kite, a Gilead company (Inst), Kyowa (Inst), Lilly (Inst), Loxo (Inst), MedImmune (Inst), Merck (Inst), Mirati Therapeutics (Inst), miRNA Therapeutics (Inst), Molecular Templates (Inst), Mologen (Inst), National Cancer Institute (Inst), Navire (Inst), Novartis (Inst), Numab (Inst), Pfizer (Inst), Seattle Genetics (Inst), Takeda (Inst), Turning Point Therapeutics (Inst), Verastem (Inst), and VM Pharma (Inst); and travel, accommodation and expenses from AACR, ASCO, Bayer Schering Pharma, Genmab, Loxo, miRNA Therapeutics, and Society for Immunotherapy of Cancer. Jordi A. Rodon reports consulting or advisory role from Ellipses Pharma, Ionctura, Kelun, Lilly, Merck Sharp & Dohme, Molecular Partners, Novartis, NovellusDx, Orion, Peptomyc, Pfizer, Roche/Genentech, SERVIER, and Spectrum Pharmaceuticals; research funding from Bayer, BioAtla, Blueprint Medicines, CytomX Therapeutics, Genmab, GlaxoSmithKline, Ipsen, Kelun, Novartis, Pfizer, Spectrum Pharmaceuticals, Symphogen, Takeda/Millennium, Tocagen; travel, accommodations, and expenses from Cancer Core Europe, Department of Defense, ESMO, Huntsman Cancer Institute, Karolinska Cancer Institute, King Abdullah International Medical Research Center, Louisiana State University, and Molecular Partners; and other relationships with Chinese University of Hong Kong; Elsevier; European Journal of Cancer; GlaxoSmithKline; SOLTI; Vall d’Hebron University Hospital Institute of Oncology. Apostolia M. Tsimberidou reports consulting or advisory role for Tempus, research funding from Agenus (Inst), Boston Biomedical (Inst), IMMATICS (Inst), Karus Therapeutics (Inst), OBI Pharma (Inst), Parker Institute for Cancer Immunotherapy (Inst), Placon (Inst), Stem Cells, Inc. (Inst), and Tempus (Inst); travel, accommodations, expenses from the Japanese Society of Clinical Oncology. Kanwal Raghav reports consulting or advisory role for AstraZeneca, Bayer, Daiichi Sankyo, and Eisai; Speakers’ Bureau for Bayer; and research funding from Bayer (Inst), Daiichi Sankyo/Lilly (Inst), Guardant Health (Inst), and Roche/Genentech (Inst). Jaffer A. Ajani reports honoraria from Acrotech Biopharma, Aduro Biotech, Amgen, Astellas Pharma, AstraZeneca, BeiGene, Boehringer Ingelheim, Bristol-Myers Squibb, Daiichi Sankyo, Dava Pharmaceuticals, Fresenius Kabi, Gilead Sciences, GRAIL, Lilly, Merck, Novartis, Oncotherics, Servier, and Zymeworks; consulting or advisory role for the American Cancer Society, BeiGene, Bristol-Myers Squibb, Insys Therapeutics, Merck, and Vaccinogen; research funding from Amgen, Astellas Pharma (Inst), Bristol-Myers Squibb, Daiichi Sankyo, Delta-Fly Pharma, Gilead Sciences, Lilly/ImClone, Merck, Novartis, ProLynx, Roche/Genentech, Taiho Pharmaceutical, Takeda, and Zymeworks; and Patents, Royalties, Other Intellectual Property: research funding from: Genentech, Roche, BMS, Taiho, MedImmune, Merck, Amgen, Lilly. Anthony P. Conley reports consulting or advisory role for Bayer, Deciphera, Genentech, Guidepoint Global, InhibRx, Medscape, Nektar, Novartis, OncLive, and Schlesinger Associates; research funding from Bavarian Nordic (Inst), Ignyta (Inst), Iovance Biotherapeutics (Inst), MedImmune (Inst), and NantHealth (Inst); and travel, accommodations, and expenses from Genentech. Frank Mott has no financial relationships to disclose. Ying Fan has no financial relationships to disclose. Jean Fan is employed by TransThera Sciences US, Inc. and has stock and other ownership interests in AstraZeneca. Peng Peng is employed by, has stock and other ownership interests in, and has patents, royalties, and other intellectual property in TransThera Sciences (Nanjing), Inc. Hui Wang is employed by TransThera Sciences (Nanjing), Inc. Shumao Ni is employed by TransThera Sciences (Nanjing), Inc. Caixia Sun is employed by TransThera Sciences (Nanjing), Inc. Xiaoyan Qiang is employed by TransThera Sciences (Nanjing), Inc. Wendy J. Levin is employed by and acts as a consultant for WJ Levin Consulting. Brenda Ngo has no financial relationships to disclose. Qinhua Cindy Ru is employed by and has stock and other ownership interests in CRC Oncology Corp. She reports consulting or advisory role for ABM Therapeutics (Inst), Cellular Biomedicine Group (Inst), Phanes Therapeutics (Inst), and Help Therapeutics. Frank Wu is employed by and has patents, royalties and other intellectual property in TransThera Sciences (Nanjing), Inc. Milind M. Javle reports consulting or advisory role for AstraZeneca, EMD Serono, Incyte, Merck, Mundipharma EDO GmbH, OncoSil, and QED Therapeutics and other relationships with Bayer, BeiGene, Incyte, Merck, Merck Serono, Novartis, Pieris Pharmaceuticals, QED Therapeutics, Rafael Pharmaceuticals, and Seattle Genetics, and advisory board or research funding from Abbvie, Array, Astellas, AstraZeneca, Bayer, Beigene, Biocartis, Boehringer Ingelheim, Bristol-Myers Squibb, Celgene, Daiichi, GSK, Halozyme, Helsinn, Incyte, Ipsen, Janssen Research, Lilly, Merck Sharp & Dohme, EMD Serono, Novartis, Transthera, Meclun, Eli Lilly, Oncosil, QED, Taiho, Servier, Agios.
Author Contributions
Conception/design: S.A.P.-P., B.X., P.P., Q.C.R., F.W., M.M.J. Provision of study material or patients: S.A.P.-P., B.X., E.E.D., S.F., D.D.K., F.M.-B., D.S.H., J.A.R., A.M.T., K.R., J.A.A., A.P.C., F.M., Y.F., J.F., P.P., H.W., C.S., W.J.L., B.N., M.M.J. Collection and/or assembly of data: S.A.P.-P., B.X., E.E.D., S.F., D.D.K., F.M.-B., D.S.H., J.A.R., A.M.T., K.R., J.A.A., A.P.C., F.M., Y.F., J.F., H.W., S.N., C.S., X.Q., M.M.J., and CRC Oncology. Data analysis and interpretation: All authors. Manuscript writing and final approval of manuscript: All authors.
Data Availability
Anonymized individual participant data that underlie the results reported in this article (text, tables, figures, and appendices) and supporting documents (study protocol) may be available upon request beginning 9 months and ending 36 months following article publication. Only requests that have a methodologically sound proposal will be considered. Proposals should be directed to [email protected].

{kind=link}
{kind=link}