






Abstract
Dolutegravir (DTG) and boosted darunavir (bDRV) are potent antiretrovirals with a high resistance barrier and might be valuable switch options for people with HIV (PWH).
DUALIS, a randomized, open-label, phase 3b, noninferiority clinical trial, compared the switch to DTG + bDRV (2DR) with continuation of 2 nucleoside reverse transcriptase inhibitors (2NRTI) + bDRV (3DR). PWH with HIV RNA <50 copies/mL taking 2NRTI + bDRV (3DR) for ≥24 weeks (1 accepted blip <200 copies/mL) were randomized to either switch to DTG 50 mg + DRV 800 mg (boosted with 100 mg of ritonavir) or continue taking 3DR. The primary end point (PE) was the proportion of HIV RNA <50 copies/mL at week (W) 48. Change in NRTI backbone was not classified as failure. The estimated sample size for PE analysis was 292; the noninferiority margin was ≤–10.0%.
In total, 263 subjects were randomized and treated (2DR n = 131, 3DR n = 132; 90.1% male; 89.7% Caucasian; median age [interquartile range], 48 [39–54] years). At W48, 86.3% (n = 113/131) of the 2DR subject and 87.9% (n = 116/132) of the 3DR subjects had HIV RNA <50 copies/mL; the difference between arms was –1.6% (95.48% CI, based on the adjusted alpha level accounting for the interim analysis at W24, –9.9% to +6.7%; discontinuations due to adverse events: 2DR, 4.6% [n = 6]; 3DR, 0.8% [n = 1]). Kaplan-Meier estimates of confirmed HIV RNA ≥50 copies/mL at W48 were 1.6% (n = 2) in the 2DR and 3.1% (n = 4) in the 3DR group. Development of treatment-emergent resistance was not observed.
Switching to DTG + bDRV was noninferior to continuing 3DR in subjects already treated with bDRV.
Nucleoside reverse transcriptase inhibitors (NRTIs) play an important role as the “antiretroviral therapy [ART] backbone” in current treatment guidelines [1]. However, NRTI use, mainly of the thymidine analogue type, can be associated with substantial side effects such as lipodystrophy, mitochondrial toxicity, or bone and kidney toxicity [2–4]. Long-term exposure is a risk factor for these often cumulative side effects [5–8], and possible cardiovascular safety issues contribute to the decision about the potential use of abacavir [9].
Alternative NRTI-sparing antiretroviral therapy options have been evaluated in different studies but were found to be associated with less virologic therapeutic success and higher rates of therapy-induced resistance compared with standard regimens in naïve HIV-1 PWH [10, 11]. While NRTI-sparing combinations of raltegravir- or lopinavir-based dual therapies were not fully capable of preventing virological failure [12, 13], more recent dual combinations of the HIV-1 integrase strand transfer inhibitor (INSTI) dolutegravir (DTG) demonstrated noninferiortiy to triple therapy.
The dual combination of dolutegravir (DTG) with lamivudine is effective and safe for treatment-naïve and pretreated people with HIV (PWH) in the absence of associated resistance [14, 15]. The dual combination of DTG and rilpivirine has also demonstrated noninferiority to 3DR for maintainance of suppression [16]. However, NRTI resistance may limit treatment options for pretreated PWH [17].
In addition, the combination of the HIV-1 protease inhibitor darunavir (DRV) with DTG is very potent, with a high barrier to resistance and favorable tolerance [18–21]. Due its well-characterized side effect profile, potential for once-daily dosing, and high virological potency, DRV is currently the most frequently used protease inhibitor in Europe and the United States [18].
The NRTI-free combination of once-daily DTG in combination with boosted DRV may therefore offer a favorable safety and efficacy profile and might be a suitable antiretroviral therapy combination for PWH. Data from a retrospective Italian cohort study indicated high efficacy rates and an acceptable safety profile [22], but no data from randomized clinical trials are available so far.
We herein report the first efficacy and safety data of a randomized, clinical switch trial of once-daily DTG in combination with boosted DRV in pretreated patients with HIV-1 who have been suppressed for at least 6 months.
METHODS
Study Design and Participants
The DUALIS study is a randomized, open-label, multicenter, noninferiority, phase 3 trial conducted at 27 active study centers in Germany. Investigators enrolled subjects aged ≥18 years with documented HIV-1 infection, who had been virologically suppressed (plasma HIV RNA <50 copies/mL) for at least 6 months before screening and were on a stable, once-daily antiretroviral therapy consisting of ritonavir-boosted DRV in combination with 2 NRTIs. One unconfirmed elevation of HIV RNA to 200 copies/mL with consecutive suppression to HIV RNA <50 copies/mL within the last 6 months before screening was allowed. Exclusion criteria were documented major darunavir or INSTI resistance, replicative hepatitis B surface antigen–positive hepatitis B infection, any evidence of active AIDS-defining disease (Centers for Disease Control and Prevention stage C HIV infection), estimated glomerular filtration rate (eGFR) <50 mL per minute, alanine aminotransferase above a 5-fold increase of the upper limit of normal, any evidence of unstable liver disease or severe hepatic impairment, and an anticipated need for interferon-based hepatitis C treatment.
It should be noted that the recruitment rate was lower than expected due to changes in the use of DRV-based ART.
This study was performed in accordance with the Declaration of Helsinki and was approved by the institutional review board of the Technical University of Munich, Munich, Germany (approval number: 162/15), and by the German Federal Institute for Drugs and Medical Devices (Bundesinstitut für Arzneimittel und Medizinprodukte, BfArM), Bonn, Germany (approval number 4 040 568). This study is registered with Eudra-CT, number 2015-000360-34. Written informed consent was obtained from all subjects before enrollment.
Procedures
Subjects were randomized 1:1 using a sequence generated with nQuery Advisor 7.0 (Statsols, Cork, Ireland) to either receive oral DTG 50 mg once daily in combination with DRV 800 mg once daily and ritonavir 100 mg daily (2DR) or continue their regimen consisting of 2 NRTIs in combination with ritonavir-boosted DRV (3DR) once a day for a total study duration of 48 weeks. Ritonavir and DRV were administered as 2 tablets. A switch of the NRTI backbone, which could be tenofovir disoproxil fumarate/emtricitabine to tenofovir alafenamide/emtricitabine, was allowed at any time during the study period at the discretion of the investigator. Both treatment groups were open-label, and no blinding was applicable.
Study visits were scheduled at baseline and weeks 4, 12, 24, 36, and 48. For protocol-requested parameters, the use of appropriate, decentralized, local, accredited laboratories was accepted. Resistance testing was performed at the discretion of the investigator according to the local guidelines and at any time of confirmed virologic failure, defined as HIV RNA >200 copies/mL in 2 consecutive measures.
