






Abstract
Disseminated gonococcal infection (DGI) is a rare complication caused by the systemic dissemination of Neisseria gonorrhoeae to normally sterile anatomical sites. Little is known about the genetic diversity of DGI gonococcal strains and how they relate to other gonococcal strains causing uncomplicated mucosal infections. We used whole genome sequencing to characterize DGI isolates (n = 30) collected from a surveillance system in Georgia, United States, during 2017–2020 to understand phylogenetic clustering among DGI as well as uncomplicated uro- and extragenital gonococcal infection (UGI) isolates (n = 110) collected in Fulton County, Georgia, during 2017–2019. We also investigated the presence or absence of genetic markers related to antimicrobial resistance (AMR) as well as surveyed the genomes for putative virulence genetic factors associated with normal human-serum (NHS) resistance that might facilitate DGI. We found that DGI strains demonstrated significant genetic variability similar to the population structure of isolates causing UGI, with sporadic incidences of geographically clustered DGI strains. DGI isolates contained various AMR markers and genetic mechanisms associated with NHS resistance. DGI isolates had a higher frequency of the porB1A allele compared with UGI (67% vs 9%, P < .0001); however, no single NHS resistance marker was found in all DGI isolates. Continued DGI surveillance with genome-based characterization of DGI isolates is necessary to better understand specific factors that promote systemic dissemination.
Disseminated gonococcal infection (DGI) is a serious condition caused by the systemic spread of Neisseria gonorrhoeae (Ng) and characterized by the presentation of septic arthritis, tenosynovitis, endocarditis, skin lesions, and various other symptoms [1]. Although the pathogenesis of DGI is poorly understood, systemic spread of the organism is thought to occur when Ng presents at mucosal sites of infection, invades epithelial tissues, and enters the bloodstream. Notably, the complement cascade plays an important role in the innate immune response to Ng and complement deficiencies result in elevated risk for dissemination [2,3]. Several genetic loci (eg, the gonococcal genetic island [GGI], the porB1A allele, the lipo-oligosaccharide [LOS] glycosyltransferase [lgt] ABCDE operon, and the opa genes) in Ng have been associated with normal human-serum (NHS) resistance [4–10]. However, no specific genetic mechanism has emerged as being required for dissemination.
Historically, DGI cases were more commonly reported in females, but recent investigations have indicated that the proportion of cases in males and females is similar [11]. In addition, studies from the 1980s indicated that isolates recovered from DGI cases shared various phenotypic characteristics such as a biochemical/nutritional requirement for arginine, hypoxanthine, and uracil [12]. Moreover, these isolates were highly susceptible to penicillin and were shown (using serological methods) to express PorB1A (previously termed protein 1A antigen). These studies suggested that specific DGI causing strains may exist within the Ng strain population, although few genetic studies have been conducted to evaluate this possibility.
In 2019, a large cluster of DGI cases was detected in southwest Michigan [13]. Of 16 cases reported, more than half (n = 9) were male and 81% (n = 13) had septic arthritis. Whole genome sequencing (WGS) analysis revealed that 11 isolates recovered from these cases were closely genetically related, sharing <50 single-nucleotide polymorphisms (SNPs) differences overall. A separate study examining a DGI isolate recovered from a prosthetic joint infection in the United States (US) revealed that the isolate belonged to a large global phylogenetic lineage of antibiotic-susceptible isolates [14]. This isolate did not cluster with various genetically distinct DGI isolates described in a study from Australia, suggesting that DGI isolates may be genetically diverse overall [15].
To account for potential genetic differences due to collection of Ng isolates from different geographical regions and time points, we sought to utilize existing surveillance systems to conduct a genetic analysis of Ng isolates from DGI and uncomplicated cases of uro- and extragenital gonococcal infection (UGI). We hypothesized that there is a population of Ng strains that only cause DGI and not UGI within a defined geographical region.