Safety was evaluated by physician assessment, including physical examination, laboratory testing, 12-lead electrocardiography, and recording of concomitant drugs. Adverse events (AEs) were coded with the Medical Dictionary for Regulatory Activities (MedDRA), version 18.0. Study treatment was to be discontinued in case of unacceptable toxic effects or harmful drug–drug interaction and if requested by the participant.
Statistical Analyses
The sample size calculation was based on the primary objective, that is, assessment of noninferiority of 2DR in terms of viral efficacy (ie, HIV RNA < 50 copies/mL, US Food and Drug Administration [FDA] snapshot analysis) compared with that of 3DR over 48 weeks. Based on 80% power to show noninferiority, a noninferiority margin of –10.0% using a 2-sided CI with an overall alpha level of .05, and the assumption of 90% efficacy rates at week 48 in both arms (based on published data), the sample size was estimated at 292 analyzable subjects (146 per treatment group). The O’Brien-Fleming adjustment, accounting for a preplanned 24-week interim analysis (with an alpha level of .01 for the interim analysis and .04519 for the final analysis), resulted in CIs of 99.00% for the interim analysis at week 24 and 95.48% for the final analysis (only provided to Data safety monitoring board DSMB). Noninferiority of the primary efficacy end point was established if the lower bound of the 95.48% CI for the difference in proportion of subjects with HIV RNA <50 copies/mL was <10%. To ensure robustness of results, a sensitivity analysis of the primary end point was also performed on the per-protocol (PP) set. As noninferiority in switch trials is no longer established based on the difference in efficacy rates using a margin of –10% but is established based on the difference in virological nonresponse using a +4% margin, post hoc analyses based on snapshot virologic nonresponse (defined as HIV RNA ≥50 copies/mL, including discontinuations for lack of efficacy or discontinuations for reasons other than AEs or death, and last HIV RNA ≥50 copies/mL) were performed on the exposed intention-to-treat (ITTe) set.
Due to preterminated recruitment, the study population size was slightly lower than the planned sample size required for an anticipated power of 80% to show noninferiority.
Analyses of the secondary outcomes were performed on the ITTe and/or the per-protocol sets in an exploratory manner. Safety-related secondary end points were performed on the safety analysis set. The safety analysis set included all randomly assigned subjects who received at least 1 dose of the study drug. Safety data were described in a summary form using all data collected up to data cutoff (May 9, 2019) or up to 30 days after the last dose of the study drug for participants who discontinued early.
In the snapshot analysis, subjects were classified according to 3 outcomes: HIV RNA <50 copies/mL at week 48, HIV RNA ≥50 copies/mL (including discontinuations due to an AE, death, or any other reason), and those with no virological data in the visit window of week 48 (±6 weeks) but still taking the study drug.
The proportion of participants with plasma HIV RNA of <200 copies/mL at weeks 24 and 48 was analyzed in the same way as the primary end point (FDA snapshot analysis).
Methods for other secondary outcomes were applied as follows: Continuous variables were summarized using descriptive statistics. The Mann-Whitney U test, where appropriate, was used to compare continuous outcomes between treatment groups. For categorical variables, the absolute frequencies and percentages of observed levels (based on the nonmissing sample sizes) were reported. The chi-square test (or Fisher exact test, if the expected frequency in any field was <5) was used for the comparison of treatment groups. The exploratory significance level was 5%. No adjustment for multiple testing was made.
For statistical analyses of all data, Stata, version 15.1 (StataCorp, College Station, TX, USA), software was used.
RESULTS
Between July 31, 2015, and June 6, 2017, PWH (subjects) at the dedicated centers were screened (n = 269) for eligibility. In total, 265 subjects were randomly assigned to the intervention group of DTG and boosted DRV (2DR; n = 131) or to continuation of antiretroviral therapy (3DR) with a combination of 2 NRTIs in combination with boosted DRV (n = 132) and received at least 1 dose of study drugs. Two subjects did not receive any dose of study medication, resulting in an exposed ITTe population of 263 subjects (Figure 1).
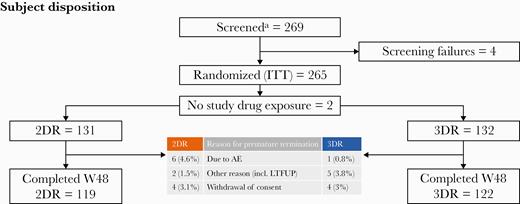
Trial profile. This figure displays the trial profile, including the total number of subjects screened and randomized, group distribution, and an overview of reasons for withdrawal. aEarly termination due to slow recruitment in June 2017 before reaching the estimated sample size of 320. Abbreviations: 2DR, dolutegravir + darunavir/ritonavir; 3DR, continuation of triple therapy of 2NRTI + DRV/ritonavir; AE, adverse event; ITT, intention-to-treat; LTFUP, loss to follow-up; W48, week 48.
Study Population
There were no significant differences in baseline and demographic characteristics between groups (Table 1). NRTI backbone in the 3DR group at baseline was as follows: 89/132 (67%) of patients took emtricitabine/tenofovir disoproxil fumarate; 22/132 (17%), abacavir/lamivudine; and 21/132 (16%), emtricitabine/tenofovir alafenamide. Numbers of historic genotypic resistance testing did not differ between the 2 groups and were available in 84 (64.1%) subjects in the 2DR group and 83 (62.9%) in the 3DR group; 20.9% in the 2DR group and 11.0% in the 3DR group had a history of ≥2 NRTI and ≥2 PI changes, respectively. Relevant rates of NRTI, non-NRTI, and major PI resistance–associated mutations (RAMs) were observed in 9.9% vs 9.1%, 15.3% vs 13.6%, and 3.8% vs 3.0% of the 2DR and 3DR groups, respectively.
Demographic and Baseline Characteristics of the Intention-to-Treat Population
| 2DR: Dolutegravir + Boosted Darunavir Group | 3DR: 2 Nucleoside Reverse Transcriptase Inhibitors + Boosted Darunavir Group | |
|---|---|---|
| Subject total, No. | 131 | 132 |
| Age, median (IQR), y | 47 (39–55) | 48 (40–53) |
| Male sex, No. (%) | 115 (88) | 122 (92) |
| Ethnicity: Caucasian, No. (%) | 118 (90) | 118 (89) |
| Documented HIV acquisition risk: MSM, No. (%) | 90 (69) | 92 (70) |
| HBsAg-negative with positive anti-HBc/negative HBsAg, No. (%) | 31/122 (25.4) | 34/121 (28.1) |
| HCV co-infection, No. (%) | 5/129 (4) | 8/131 (6) |
| eGFR MDRD, median (IQR), mL per min | 92.1 (80.1–103.7) | 91.8 (81.2–105.7) |
| HIV RNA <50 copies/mL, No. (%) | 128 (98) | 129 (98) |
| CD4 count at baseline, median (IQR), cells per µL | 609 (401–818) | 585 (453–823) |
| CD4 cell count at baseline, median (%) | ||
| <200/µL | 3 (2) | 1 (0.8) |
| 200–349/µL | 17 (13) | 17 (13) |
| 350–500/µL | 28 (21) | 24 (18) |
| >500/µL | 83 (63) | 89 (68) |
| CDC stage at HIV diagnosis, No. (%) | ||
| A | 57/121 (47) | 51/125 (41) |
| B | 33/121 (27) | 35/125 (28) |
| C | 31/121 (26) | 39/125 (31) |
| CD4-nadir <200/µL, No. (%) | 50/107 (47) | 52/110 (47) |
| Time since HIV diagnosis, median (IQR), y | 7.0 (4.4–12.0) | 7.6 (3.8–12.8) |
| Time on previous ART, median (IQR), mo | 50 (27–69) | 38 (17–65) |
| NRTI-based ART combination before study entry, No. (%) | ||
| FTC/TDF | 110 (84) | 94 (71)a |
| FTC/TAF | 11 (8) | 13 (10)a |
| 3TC/ABC | 9 (7) | 23 (17)a |
| 2DR: Dolutegravir + Boosted Darunavir Group | 3DR: 2 Nucleoside Reverse Transcriptase Inhibitors + Boosted Darunavir Group | |
|---|---|---|
| Subject total, No. | 131 | 132 |
| Age, median (IQR), y | 47 (39–55) | 48 (40–53) |
| Male sex, No. (%) | 115 (88) | 122 (92) |
| Ethnicity: Caucasian, No. (%) | 118 (90) | 118 (89) |
| Documented HIV acquisition risk: MSM, No. (%) | 90 (69) | 92 (70) |
| HBsAg-negative with positive anti-HBc/negative HBsAg, No. (%) | 31/122 (25.4) | 34/121 (28.1) |
| HCV co-infection, No. (%) | 5/129 (4) | 8/131 (6) |
| eGFR MDRD, median (IQR), mL per min | 92.1 (80.1–103.7) | 91.8 (81.2–105.7) |
| HIV RNA <50 copies/mL, No. (%) | 128 (98) | 129 (98) |
| CD4 count at baseline, median (IQR), cells per µL | 609 (401–818) | 585 (453–823) |
| CD4 cell count at baseline, median (%) | ||
| <200/µL | 3 (2) | 1 (0.8) |
| 200–349/µL | 17 (13) | 17 (13) |
| 350–500/µL | 28 (21) | 24 (18) |
| >500/µL | 83 (63) | 89 (68) |
| CDC stage at HIV diagnosis, No. (%) | ||
| A | 57/121 (47) | 51/125 (41) |
| B | 33/121 (27) | 35/125 (28) |
| C | 31/121 (26) | 39/125 (31) |
| CD4-nadir <200/µL, No. (%) | 50/107 (47) | 52/110 (47) |
| Time since HIV diagnosis, median (IQR), y | 7.0 (4.4–12.0) | 7.6 (3.8–12.8) |
| Time on previous ART, median (IQR), mo | 50 (27–69) | 38 (17–65) |
| NRTI-based ART combination before study entry, No. (%) | ||
| FTC/TDF | 110 (84) | 94 (71)a |
| FTC/TAF | 11 (8) | 13 (10)a |
| 3TC/ABC | 9 (7) | 23 (17)a |
All percent data refer to the given number of subjects within subgroups, unless otherwise stated.
Abbreviations: 2DR, dolutegravir + darunavir/ritonavir; 3DR, continuation of triple therapy of 2NRTI + DRV/ritonavir; 3TC, lamivudine; ABC, abacavir; ART, antiretroviral therapy; CDC, Centers for Disease Control and Prevention; eGFR, estimated glomerular filtration rate (by Modification of Diet in Renal Disease); FTC, emtricitabine; HBsAg, hepatitis B surface antigen; HBV, hepatitis B virus; HCV, hepatitis C virus; MSM, men who have sex with men; NRTI, nucleoside reverse transcriptase inhibitor; TDF, tenofovir disoproxil fumarate; TAF, tenofovir alafenamide.
aOf note, at baseline there were few changes in NRTI backbone, resulting in slight differences to the NRTIs at baseline mentioned in the manuscript body.
Demographic and Baseline Characteristics of the Intention-to-Treat Population
| 2DR: Dolutegravir + Boosted Darunavir Group | 3DR: 2 Nucleoside Reverse Transcriptase Inhibitors + Boosted Darunavir Group | |
|---|---|---|
| Subject total, No. | 131 | 132 |
| Age, median (IQR), y | 47 (39–55) | 48 (40–53) |
| Male sex, No. (%) | 115 (88) | 122 (92) |
| Ethnicity: Caucasian, No. (%) | 118 (90) | 118 (89) |
| Documented HIV acquisition risk: MSM, No. (%) | 90 (69) | 92 (70) |
| HBsAg-negative with positive anti-HBc/negative HBsAg, No. (%) | 31/122 (25.4) | 34/121 (28.1) |
| HCV co-infection, No. (%) | 5/129 (4) | 8/131 (6) |
| eGFR MDRD, median (IQR), mL per min | 92.1 (80.1–103.7) | 91.8 (81.2–105.7) |
| HIV RNA <50 copies/mL, No. (%) | 128 (98) | 129 (98) |
| CD4 count at baseline, median (IQR), cells per µL | 609 (401–818) | 585 (453–823) |
| CD4 cell count at baseline, median (%) | ||
| <200/µL | 3 (2) | 1 (0.8) |
| 200–349/µL | 17 (13) | 17 (13) |
| 350–500/µL | 28 (21) | 24 (18) |
| >500/µL | 83 (63) | 89 (68) |
| CDC stage at HIV diagnosis, No. (%) | ||
| A | 57/121 (47) | 51/125 (41) |
| B | 33/121 (27) | 35/125 (28) |
| C | 31/121 (26) | 39/125 (31) |
| CD4-nadir <200/µL, No. (%) | 50/107 (47) | 52/110 (47) |
| Time since HIV diagnosis, median (IQR), y | 7.0 (4.4–12.0) | 7.6 (3.8–12.8) |
| Time on previous ART, median (IQR), mo | 50 (27–69) | 38 (17–65) |
| NRTI-based ART combination before study entry, No. (%) | ||
| FTC/TDF | 110 (84) | 94 (71)a |
| FTC/TAF | 11 (8) | 13 (10)a |
| 3TC/ABC | 9 (7) | 23 (17)a |
| 2DR: Dolutegravir + Boosted Darunavir Group | 3DR: 2 Nucleoside Reverse Transcriptase Inhibitors + Boosted Darunavir Group | |
|---|---|---|
| Subject total, No. | 131 | 132 |
| Age, median (IQR), y | 47 (39–55) | 48 (40–53) |
| Male sex, No. (%) | 115 (88) | 122 (92) |
| Ethnicity: Caucasian, No. (%) | 118 (90) | 118 (89) |
| Documented HIV acquisition risk: MSM, No. (%) | 90 (69) | 92 (70) |
| HBsAg-negative with positive anti-HBc/negative HBsAg, No. (%) | 31/122 (25.4) | 34/121 (28.1) |
| HCV co-infection, No. (%) | 5/129 (4) | 8/131 (6) |
| eGFR MDRD, median (IQR), mL per min | 92.1 (80.1–103.7) | 91.8 (81.2–105.7) |
| HIV RNA <50 copies/mL, No. (%) | 128 (98) | 129 (98) |
| CD4 count at baseline, median (IQR), cells per µL | 609 (401–818) | 585 (453–823) |
| CD4 cell count at baseline, median (%) | ||
| <200/µL | 3 (2) | 1 (0.8) |
| 200–349/µL | 17 (13) | 17 (13) |
| 350–500/µL | 28 (21) | 24 (18) |
| >500/µL | 83 (63) | 89 (68) |
| CDC stage at HIV diagnosis, No. (%) | ||
| A | 57/121 (47) | 51/125 (41) |
| B | 33/121 (27) | 35/125 (28) |
| C | 31/121 (26) | 39/125 (31) |
| CD4-nadir <200/µL, No. (%) | 50/107 (47) | 52/110 (47) |
| Time since HIV diagnosis, median (IQR), y | 7.0 (4.4–12.0) | 7.6 (3.8–12.8) |
| Time on previous ART, median (IQR), mo | 50 (27–69) | 38 (17–65) |
| NRTI-based ART combination before study entry, No. (%) | ||
| FTC/TDF | 110 (84) | 94 (71)a |
| FTC/TAF | 11 (8) | 13 (10)a |
| 3TC/ABC | 9 (7) | 23 (17)a |
All percent data refer to the given number of subjects within subgroups, unless otherwise stated.