METHODS
Isolate Selection
The Active Bacterial Core surveillance system is a core component of the Centers for Disease Control and Prevention’s (CDC) Emerging Infections Program and monitors epidemiological trends for various pathogens including Haemophilus influenzae, Neisseria meningitidis, group A and B Streptococcus, and Streptococcus pneumoniae at 10 sites (including state health departments or academic institutions) across the US [16]. Special studies can also utilize this infrastructure, and since 2017, active surveillance for DGI has been conducted among residents in the 3-county San Francisco Bay area in California, the 20-county metro Atlanta, Georgia area (GA-MSA), and all counties outside the Atlanta metropolitan area (GA-DPH). Available DGI isolates from normally sterile anatomical sites from DGI cases identified among residents in these participating surveillance areas were sent to CDC for antimicrobial sensitivity testing and WGS. Most DGI isolates (n = 60) were from cases identified in GA-MSA and GA-DPH, and as a result, this genomic analysis was restricted to DGI isolates from those 2 surveillance areas. Weston et al [17] further describe details regarding DGI isolate collection and selection.
We further compared these genomes to those from UGI isolates collected in the Atlanta area as part of the Gonococcal Isolate Surveillance Project (GISP) or Strengthening the US Response to Resistant Gonorrhea (SURRG) program for calendar years 2017–2019 [18, 19]. GISP is a sentinel surveillance program that performs antimicrobial sensitivity testing on gonococcal urethral isolates from symptomatic men presenting to select sexually transmitted disease clinics across the US, data from which are used to inform CDC’s gonorrhea treatment guidelines [18]. SURRG is a rapid detection and response capacity building project that includes collecting specimens (including from patients of all genders and from genital and extragenital sites of infection) in specific public health jurisdictions, performing local rapid antimicrobial sensitivity testing, and investigating patients identified as having Ng infections with resistance or reduced antimicrobial susceptibility [19]. The availability of gonococcal isolates from these programs representing overlapping geographical regions and time of collection compared to the DGI isolates provided an ideal opportunity to explore the relationship of strains recovered from DGI and uncomplicated gonorrhea with limited introduction of confounding variables (eg, potential differences in locally circulating strain populations) associated with comparisons of isolates across distant geographical regions.
Thirty viable isolates obtained from normally sterile sites (blood, synovial fluid, etc) collected from either GA-MSA or GA-DPH from June 2017 to May 2020 underwent WGS. For comparison, 110 gonococcal isolates associated with uncomplicated gonococcal infections from nonsterile sites (UGI) were included. These isolates were collected in Fulton County, Georgia, between 2017 and 2019 by GISP and/or SURRG according to established protocols [16, 17].
Isolate Growth, DNA Extraction, and Whole Genome Sequencing
DGI isolates were stored at −80°C at CDC and subcultured on GC medium base + 1% isovitalex + 5% fetal bovine serum at 37°C and 5% carbon dioxide for 18–22 hours. DNA was isolated from fresh overnight cultures using the Wizard DNA Purification Kit (Promega, Madison, Wisconsin). WGS was performed on the MiSeq (Illumina, San Diego, California) platform using V2 reagents (2 × 250 bp). A subset of UGI isolates from GISP and SURRG was sequenced at the Tennessee State Public Health Laboratory, which is 1 of 4 regional laboratories in the CDC’s Antibiotic Resistance Laboratory Network that conducts WGS on Ng specimens. Long-read WGS was performed as described previously on a subset of 22 DGI isolates using the PacBio RSII (Pacific Biosciences, Menlo Park, California) at the CDC Biotechnology Core Facility Branch (Atlanta, Georgia) [20].
Genome Data and Phylogenetic Analysis
The quality of the Illumina reads was assessed using FastQC. Kraken version 0.10.5 was used to screen for contaminant reads and exclude samples with >10% reads matching to a nongonococcal organism [21]. Reads were trimmed using CutAdapt version 2.3 to remove adapter sequences and bases using a minimum quality cutoff of <Q30 and minimum length of 19 bp [22]. High-quality reads were de novo assembled using Spades version 3.9.0 using the “careful” option [23] (National Center for Biotechnology Information Sequence Read Archive accession numbers can be found in Supplementary Table 1).
To accurately observe nucleotide repeat regions that might not show in short-read sequencing (described below), the PacBio sequenced DGI isolates (n = 22) were assembled using the long-read assembler Flye version 2.8 and further polished by mapping Illumina reads by snippy version 4.3.8 [24].
For phylogenetic analyses, a full-length whole genome alignment of the 30 DGI isolates was first generated using snippy version 4.3.8 with FA19 (GenBank accession number CP012026.1) as the reference. Gubbins was used on the full-length whole genome alignment to identify and filter regions of homologous recombination [25]. The recombination masked SNP alignment was used as input for RAxML version 8.2.12 under the GTR + GAMMA model of nucleotide substitution with a majority-rule consensus convergence criterion to reconstruct an ascertainment-bias corrected (Stamatakis method) maximum likelihood (ML) phylogeny [26]. Fastbaps version 1.0.4 was used to define clades within the final ML phylogenetic tree [27]. A second phylogenetic analysis was conducted using the 30 DGI isolates in addition to the 110 UGI isolates as described above. Pairwise SNP distances between isolates were determined from the final core-genome SNP alignments using snp-dists version 0.7 (https://github.com/tseemann/snp-dists). A single gene phylogeny of all porB sequences from both the DGI and UGI isolates was also estimated by generating porB alignment using Muscle version 2.8 [28], inferred the evolutionary distances using the Hasegawa, Kishino, and Yan 85 substitution model followed by reconstructing a neighbor-joining tree using the PHYLIP package [29]. All phylogenetic trees were visualized and annotated using the R package ggtree [30].
Antimicrobial resistance (AMR) genotypic profiles were determined using the N. gonorrhoeae Genome Profiler (CDC), an in-house customized python script [31]. In brief, this script takes preprocessed sequencing reads and maps to genes of interest on the FA19 reference genome using breseq version 0.30 [32]. Multilocus sequence typing (MLST) and N gonorrhoeae multiantigen sequence typing schemes were determined using StringMLST [33] and NG-MASTER [34], respectively. NHS resistance genetic markers including porB, the GGI, the lgtABCDE operon, and the opa genes were also investigated within the DGI isolates. The NG-STAR database was used to determine porB allele types [35]. The Illumina contigs were used to determine the GGI allele type by comparing them against a customized Basic Local Alignment Search Tool (BLAST) database based on the GGI of gonococcal strain MS11 (AY803022.1) as the reference [36]. Similarly, the length of the homopolymer guanine (G) nucleotide repeats present in the lgtABCDE operon were determined by BLAST search of the Illumina-polished PacBio data against the reference FA19 lgtABCDE operon sequences. To further investigate the phase variation occurring in the lgtABCDE operon, we extracted the lgtA, lgtC, and lgtD genes from the 22 Illumina-polished PacBio DGI genomes. The sequences were translated, and the predicted protein (glycosyltransferase) was determined using Hidden Markov Model searches and aligning against the Pfam database [37]. Genes were determined to be phase-on and in-frame if the translated protein sequence produced a significant Hidden Markov Model motif alignment (E-value ≈ 0) with a functional glycosyltransferase protein matched in the Pfam database. Gene phase variation was determined by searching for presence or absence of premature stop codons and/or complete/incomplete functional glycosyltransferase protein. To determine the number of pentameric repeat units present in the opa genes, a BLAST search was conducted on the Illumina-polished PacBio data against reference FA1090 opa sequences. A multiple-sequence alignment was created in Geneious version 2019.1.1 to calculate pairwise sequence similarity and opaD phase variation was determined as described above. Fisher exact test was used to determine if significant associations existed when comparing the DGI isolates to the UGI isolates and the presence or absence of NHS genetic marker mentioned above.
RESULTS
DGI Isolates Were Genetically Diverse and Were Not Distinct From Isolates Collected From Uncomplicated Mucosal Gonorrhea
DGI isolates included in this study were recovered from blood (n = 21), synovial fluid (n = 8), or peritoneal fluid (n = 1) (Supplementary Table 1). A phylogenetic tree containing the 30 DGI isolates is shown in Figure 1. The average pairwise SNP distance across the entire phylogeny was 1999 ± 743 (average ± standard deviation) SNPs, indicating a high degree of genetic diversity among DGI isolates.
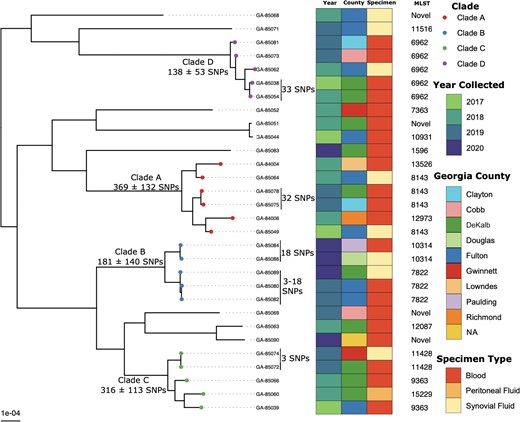
Maximum-likelihood phylogenetic tree based on core-genome single-nucleotide polymorphisms of 30 Neisseria gonorrhoeae disseminated gonococcal infection isolates collected from 2017 to 2020 in Georgia, United States. Abbreviations: GA, Georgia; MLST, multilocus sequence type; NA, not available; SNP, single-nucleotide polymorphism.