Abbreviations: 2DR, dolutegravir + darunavir/ritonavir; 3DR, continuation of triple therapy of 2NRTI + DRV/ritonavir; 3TC, lamivudine; ABC, abacavir; ART, antiretroviral therapy; CDC, Centers for Disease Control and Prevention; eGFR, estimated glomerular filtration rate (by Modification of Diet in Renal Disease); FTC, emtricitabine; HBsAg, hepatitis B surface antigen; HBV, hepatitis B virus; HCV, hepatitis C virus; MSM, men who have sex with men; NRTI, nucleoside reverse transcriptase inhibitor; TDF, tenofovir disoproxil fumarate; TAF, tenofovir alafenamide.
aOf note, at baseline there were few changes in NRTI backbone, resulting in slight differences to the NRTIs at baseline mentioned in the manuscript body.
Virologic Outcomes
The median duration of overall treatment in both arms (interquartile range [IQR]) was 48.1 (47.9–49.3) weeks. At week 48, an almost equal proportion of subjects in both study arms, 86.3% in the 2DR group and 87.9% in the 3DR group, had an HIV RNA level <50 copies/mL (Table 2), resulting in a difference in response rates between the 2DR and 3DR study groups of –1.6%. Based on the lower bound of the 95.48% CI (–9.9% to +6.7%), the combination of DTG and boosted DRV is considered noninferior to the continuation of treatment of 2 NRTIs in combination with boosted DRV (Figure 2A/B).
Virological Outcomes at Week 48 in Intention-to-Treat Analysis Population
| 2DR: Dolutegravir + Boosted Darunavir Group (n = 131), No. (%) | 3DR: 2 NRTI + Boosted Darunavir Group (n = 132), No. (%) | |
|---|---|---|
| Subject total | 131 | 132 |
| Primary end point | ||
| HIV RNA <50 copies/mL | 113 (86.3)a | 116 (87.9)a |
| HIV RNA <200 copies/mL (secondary outcome) | 118 (90.1) | 121 (91.7) |
| Snapshot virologic nonresponse | 5 (3.8)b | 7 (5.3)b |
| HIV RNA ≥50 copies/mL | 5 (3.8) | 6 (4.5) |
| Discontinuation due to lack of efficacy | 0 (0.0) | 0 (0.0) |
| Discontinuation for other reasons while not <50 cp/mL | 0 (0.0) | 1 (0.8) |
| No virologic data | 13 (9.9) | 9 (6.8) |
| Discontinuation due to adverse event | 6 (4.6) | 1 (0.8) |
| Discontinuation due to loss to follow-up with last HIV RNA <50 copies/mL | 2 (1.5) | 4 (3.0) |
| Discontinuation for other reasonsc with last HIV RNA <50 copies/mL | 4 (3.1) | 4 (3.0) |
| Missing HIV RNA in window (but on study) | 1 (0.8)d | 0 (0.0) |
| 2DR: Dolutegravir + Boosted Darunavir Group (n = 131), No. (%) | 3DR: 2 NRTI + Boosted Darunavir Group (n = 132), No. (%) | |
|---|---|---|
| Subject total | 131 | 132 |
| Primary end point | ||
| HIV RNA <50 copies/mL | 113 (86.3)a | 116 (87.9)a |
| HIV RNA <200 copies/mL (secondary outcome) | 118 (90.1) | 121 (91.7) |
| Snapshot virologic nonresponse | 5 (3.8)b | 7 (5.3)b |
| HIV RNA ≥50 copies/mL | 5 (3.8) | 6 (4.5) |
| Discontinuation due to lack of efficacy | 0 (0.0) | 0 (0.0) |
| Discontinuation for other reasons while not <50 cp/mL | 0 (0.0) | 1 (0.8) |
| No virologic data | 13 (9.9) | 9 (6.8) |
| Discontinuation due to adverse event | 6 (4.6) | 1 (0.8) |
| Discontinuation due to loss to follow-up with last HIV RNA <50 copies/mL | 2 (1.5) | 4 (3.0) |
| Discontinuation for other reasonsc with last HIV RNA <50 copies/mL | 4 (3.1) | 4 (3.0) |
| Missing HIV RNA in window (but on study) | 1 (0.8)d | 0 (0.0) |
Abbreviations: 2DR, dolutegravir + darunavir/ritonavir; 3DR, continuation of triple therapy of 2NRTI + DRV/ritonavir; NRTI, nucleoside reverse transcriptase inhibitor.
a95.48% CI, –1.6% (–9.9% to +6.7%); CI based on the adjusted alpha-level accounting for the interim analysis.
b95% CI, –1.4% (–6.5% to +3.6%); post hoc analysis.
cOther reasons include withdrawal of consent and discontinuation due to drug–drug interactions.
dLast HIV RNA <50 copies/mL.