The DGI specific phylogeny was partitioned into 4 monophyletic clades with >2 isolates (clades A, B, C, and D; Figure 1). The largest clade, clade A (n = 6; 369 ± 132 SNPs), consisted of a genetically diverse group of isolates collected from different counties in Georgia during 2017–2019. There were 2 isolates in clade A, GA-85075 and GA-85078 (both MLST ST-8143), that were separated by only 32 SNPs and collected from 2 different counties (Clayton and DeKalb) within the Atlanta metropolitan area. In clade B, we observed 2 MLST types, ST-7822 (n = 3) and ST-10314 (n = 2), that formed separate subclades with intra-clade SNP distances of 181 ± 140 SNPs. Within this clade, we observed a cluster of 3 ST-7822 isolates (GA-85080, GA-85082, and GA-85089) that were separated by only 12 ± 7 SNPs and were collected from neighboring Fulton and DeKalb counties from December 2019 to May 2020. Similarly, clade C (n = 5) contained a subclade of 2 highly similar isolates (GA-85072 and GA-85074; 3 SNPs difference), both belonging to MLST ST-11428 and were isolated in May 2019 from neighboring DeKalb and Gwinnett counties.
To better understand the population structure of DGI isolates compared to UGI cases in Georgia, we reconstructed a phylogeny consisting of 30 DGI isolates and 110 UGI isolates collected from 2017–2019 (Figure 2). Fastbaps clustered the phylogeny into 16 different clades. The average pairwise SNP distance across the entire phylogeny was 1895 ± 630 SNPs. Of the 16 clades defined, 10 clades (63%) contained both DGI and UGI isolates that were phylogenetically clustered together, but the within-clade genetic diversity were highly variable. Overall, there were no distinct clades that contained only DGI isolates; instead, they were dispersed throughout the phylogeny. Clade 10 (n = 28) was the largest clade with both DGI (n = 6) and UGI (n = 22) isolates that formed 2 subclades. The larger subclade within clade 10 consisted of MLST ST-13526 with 16 UGI and 2 DGI isolates but has a high within-subclade genetic diversity (193 ± 129 SNPs). Similarly, the second MLST ST-8143 subclade consisted of 6 and 4 UGI and DGI isolates, respectively, but with a wide within-subclade genetic variability (234 ± 114 SNPs).
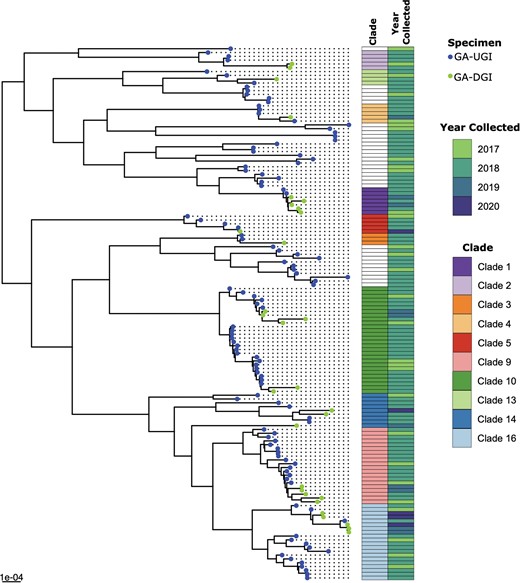
Maximum likelihood phylogenetic tree based on core-genome single-nucleotide polymorphisms (SNPs) of 30 disseminated gonococcal infection (DGI; 2017–2020) and 110 background urogenital and extragenital gonococcal infection (UGI; 2017–2019) isolates collected in Georgia (GA), United States.
Clades with both DGI and UGI isolates that belonged to the same MLST type were also observed. For example, clade 1 consisted of 7 MLST ST-6962 isolates, which were both DGI (n = 5) and UGI (n = 2), with an average pairwise SNP distance of 108 ± 45. All the clade 1 DGI isolates that formed a monophyletic clade in the DGI-only phylogenetic tree (clade D; Figure 1) also clustered similarly in the phylogenetic tree with these UGI isolates (clade 1; Figure 2) and maintained a similar SNP difference between the GA-85038 and GA-85054 DGI isolates (39 vs 33 SNPs) in both the phylogenetic trees. Similarly, clade 4 contained both DGI (n = 1) and UGI (n = 4) isolates that belonged to MLST types ST-11516 (n = 4) and ST-12905 (n = 1) and separated on average by a pairwise SNP distance of 212 ± 140 SNPs. Clade 5 also contained 5 MLST ST-1596 isolates, DGI (n = 1) and UGI (n = 4), with an average pairwise SNP distance of 291 ± 153 SNPs. However, this clade did contain a pair of DGI (GA-85083) and UGI (GCWGS-1269) isolates that were most closely related based on pairwise SNP distance (20 SNPs). In addition to sharing ≤20 SNPs, DGI isolate GA-85083 collected in 2020 and UGI isolate GCWGS-1269 collected in 2018 were from individuals in the metropolitan Atlanta region, suggesting a potential locally circulating strain associated with both DGI and UGI [38]. Monophyletic clades of DGI isolates from the DGI-only phylogenetic tree (Figure 1) maintained similar SNP differences in the UGI phylogeny as well. For example, genetically similar DGI isolates identified in clade A (GA-85078 vs GA-85075), 2 subclades in clade B (GA-85084 vs GA-85086 and GA-85089 vs GA-85080 vs GA-85082), and clade C (GA-85074 vs GA-85072) maintained low SNP differences of 37, 18, 11, and 2, respectively, when compared to the UGI isolates, suggesting that there might be multiple highly similar DGI strains circulating in GA.
DGI Isolates Harbored Various AMR Markers
AMR markers associated with reduced susceptibility to ceftriaxone, such as mosaic penA alleles, were not detected in the DGI isolates in this study, suggesting that all of the DGI isolates were susceptible to the current recommended treatment for gonorrhea in the US [39]. However, other AMR markers were identified in some DGI isolates that might confer reduced susceptibility for macrolides, penicillin, quinolones, and tetracyclines (Supplementary Table 1) [40]. Minimum inhibitory concentration (MIC) data for the DGI isolates described here has been previously published [17].
Important AMR markers associated with reduced susceptibility to macrolides and penicillin were observed in the DGI isolates studied here, such as the mtrR mosaic allele (GA-85066), the single nucleotide (A) deletion in the 13-bp inverted repeat sequence (GA-85044 and GA-85051), and the mtrR premature stop codon (n = 6) [40–43]. Mutations in mtrR such as A39T (n = 2), G45D (n = 2), and H105Y (n = 1) were also observed. Seven DGI isolates carried the mtrD K823E mutation which has also been shown to increase MICs to macrolides (Supplementary Table 1) [42, 43]. Eleven DGI isolates carried the gyrA S91F mutation, a known variant that confers resistance to fluroquinolones [40]. Two of those isolates, GA-85052 and GA-85069, carried both gyrA S91F and A95G mutations. A single DGI isolate, GA-85069, carried the TetM-encoding plasmid, which is known to confer resistance to tetracyclines [40]. Seven DGI isolates carried the β-lactamase–encoding plasmid that is associated with high-level resistance to penicillin [40]. Overall, the AMR markers detected within the DGI isolates studied here were also detected in the AMR profiles extracted from the genome sequences of the UGI isolates.
Multiple Markers Associated With Normal Human-Serum Resistance Were Detected in DGI Isolates
Multiple genetic alleles that might confer resistance to NHS have been proposed to be associated with invasive infection, including serotype A of porB (porB1A), the GGI, the lgtABCDE operon, and the opa genes [4–10]. Our analysis also identified invasive gonococcal genetic markers among the GA-DGI isolates suggesting resistance to NHS, which facilitates dissemination of the gonococci. A total of 20 of the 30 (67%) DGI genome sequences examined in this study possessed a porB1A allele, while only 10 of 110 UGI genome sequences contained the porB1A allele (9%), which indicated a statistically significant association of the presence of the porB1A allele among DGI isolates. (Fisher exact test, P < .0001) (Table 1). The same trend was also observed in the reconstructed phylogenetic tree using all of the porB sequences extracted from both the DGI and UGI isolates (Figure 3), where the isolates clustered into 2 distinct clades representing the 2 different serotypes of porB, of which the porB1A clade contained the majority (20/30) of the DGI isolates while the porB1B clade was enriched with UGI isolates (100/110) (Table 1).