Virological Outcomes at Week 48 in Intention-to-Treat Analysis Population
| 2DR: Dolutegravir + Boosted Darunavir Group (n = 131), No. (%) | 3DR: 2 NRTI + Boosted Darunavir Group (n = 132), No. (%) | |
|---|---|---|
| Subject total | 131 | 132 |
| Primary end point | ||
| HIV RNA <50 copies/mL | 113 (86.3)a | 116 (87.9)a |
| HIV RNA <200 copies/mL (secondary outcome) | 118 (90.1) | 121 (91.7) |
| Snapshot virologic nonresponse | 5 (3.8)b | 7 (5.3)b |
| HIV RNA ≥50 copies/mL | 5 (3.8) | 6 (4.5) |
| Discontinuation due to lack of efficacy | 0 (0.0) | 0 (0.0) |
| Discontinuation for other reasons while not <50 cp/mL | 0 (0.0) | 1 (0.8) |
| No virologic data | 13 (9.9) | 9 (6.8) |
| Discontinuation due to adverse event | 6 (4.6) | 1 (0.8) |
| Discontinuation due to loss to follow-up with last HIV RNA <50 copies/mL | 2 (1.5) | 4 (3.0) |
| Discontinuation for other reasonsc with last HIV RNA <50 copies/mL | 4 (3.1) | 4 (3.0) |
| Missing HIV RNA in window (but on study) | 1 (0.8)d | 0 (0.0) |
| 2DR: Dolutegravir + Boosted Darunavir Group (n = 131), No. (%) | 3DR: 2 NRTI + Boosted Darunavir Group (n = 132), No. (%) | |
|---|---|---|
| Subject total | 131 | 132 |
| Primary end point | ||
| HIV RNA <50 copies/mL | 113 (86.3)a | 116 (87.9)a |
| HIV RNA <200 copies/mL (secondary outcome) | 118 (90.1) | 121 (91.7) |
| Snapshot virologic nonresponse | 5 (3.8)b | 7 (5.3)b |
| HIV RNA ≥50 copies/mL | 5 (3.8) | 6 (4.5) |
| Discontinuation due to lack of efficacy | 0 (0.0) | 0 (0.0) |
| Discontinuation for other reasons while not <50 cp/mL | 0 (0.0) | 1 (0.8) |
| No virologic data | 13 (9.9) | 9 (6.8) |
| Discontinuation due to adverse event | 6 (4.6) | 1 (0.8) |
| Discontinuation due to loss to follow-up with last HIV RNA <50 copies/mL | 2 (1.5) | 4 (3.0) |
| Discontinuation for other reasonsc with last HIV RNA <50 copies/mL | 4 (3.1) | 4 (3.0) |
| Missing HIV RNA in window (but on study) | 1 (0.8)d | 0 (0.0) |
Abbreviations: 2DR, dolutegravir + darunavir/ritonavir; 3DR, continuation of triple therapy of 2NRTI + DRV/ritonavir; NRTI, nucleoside reverse transcriptase inhibitor.
a95.48% CI, –1.6% (–9.9% to +6.7%); CI based on the adjusted alpha-level accounting for the interim analysis.
b95% CI, –1.4% (–6.5% to +3.6%); post hoc analysis.
cOther reasons include withdrawal of consent and discontinuation due to drug–drug interactions.
dLast HIV RNA <50 copies/mL.
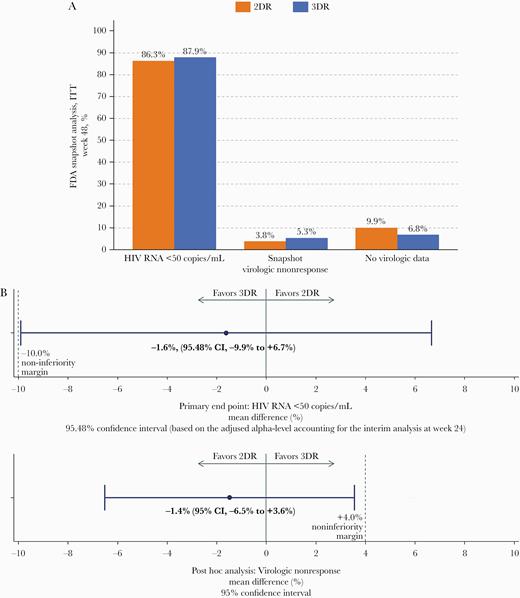
A, Study outcome. This figure displays the overall study outcomes of the ITTe population, including HIV RNA <50 cps/mL, proportion with virologic nonresponse, and proportion with no virologic data in the window. B, Study outcome and noninferiority margin for primary end point and post hoc analyses. This figure displays a forest plot of the primary outcome and a post hoc analysis indicating noninferiority of 2DR vs 3DR. Abbreviations: 2DR, dolutegravir + darunavir/ritonavir; 3DR, continuation of triple therapy of 2NRTI + DRV/ritonavir; AE, adverse event; FDA, Food and Drug Administration; ITT, intention-to-treat; LTFUP, loss to follow-up; W48, week 48.
It should be noted that a change of NRTI backbone was permitted as per protocol at any time point and was not classified as failure. During study follow-up, 14/132 subjects (11%) were switched to tenofovir alafenamide (TAF).
The per-protocol analysis set consisted of 241 patients (2DR n = 119, 3DR n = 122); primary end point analysis in the PP set resulted in 48-week response rates of 95.8% (2DR; 113/118) and 95.1% (3DR; 116/122), with a mean difference 0.7% (95% CI, –4.6% to +6.0%). Rates of virologic nonresponse at month 48 were 3.8% and 5.3% in the 2DR and 3DR arms, respectively (Table 2), with a difference between arms of –1.4% (95% CI, –6.5% to +3.6%). It is noteworthy that of patients with documented major PI RAMs, no patient in either group had virologic nonresponse or confirmed virologic failure. No virologic failure with treatment-emergent resistance to any of the study component drugs was documented during the entire study period.
Consistent results concerning treatment responses in the 2DR vs 3DR groups were observed when stratifying for sex, age (≤50 years vs >50 years), CDC stage (A/B vs C), and CD4 nadir (≥200 cells/µL vs <200 cells/µL) (Supplementary Table 1).
The median changes in the absolute CD4 cell counts from baseline (IQR) were not significantly different (P = .8414) between the 2 groups (+30.5 [+9.5 to +24.0] cells/µL in the 2DR group and +32.0 [–9.0 to +12.0] cells/µL in the 3DR group).
Kaplan-Meier estimates of confirmed HIV RNA ≥50 copies/mL were 0.8% (1 subject) at week 24 and 1.6% (2 subjects) at week 48 in the 2DR group and 1.5% (2 subjects) at week 24 and 3.1% (4 subjects) at week 48 in the 3DR group, respectively.