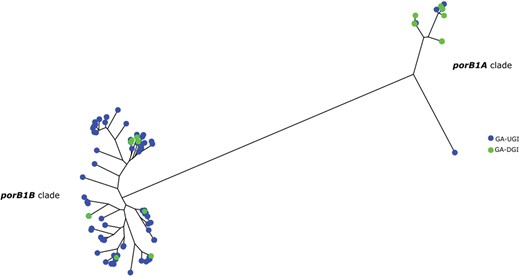
Neighbor-joining phylogenetic tree of porB sequences from the 30 Georgia (GA) disseminated gonococcal infection (DGI) isolates and the background comparative 110 GA urogenital and extragenital gonococcal infection (UGI) isolates. The porB1A cluster is represented by the right cluster and the porB1B cluster is represented by the left cluster. There are 20 DGI isolates that carry the porB1A allele and 10 DGI isolates that carry the porB1B allele. The majority of UGI (100/110) isolates were in the porB1B cluster while the remaining (10/110) isolates were in the porB1A cluster.
Presence or Absence of Various Genetic Markers Found Within the Disseminated Gonococcal Infection and Urogenital and Extragenital Gonococcal Infection Isolates
| Isolate | porB1Aa | porB1Ba | GGIb | GGI(atlA+ and traG[sac-4]+)b |
|---|---|---|---|---|
| DGI | 67%c (20/30) | 33% (10/30) | 57% (17/30) | 41% (7/17) |
| UGI | 9% (10/110) | 91% (100/110) | 63% (69/110) | 39% (27/69) |
| Isolate | porB1Aa | porB1Ba | GGIb | GGI(atlA+ and traG[sac-4]+)b |
|---|---|---|---|---|
| DGI | 67%c (20/30) | 33% (10/30) | 57% (17/30) | 41% (7/17) |
| UGI | 9% (10/110) | 91% (100/110) | 63% (69/110) | 39% (27/69) |
Abbreviations: DGI, disseminated gonococcal infection; GGI, gonococcal genetic island; GGI(atlA+ and traG[sac-4]+), gonococcal genetic island containing the atlA and traG allele combination; porB1A, porin B 1A allele; porB1B, porin B 1B allele; UGI, urogenital and extragenital gonococcal infection.
The porB allele type was determined using the NG-STAR database.
The GGI presence or absence and allele combination were determined using a customized BLAST database of MS11 reference GGI allele.
Fisher exact test; statistically significant at P < .0001.
Presence or Absence of Various Genetic Markers Found Within the Disseminated Gonococcal Infection and Urogenital and Extragenital Gonococcal Infection Isolates
| Isolate | porB1Aa | porB1Ba | GGIb | GGI(atlA+ and traG[sac-4]+)b |
|---|---|---|---|---|
| DGI | 67%c (20/30) | 33% (10/30) | 57% (17/30) | 41% (7/17) |
| UGI | 9% (10/110) | 91% (100/110) | 63% (69/110) | 39% (27/69) |
| Isolate | porB1Aa | porB1Ba | GGIb | GGI(atlA+ and traG[sac-4]+)b |
|---|---|---|---|---|
| DGI | 67%c (20/30) | 33% (10/30) | 57% (17/30) | 41% (7/17) |
| UGI | 9% (10/110) | 91% (100/110) | 63% (69/110) | 39% (27/69) |
Abbreviations: DGI, disseminated gonococcal infection; GGI, gonococcal genetic island; GGI(atlA+ and traG[sac-4]+), gonococcal genetic island containing the atlA and traG allele combination; porB1A, porin B 1A allele; porB1B, porin B 1B allele; UGI, urogenital and extragenital gonococcal infection.
The porB allele type was determined using the NG-STAR database.
The GGI presence or absence and allele combination were determined using a customized BLAST database of MS11 reference GGI allele.
Fisher exact test; statistically significant at P < .0001.
The presence or absence of the GGI, a 57-kb DNA element with considerable genetic variability found in approximately 80% of gonococcal isolates [6], among the DGI and UGI genomes revealed that 17 of 30 (57%) DGI isolates, compared to 69 of 110 (63%) UGI isolates, carried the GGI (Table 1). It has been previously reported that a specific allele combination of the GGI containing both a peptidoglycan hydrolase atlA and serum resistance allele traG (sac-4) was enriched among DGI isolates [6]. Similarly, we also detected the atlA+ and traG (sac-4)+–specific allele combination within the GGI among 7 of 17 (41%) DGI isolates while 27 of 69 (39%) UGI isolates contained the specific allele combination within the GGI (Table 1). The association of this allele to DGI was not statistically significant (P = .34) in our analysis.