One of the 4 subjects in the 3DR group reported incomplete adherence, while the other subjects, including the 2 subjects from the 2DR group, reported favorable adherence, as measured by pill count.
Safety
Treatment was well tolerated in both groups, and most AEs were mild or moderate (Table 3). Overall, AEs were observed in 104 (78.2%) subjects in the 2DR group and 100 (75.2%) subjects in the 3DR group during the entire study period. The number of AEs was comparable between both study groups (306 AEs [2DR] vs 315 AEs [3DR]). Significantly more patients experienced drug-related adverse reactions in the 2DR group (2DR: 34 events in 19 patients [14.3%]; vs 3DR: 10 events in 7 patients [5.3%]; P = .02; post hoc analysis). A total of 24 serious AEs were documented, with 12 in each group (affecting 5.3% of subjects in both groups). No deaths were reported during the study period.
Summary of Incidence of Nonserious and Serious Adverse Events in the Safety Set
| 2DR: Dolutegravir + Boosted Darunavir Group, No. (%) | 3DR: 2 NRTI + Boosted Darunavir Group, No. (%) | |
|---|---|---|
| Number of (nonserious or serious) adverse events | 306 | 315 |
| Patients with AEs or SAEs | 104/133 (78.2) | 100/133 (75.2) |
| Number of severe adverse events (grade 3 or 4) | 11/306 (3.6) | 6/315 (1.9) |
| Patients with severe adverse event (grade 3 or 4) | 6/133 (4.5) | 6/133 (4.5) |
| Number of SAEs | 12/306 (3.9) | 12/315 (3.8) |
| Patients with SAEs | 7/133 (5.3) | 7/133 (5.3) |
| Number of treatment-related adverse events (AEs or SAEs) | 34/306 (11.1) | 10/315 (3.2) |
| Patients with treatment-related AEs or SAEs | 19/133 (14.3) | 7/133 (5.3) |
| Count of treatment-related SAEs) | 1/306 (0.3) | 0/315 (0.0) |
| Patients with treatment-related SAE | 1/133 (0.8) | 0 (0.0) |
| Adverse events leading to study discontinuation | 14/305 (4.6)a | 5/313 (1.6)a |
| Death | 0 (0.0) | 0 (0.0) |
| Most common adverse events (per patient; MedDRA SOC terms in >10% subjects in at least 1 study group) | ||
| Infections and infestations | 66 (49.6) | 67 (50.4) |
| Gastrointestinal | 22 (16.5) | 25 (18.8) |
| Musculoskeletal | 21 (15.8) | 20 (15.0) |
| Skin | 17 (12.8) | 13 (9.8) |
| Nervous system | 12 (9.0) | 16 (12.0) |
| Most common drug-related adverse events (per patient; MedDRA SOC terms in >3% subjects in at least 1 study group) | ||
| Gastrointestinal | 6 (4.5) | 1 (0.8) |
| Skin | 5 (3.8) | 0 (0.0) |
| Psychiatric | 4 (3.0) | 1 (0.8) |
| Nervous system | 4 (3.0) | 1 (0.8) |
| 2DR: Dolutegravir + Boosted Darunavir Group, No. (%) | 3DR: 2 NRTI + Boosted Darunavir Group, No. (%) | |
|---|---|---|
| Number of (nonserious or serious) adverse events | 306 | 315 |
| Patients with AEs or SAEs | 104/133 (78.2) | 100/133 (75.2) |
| Number of severe adverse events (grade 3 or 4) | 11/306 (3.6) | 6/315 (1.9) |
| Patients with severe adverse event (grade 3 or 4) | 6/133 (4.5) | 6/133 (4.5) |
| Number of SAEs | 12/306 (3.9) | 12/315 (3.8) |
| Patients with SAEs | 7/133 (5.3) | 7/133 (5.3) |
| Number of treatment-related adverse events (AEs or SAEs) | 34/306 (11.1) | 10/315 (3.2) |
| Patients with treatment-related AEs or SAEs | 19/133 (14.3) | 7/133 (5.3) |
| Count of treatment-related SAEs) | 1/306 (0.3) | 0/315 (0.0) |
| Patients with treatment-related SAE | 1/133 (0.8) | 0 (0.0) |
| Adverse events leading to study discontinuation | 14/305 (4.6)a | 5/313 (1.6)a |
| Death | 0 (0.0) | 0 (0.0) |
| Most common adverse events (per patient; MedDRA SOC terms in >10% subjects in at least 1 study group) | ||
| Infections and infestations | 66 (49.6) | 67 (50.4) |
| Gastrointestinal | 22 (16.5) | 25 (18.8) |
| Musculoskeletal | 21 (15.8) | 20 (15.0) |
| Skin | 17 (12.8) | 13 (9.8) |
| Nervous system | 12 (9.0) | 16 (12.0) |
| Most common drug-related adverse events (per patient; MedDRA SOC terms in >3% subjects in at least 1 study group) | ||
| Gastrointestinal | 6 (4.5) | 1 (0.8) |
| Skin | 5 (3.8) | 0 (0.0) |
| Psychiatric | 4 (3.0) | 1 (0.8) |
| Nervous system | 4 (3.0) | 1 (0.8) |
Data consist of the safety analysis population of the study (n = 266).
Abbreviations: 2DR, dolutegravir + darunavir/ritonavir; 3DR, continuation of triple therapy of 2NRTI + DRV/ritonavir; AE, adverse event; MedDRA SOC, Medical Dictionary for Regulatory Activities system organ class; SAE, serious adverse event.
aFisher exact test: P = .036.
Summary of Incidence of Nonserious and Serious Adverse Events in the Safety Set
| 2DR: Dolutegravir + Boosted Darunavir Group, No. (%) | 3DR: 2 NRTI + Boosted Darunavir Group, No. (%) | |
|---|---|---|
| Number of (nonserious or serious) adverse events | 306 | 315 |
| Patients with AEs or SAEs | 104/133 (78.2) | 100/133 (75.2) |
| Number of severe adverse events (grade 3 or 4) | 11/306 (3.6) | 6/315 (1.9) |
| Patients with severe adverse event (grade 3 or 4) | 6/133 (4.5) | 6/133 (4.5) |
| Number of SAEs | 12/306 (3.9) | 12/315 (3.8) |
| Patients with SAEs | 7/133 (5.3) | 7/133 (5.3) |
| Number of treatment-related adverse events (AEs or SAEs) | 34/306 (11.1) | 10/315 (3.2) |
| Patients with treatment-related AEs or SAEs | 19/133 (14.3) | 7/133 (5.3) |
| Count of treatment-related SAEs) | 1/306 (0.3) | 0/315 (0.0) |
| Patients with treatment-related SAE | 1/133 (0.8) | 0 (0.0) |
| Adverse events leading to study discontinuation | 14/305 (4.6)a | 5/313 (1.6)a |
| Death | 0 (0.0) | 0 (0.0) |
| Most common adverse events (per patient; MedDRA SOC terms in >10% subjects in at least 1 study group) | ||
| Infections and infestations | 66 (49.6) | 67 (50.4) |
| Gastrointestinal | 22 (16.5) | 25 (18.8) |
| Musculoskeletal | 21 (15.8) | 20 (15.0) |
| Skin | 17 (12.8) | 13 (9.8) |
| Nervous system | 12 (9.0) | 16 (12.0) |
| Most common drug-related adverse events (per patient; MedDRA SOC terms in >3% subjects in at least 1 study group) | ||
| Gastrointestinal | 6 (4.5) | 1 (0.8) |
| Skin | 5 (3.8) | 0 (0.0) |
| Psychiatric | 4 (3.0) | 1 (0.8) |
| Nervous system | 4 (3.0) | 1 (0.8) |
| 2DR: Dolutegravir + Boosted Darunavir Group, No. (%) | 3DR: 2 NRTI + Boosted Darunavir Group, No. (%) | |
|---|---|---|
| Number of (nonserious or serious) adverse events | 306 | 315 |
| Patients with AEs or SAEs | 104/133 (78.2) | 100/133 (75.2) |
| Number of severe adverse events (grade 3 or 4) | 11/306 (3.6) | 6/315 (1.9) |
| Patients with severe adverse event (grade 3 or 4) | 6/133 (4.5) | 6/133 (4.5) |
| Number of SAEs | 12/306 (3.9) | 12/315 (3.8) |
| Patients with SAEs | 7/133 (5.3) | 7/133 (5.3) |
| Number of treatment-related adverse events (AEs or SAEs) | 34/306 (11.1) | 10/315 (3.2) |
| Patients with treatment-related AEs or SAEs | 19/133 (14.3) | 7/133 (5.3) |
| Count of treatment-related SAEs) | 1/306 (0.3) | 0/315 (0.0) |
| Patients with treatment-related SAE | 1/133 (0.8) | 0 (0.0) |
| Adverse events leading to study discontinuation | 14/305 (4.6)a | 5/313 (1.6)a |
| Death | 0 (0.0) | 0 (0.0) |
| Most common adverse events (per patient; MedDRA SOC terms in >10% subjects in at least 1 study group) | ||
| Infections and infestations | 66 (49.6) | 67 (50.4) |
| Gastrointestinal | 22 (16.5) | 25 (18.8) |
| Musculoskeletal | 21 (15.8) | 20 (15.0) |
| Skin | 17 (12.8) | 13 (9.8) |
| Nervous system | 12 (9.0) | 16 (12.0) |
| Most common drug-related adverse events (per patient; MedDRA SOC terms in >3% subjects in at least 1 study group) | ||
| Gastrointestinal | 6 (4.5) | 1 (0.8) |
| Skin | 5 (3.8) | 0 (0.0) |
| Psychiatric | 4 (3.0) | 1 (0.8) |
| Nervous system | 4 (3.0) | 1 (0.8) |
Data consist of the safety analysis population of the study (n = 266).
Abbreviations: 2DR, dolutegravir + darunavir/ritonavir; 3DR, continuation of triple therapy of 2NRTI + DRV/ritonavir; AE, adverse event; MedDRA SOC, Medical Dictionary for Regulatory Activities system organ class; SAE, serious adverse event.
aFisher exact test: P = .036.
The overall cumulative incidence of grade 3–4 (serious) AEs (Common Terminology Criteria for Adverse Events catalogue) was calculated by dividing the number of new grade 3–4 (serious) AEs through 48 week by the number of subjects at risk in the safety analysis set, with 11/133 (8.3%) in the 2DR group and 6/133 (4.5%) in the 3DR group. The cumulative incidence of (serious) adverse reactions (ARs), which was calculated by dividing the number of new (serious) adverse reactions through 48 weeks by the number of subjects at risk in the safety analysis set, was higher in the 2DR group compared with the 3DR group (Table 3).
Laboratory grade 3/4 abnormalities were reported in 1 (<1%) of the safety analysis subjects in the 2DR group and 0 (0%) in the 3DR group.
Changes from baseline to week 48 in median concentrations of total cholesterol, low-density lipoprotein (LDL) and high-density lipoprotein (HDL) cholesterol, and triglycerides were significantly different between both groups, favoring the 3DR group, except for changes in HDL and triglycerides. Absolute lipid parameters are displayed in Supplementary Table 2.
DISCUSSION
Switching to DTG in combination with boosted DRV was noninferior to continuing a previous therapy consisting of boosted DRV in combination with 2 NRTIs in the present study. Maintenance of suppression remained high 48 weeks after switch to either arm, and no virologic failure with emergence of resistance was documented. The overall tolerability of both treatment strategies was acceptable, with comparable rates of AEs. Although open-label switch studies might be at risk for increased AE rates in the intervention arm, the difference was not significant between arms in this study [15, 16]. The observed drug-related AEs (most commonly gastrointestinal and skin or neurological system disorders) were more frequent in the open-label intervention 2DR group; all have been associated in the past with DTG and DRV [23, 24]. No renal AEs leading to discontinuation have been observed.
With regard to changes in lipid metabolism, the use of protease inhibitors has been associated with many challenges in PWH. Protease inhibitor–free strategies may be a helpful approach in PWH and cardiovascular risk [25]. However, clinical and resistance conditions may not always allow a protease inhibitor–free strategy. In all cases, changes in lipid metabolism due to protease inhibitors may also be influenced by the lipid-lowering effects of tenofovir disoproxil and the fact that the majority of subjects had a tenofovir disoproxil–containing ART regimen before being enrolled in the study [26].
Except for the lipid elevations and the previously discussed higher, albeit still low, rates of drug-related AEs, the study confirmed a switch to DTG and boosted DRV as a valuable option for previously virally suppressed people with HIV. It is important to emphasize that the trial population was of older age, on longer pretreatment, and had no restrictions in terms of prior virologic failure and non-DTG- or non-DRV-associated resistance mutations, making this study population different from those of other registrational trials [14, 15].
This study has some limitations: The open-label design is associated with a potential bias in participants and investigators. Moreover, the change of treatment practice with decreasing rates of protease inhibitors was associated with a low recruitment speed and, therefore, resulted in the early termination of patient recruitment. Noninferiority in terms of efficacy of the 2DR regimen could be established in DUALIS. However, the confidence interval was wide, although not reaching the predefined margin of –10.0%. Moreover, the novel noninferiority margin of the FDA of +4%, which is based on virologic nonresponse, is even more challenging for investigational studies. However, in a post hoc analysis, we could confirm noninferiority for snapshot virologic nonresponse as well. Nonetheless, despite no restrictions in previous virologic failures and an incomplete history of genotypic resistance documentation, no single virologic failure with treatment-emergent resistance has been observed.
In conclusion, a switch to DTG in combination with boosted DRV is effective and has an acceptable safety profile in previously suppressed people with HIV. Further investigations of this combination as a switch strategy in people with HIV and treatment failure are of interest and are underway.
Supplementary Data
Supplementary materials are available at Open Forum Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
Acknowledgment
We thank the involved participants, sites, and staff, as well as the clinical research organizations Munich Study Center (Helen Bidner for study coordination) and MUC Research (Annamaria Balogh for data management and statistical analyses), Munich, Germany. We thank the members of the Data Safety Monitoring Board (Georg Behrens, Armin Koch, and Christian Hoffmann), as well as the study coordinators Marcus Kosch and Helen Bidner (both Technical University Munich) for their overall assistance.
DUALIS Study Group Members: Dr Christoph D. Spinner (Technical University of Munich, University Hospital rechts der Isar, München), Dr Björn Jensen (Universitätsklinikum Duesseldorf Klinik für Gastroenterologie, Hepatologie und Infektiologie Leber und Infektionszentrum, MX-Ambulanz, Düsseldorf), Dr Thomas Lutz (Infektiologikum Frankfurt, Frankfurt am Main), Dr Petra Spornraft-Ragaller (Universitätsklinikum Carl Gustav Carus an der Technischen Universität Dresden Anstalt des öffentlichen Rechts des Freistaates Sachsen, Dresden), Prof. Dr Stellbrink (ICH Study Center GmbH, Hamburg), Martin Hower (Klinikum Dortmund gGmbH ID-Ambulanz, Dortmund), Dr Ulrich Bohr (Praxis Kaiserdamm, Berlin), Dr Wilfried Obst (Universitätsklinikum Magdeburg A.ö.R. Klinik für Gastroenterologie, Hepatologie und Infektiologie, Magdeburg), Dr Ivanka Krznaric (Zentrum für Infektiologie Berlin Prenzlauer Berg GmbH, Berlin), Dr Martin Sprinzl (Universitätsmedizin Mainz I. Med. Klink, Mainz), Dr Franz Audebert (Praxiszentrum Alte Mälzerei, Regensburg), Dr Tim Kuemmerle (Praxis am Ebertplatz, Köln), Dr Stefan Scholten (Praxis Hohenstaufenring, Köln), Dr Heribert Hillenbrand (Medizinisches Versorgungszentrum Berlin-Friedrichshain, Berlin), Dr Christiane Cordes (Praxis Cordes, Berlin Friedrichshain), Dr Heribert Knechten (Dr. Heribert Knechten, Aachen), Dr Birger Kuhlmann (Praxis Kuhlmann, Hannover), Dr Heiko Jessen (Praxis Jessen2 + Kollegen, Berlin), Dr Patrick Beck (Gemeinschaftspraxis -infectomed-, Stuttgart), Prof. Dr Gerd Fätkenheuer (Uniklinik Köln Klinik I für Innere Medizin, Köln), Prof. Dr Hartwig HF Klinker (Universitätsklinikum Würzburg Medizinische Klinik und Poliklinik II, Würzburg), Prof. Dr Juergen K. Rockstroh (Universitätsklinikum Bonn Medizinische Klinik und Poliklinik I, Bonn), Dr Stefan Esser (Universitätsklinikum Essen Klinik und Poliklinik für Dermatologie, Essen), Prof. Dr Christoph Stephan (Klinikum der Johann Wolfgang Goethe Universität Zentrum der Inneren Medizin - HIV-Center, Frankfurt), Dr Olaf Degen (Universitätsklinikum Hamburg-Eppendorf Ambulanzzentrum Infektiologie, Hamburg), Dr Andreas Bellmunt-Zschäpe (Allgemeinmedizinische Praxis Dr. med. Dipl.-Biochem. Bellmunt Zschäpe, Dortmund), Dr Pavel Khaykin (MainFachArzt, Frankfurt/Main), Prof Norbert Brockmeyer (Klinikum Bochum WIR “Walk In Ruhr” im St. Elisabeth-Hospital, Bochum), Dr Albrecht Stoehr (ifi - Institut für interdisziplinäre Medizin, Zentrum für Infektiologie und Tagesklinik an der Asklepios Klinik St. Georg, Hamburg).
Financial support. This work was supported by Technical University Munich (TUM) with grants from Janssen-Cilag and ViiV Healthcare.
Potential conflicts of interest. Dr. Spinner reports grants and personal fees from Jansen-Cilag and grants and personal fees from ViiV Healthcare during the conduct of the study; grants and personal fees from Gilead Sciences, personal fees from AbbVie, and personal fees from MSD outside the submitted work. Dr. Heiken reports personal fees from TUM and IZAR during the conduct of the study; personal fees from Janssen Cilag, personal fees from Gilead, personal fees from MSD, personal fees from ViiV Healthcare, and personal fees from AbbVie outside the submitted work. Dr. Jensen reports personal fees from Gilead, personal fees from ViiV, and personal fees from Janssen-Cilag outside the submitted work. Dr. Lehmann receives funding from the Thematic Translational Unit HIV (TTU HIV) of the German Center of Infection Research (DZIF). C.L. has received honoraria for speaking at educational events or consulting for ViiV, Gilead, Janssen, and MSD. Dr. Jessen reports personal fees from Janssen-Cilag GmbH, personal fees from ViiV Helathcare GmbH, personal fees from MSD GmbH, grants and personal fees from Gilead Sciences GmbH, grants from the US Military HIV Research Program (MHRP), personal fees from Hormosan Pharma GmbH, personal fees from Ifi-Medizin GmbH, personal fees from CIP Clinic GmbH, and personal fees from GlaxoSmithKline GmbH & Co., KG, outside the submitted work. Dr. Spornraft-Ragaller reports that Janssen-Cilag and ViiV Healthcare, among other companies, support the Dresden AIDS Symposium every year; thus, some honoraria are provided for speakers, including herself. Dr. Boesecke reports personal fees from Abbvie, Gilead, Janssen, MSD, and ViiV outside the submitted work. All other authors have nothing disclosed. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
Patient consent. Written informed consent was obtained from all subjects before enrollment. This study was performed in accordance with the Declaration of Helsinki and was approved by the institutional review board of the Technical University of Munich, Munich, Germany (approval number: 162/15), and by the German Federal Institute for Drugs and Medical Devices (Bundesinstitut für Arzneimittel und Medizinprodukte, BfArM), Bonn, Germany (approval number 4040568). This study is registered with Eudra-CT number 2015-000360-34.
Prior presentation. Parts of this study were presented at the International AIDS Society Conference in Mexico 2019 and the European AIDS Society Conference in Basel 2019.
References
European AIDS Clinical Society.


{kind=link}
{kind=link}
Comments