The lgtABCDE operon, which encodes for various glycosyl transferases and mediates the biosynthesis of LOS [7,8], has also been associated with gonococcal NHS resistance. It has been previously shown that the various genes of the lgtABCDE operon are phase variable, meaning that they can be switched on or off via contraction or expansion of the poly G repeat tract in those genes, which enables the gonococci to express several unique LOS structures that differ in their glycan composition [7, 8]. The differing compositions can play a role in virulence and immune evasion with some compositions possibly conferring the resistance of the gonococci to NHS [8]. We observed varying lengths of the poly G repeat track present in the homopolymer site in the lgtA/C/D genes (Supplementary Table 1), where 22 DGI hybrid assemblies contained a range of 10–20 G’s at the homopolymer repeat region within lgtA, while the G repeat ranged from 9 to 16 and 8 to 15 in the lgtC and lgtD genes, respectively. Based on various lgtA/C/D on/off combinations identified from the sequence data (as described in the Materials and Methods), we observed 7 DGI genotypes (Figure 4), of which 10 of 22 (45%) DGI isolates belonged to the combination of lgtA phase off, lgtC phase on, and lgtD phase off (group 1). Interestingly, the LOS structure resulting from the group 1 genotype has been shown to be associated with NHS resistance [8].
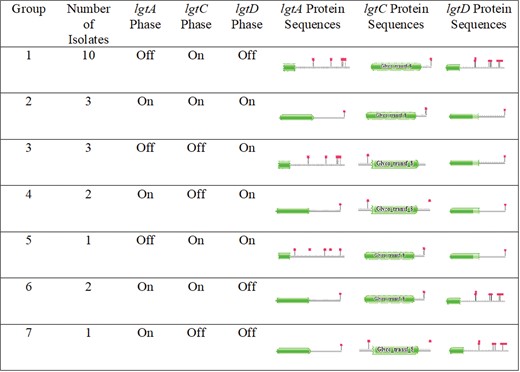
Groupings of the 22 disseminated gonococcal infection hybrid PacBio/Illumina genomes based on the phase on/off combinations of the lgtAC/D genes within the lgtABCDE operon. The green shapes represent functional protein domains with the red flags representing stop codons. The gene was determined to be phase on/off based on the translated protein sequence and the subsequent protein motif Hidden Markov Model matches found in Pfam. lgtA and lgtC were considered phase on when the protein showed a completed functional protein sequence with a stop codon at the end of the translated sequence. We determined lgtD to be considered phase on (ie, group 2) when the partial protein sequence was observed with a stop codon at the end of the sequence. The partial functional protein sequence in lgtD observed might be due to the absence of Neisseria species–specific sequences possibly absent in the Pfam database.
The opacity (Opa) proteins are outer membrane proteins that interact with 1 or more human carcinoembryonic antigen-related cell adhesion molecules (CEACAMs) [9, 10]. The Ng genome has approximately 11 opa genes and translation of these genes is controlled by phase variation based on the number of pentameric (CTCTT) repeat units in the 5′ coding region of each opa gene [9, 10]. When switched on, various Opa proteins will interact with CEACAMs, affecting host neutrophil antibacterial activities. In contrast, when switched off, Opa-negative Ng failed to induce phagocytosis and killing by human polymorphonuclear leukocytes (PMNs) [10]. The pairwise sequence identities of all 11 opa genes within the 22 DGI hybrid assemblies ranged from 79% to 90%, and the pentameric repeat (CTCTT) counts varied from 1 to 20, suggesting inherent genetic variability across DGI isolates (Supplementary Table 1). Phase variations in the Ng opaD gene has been specifically associated with NHS resistance where opaD phase-on genotype stimulated a strong oxidated burst in PMNs but opaD phase-off genotype did not, which could lead to invasive infection. However, we found only 3 of 22 (14%) DGI hybrid assemblies with an opaD phase-off genotype (Supplementary Table 1).
DISCUSSION
We have presented an in-depth genomic analysis of Ng isolates obtained from DGI cases in Georgia. Few studies have utilized WGS data of DGI isolates to investigate their evolution and relatedness to UGI isolates. Using existing surveillance systems, we compared DGI and UGI isolates in a specific geographical region during a defined period (2017–2020).
Overall, DGI strains in our analysis showed significant genetic variability that appears to mimic the population structure of uncomplicated gonococcal infections with sporadic instances of clustered strains based on genetic similarity. It has been shown that gonococcal isolates from potential transmission pair partners differ by ≤6 SNPs, and we observed a few subclades of DGI isolates that were differentiated by a similar SNP range [38, 44]. These DGI isolates were collected in neighboring counties and within the same year, suggesting evidence of a possible transmission cluster of DGI isolates. Such clustered DGI strains might also represent multiple distinct DGI strains circulating in Georgia, which might have caused sporadic cases, with or without epidemiological links. This warrants continued surveillance to understand if such Ng strains are associated with invasive infection. The DGI isolates represented most of the MLST strain types observed among the gonococcal isolates that caused uncomplicated gonorrhea within the same geographical area and time. However, they were genetically distinct (44–3077 pairwise SNPs distance) with 1 exception where a pair of DGI (GA-85083) and UGI (GCWGS-1269) isolates detected within the Atlanta metropolitan area was closely related with a pairwise SNP distance of 20 SNPs with no known epidemiological links. We also did not observe any particular gonococcal MLST strain type to be more frequently associated to an invasive infection. Similarly, all DGI isolates were susceptible to ceftriaxone but have varied susceptibility to other antibiotics used in gonococcal treatments, similar to what we typically observe in noninvasive urogenital gonococcal strains [45].
Even though we observed genetic markers associated with NHS resistance within the majority of DGI isolates, not all the NHS resistance genetic markers investigated in this study were found within all the DGI isolates. Nevertheless, a variety of NHS resistance genetic mechanisms were found throughout the DGI population and to a lesser degree were also detected among UGI isolates, suggesting that DGI may be facilitated by circulating gonococcus strains that have 1 or several mechanisms to evade serum killing. It is likely that a combination of strain and host factors might lead to the dissemination of the infection.
There are a few limitations in this genomic comparative analysis. Most notably, the number of DGI isolates was limited by the small number of cases identified through active surveillance. Second, all of the UGI isolates were from Fulton County, Georgia, whereas the DGI isolates were collected across the entire state, hence the diversity of circulating UGI strains from more distant locations within the state may not have been captured. Additionally, the in silico phase variation analysis performed on various genes of the lgtABCDE operon and the opa genes could also differ from results of a phenotypic analysis using a viable DGI strain that could undergo further changes during laboratory passage.
Though our analysis identifies specific genetic markers associated with dissemination, we do not identify specific markers necessary for dissemination. Hence, further research into the molecular and host factor mechanisms of the pathogenesis of dissemination is needed. Nevertheless, continued surveillance for DGI cases with isolate collection paired with large-scale comparative genomic analysis of DGI and UGI isolates will help monitor genetic variability to the DGI and UGI strain population over time, facilitate detection of related DGI cases, and ultimately contribute to the identification of specific mechanisms that promote systemic gonococcal dissemination within the host.
Supplementary Data
Supplementary materials are available at Open Forum Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
Acknowledgments
The authors acknowledge the following individuals for their key contribution to the establishment and maintenance of the Active Bacterial Core surveillance disseminated gonococcal infection system: Mirasol Apostol, Art Reingold, and Gretchen Rothrock (California Emerging Infections Program); Charletta Cloud, Shanita Shack, and Stepy Thomas (Georgia Emerging Infections Program); Melissa Arvay (Division of Bacterial Diseases, Centers for Disease Control and Prevention [CDC]); and Gail Bolan, Brad Roland, Ellen Kersh, and Samera Sharpe (Division of STD Prevention, CDC). We also thank the Gonococcal Isolate Surveillance Project and US Response to Resistant Gonorrhea site at the Fulton County STD clinic and the Tennessee Department of Health Laboratory Services, which is part of the Antibiotic Resistance Laboratory Network, for their valuable contribution of isolate genome data. We would also like to thank William M. Shafer from Emory University for his valued insight and critical review of the manuscript.
Notes
Data availability. Supplementary Table 1 lists individual accession numbers for the whole genome sequencing data for all the isolates along with associated metadata and antimicrobial resistance data used in this study.
Disclaimer. The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the CDC.
Potential conflicts of interest. All authors: No reported conflicts of interest.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments