






Abstract
Febrile neutropenia (FN) is a frequent complication in immunocompromised patients. However, causative microorganisms are detected in only 10% of patients. This study aimed to detect the microorganisms that cause FN using next-generation sequencing (NGS) to identify the genome derived from pathogenic microorganisms in the bloodstream. Here, we implemented a metagenomic approach to comprehensively analyze microorganisms present in clinical samples from patients with FN.
FN is defined as a neutrophil count <500 cells/µL and fever ≥37.5°C. Plasma/serum samples of 112 pediatric patients with FN and 10 patients with neutropenia without fever (NE) were sequenced by NGS and analyzed by a metagenomic pipeline, PATHDET.
The putative pathogens were detected by NGS in 5 of 10 FN patients with positive blood culture results, 15 of 87 FN patients (17%) with negative blood culture results, and 3 of 8 NE patients. Several bacteria that were common in the oral, skin, and gut flora were commonly detected in blood samples, suggesting translocation of the human microbiota to the bloodstream in the setting of neutropenia. The cluster analysis of the microbiota in blood samples using NGS demonstrated that the representative bacteria of each cluster were mostly consistent with the pathogens in each patient.
NGS technique has great potential for detecting causative pathogens in patients with FN. Cluster analysis, which extracts characteristic microorganisms from a complex microbial population, may be effective to detect pathogens in minute quantities of microbiota, such as those from the bloodstream.
Febrile neutropenia (FN) is a frequent complication among patients with solid tumors and hematologic malignancies undergoing cytotoxic chemotherapy and hematopoietic stem cell transplantation [1] that occurs in approximately 10%–60% of cases [2]. The mortality rate of FN is high if proper antibiotic treatment is not given [3], and prompt empirical antibiotic administration is recommended for initial treatment [4]. Knowledge of expected pathogens and antimicrobial susceptibility patterns is required to optimize empiric treatment. In pediatric patients with FN, however, only 10% of patients had documented bacteremia whereas approximately 80% of patients exhibited fever of unknown origin [2]. Similarly, for adult FN, clinically documented infections occur in only 20%–30% of cases [4]. Poor microorganism detection is due to the lack of a nonexclusive culture system for covering all possible pathogens present in blood samples. Therefore, blood culture tests need to be assisted by more sensitive and comprehensive systems such as molecular biological tests. Establishing a method for the detection of bloodstream pathogens contributes to the improvement of disease prognosis and prevents the occurrence of drug-resistant bacteria.
Next-generation sequencing (NGS) is suitable for the comprehensive analysis of nucleic acids within a sample. In the field of clinical microbiology, NGS is considered to have the potential to detect traces of microbial genetic material from clinical samples of patients with infectious diseases [5]. In comparison with the 16S ribosomal DNA (rDNA) method, which is exclusively used for bacterial profiling, metagenomic NGS allows us to obtain entire genomic information of all microorganisms, including bacteria, viruses, fungi, and parasites, present in a clinical sample [6, 7]. Recently, metagenomic NGS has been reported as a clinical application for diagnosing infectious diseases [8–11], and we also previously reported that metagenomic NGS can be used to detect causative pathogens of bloodstream infections [12], acute encephalitis/encephalopathy [13], acute liver failure [14], severe respiratory failure [15] and acute myocarditis [16]. The present study aimed to detect causative pathogens in blood samples from pediatric patients with FN using metagenomic NGS analysis. Additionally, contamination from the experimental and bioinformatic analytical processes and translocation from the microbiota of the gut, oral cavity, skin, and vagina were analyzed. Moreover, clustering analysis was proposed to determine the pathogens in patients with FN. Metagenomic NGS has great potential for detecting putative pathogens in patients with FN.
MATERIALS AND METHODS
Patient Consent Statement
The study design and methods were approved by the Institutional Review Board of Nagoya University Hospital (IRB number 9069). The methods were carried out in accordance with approved guidelines. Written informed consent was obtained from all patients or their guardians.
Patients and Samples
We enrolled 122 pediatric patients at Nagoya University Hospital between August 2016 and October 2016 and between April 2018 and July 2018. A total of 112 FN patients were diagnosed using the following criteria: (1) neutrophil count <500 cells/µL or a higher count with a predicted decrease to <500/µL within 24 hours; and (2) fever ≥37.5°C. The remaining 10 patients showed neutropenia of <500 cells/µL without fever (hereafter “NE”). Patients’ characteristics are shown in Table 1. All enrolled patients were under prophylactic use of antimicrobial agents: 99% of the patients took sulfamethoxazole-trimethoprim and fluconazole. All FN patients received additional antibiotic treatment (Supplementary Tables 1 and 2). Finally, their neutrophil count recovered (>500 cells/µL) and they were in the defervescence phase (<37.5°C). Blood samples were collected at the same time as blood culture when patients were diagnosed with FN, and 8 patients were administered antibiotics before sampling. The samples were stored at −30°C until further analysis. Blood culture was performed according to the method described previously [12].
Characteristics of the 104 Patients
| Characteristic | Febrile Neutropenia (n = 97) | Neutropenia Without Fever (n = 7) | ||
|---|---|---|---|---|
| Age, y, median | 7 | … | 6 | … |
| Age distribution, No. (%) | ||||
| ≤2 y | 17 | (17.5) | 1 | (16.7) |
| 3–5 y | 24 | (24.7) | 2 | (33.3) |
| 6–10 y | 22 | (22.7) | 3 | (50.0) |
| 11–15 y | 27 | (27.8) | 1 | (16.7) |
| ≥16 y | 7 | (7.2) | 0 | (0) |
| Sex, male, No. (%) | 67 | (69.0) | 4 | (57.1) |
| Cause of neutropenia, No. (%) | ||||
| Cancer chemotherapy | 80 | (82.5) | 5 | (71.4) |
| HSCT | 12 | (12.4) | 1 | (14.3) |
| Underlying disease | 5 | (5.2) | 1 | (14.3) |
| Primary medical condition, No. (%) | ||||
| Leukemia | 43 | (44.3) | 3 | (50.0) |
| Solid tumors | 34 | (35.1) | 2 | (33.3) |
| Bone marrow failure | 9 | (9.3) | 1 | (16.7) |
| Lymphoma | 5 | (5.2) | 0 | (0) |
| CNS tumors | 4 | (4.1) | 1 | (16.7) |
| Primary immunodeficiency | 2 | (2.1) | 0 | (0) |
| Clinical symptoms, No. (%) | ||||
| Oral | 16 | (16.5) | 1 | (14.3) |
| Skin | 5 | (5.2) | 0 | (0) |
| Gastrointestinal | 20 | (20.6) | 0 | (0) |
| Other(s) | 5 | (5.2) | 0 | (0) |
| Blood culture test positive, No. (%) | 10 | (10.3) | … | … |
| Characteristic | Febrile Neutropenia (n = 97) | Neutropenia Without Fever (n = 7) | ||
|---|---|---|---|---|
| Age, y, median | 7 | … | 6 | … |
| Age distribution, No. (%) | ||||
| ≤2 y | 17 | (17.5) | 1 | (16.7) |
| 3–5 y | 24 | (24.7) | 2 | (33.3) |
| 6–10 y | 22 | (22.7) | 3 | (50.0) |
| 11–15 y | 27 | (27.8) | 1 | (16.7) |
| ≥16 y | 7 | (7.2) | 0 | (0) |
| Sex, male, No. (%) | 67 | (69.0) | 4 | (57.1) |
| Cause of neutropenia, No. (%) | ||||
| Cancer chemotherapy | 80 | (82.5) | 5 | (71.4) |
| HSCT | 12 | (12.4) | 1 | (14.3) |
| Underlying disease | 5 | (5.2) | 1 | (14.3) |
| Primary medical condition, No. (%) | ||||
| Leukemia | 43 | (44.3) | 3 | (50.0) |
| Solid tumors | 34 | (35.1) | 2 | (33.3) |
| Bone marrow failure | 9 | (9.3) | 1 | (16.7) |
| Lymphoma | 5 | (5.2) | 0 | (0) |
| CNS tumors | 4 | (4.1) | 1 | (16.7) |
| Primary immunodeficiency | 2 | (2.1) | 0 | (0) |
| Clinical symptoms, No. (%) | ||||
| Oral | 16 | (16.5) | 1 | (14.3) |
| Skin | 5 | (5.2) | 0 | (0) |
| Gastrointestinal | 20 | (20.6) | 0 | (0) |
| Other(s) | 5 | (5.2) | 0 | (0) |
| Blood culture test positive, No. (%) | 10 | (10.3) | … | … |
Fifteen FN patients and three NE patients were excluded due to low quality of their samples for sequencing.
Abbreviations: CNS, central nervous system; HSCT, hematopoietic stem cell transplantation.
Characteristics of the 104 Patients
| Characteristic | Febrile Neutropenia (n = 97) | Neutropenia Without Fever (n = 7) | ||
|---|---|---|---|---|
| Age, y, median | 7 | … | 6 | … |
| Age distribution, No. (%) | ||||
| ≤2 y | 17 | (17.5) | 1 | (16.7) |
| 3–5 y | 24 | (24.7) | 2 | (33.3) |
| 6–10 y | 22 | (22.7) | 3 | (50.0) |
| 11–15 y | 27 | (27.8) | 1 | (16.7) |
| ≥16 y | 7 | (7.2) | 0 | (0) |
| Sex, male, No. (%) | 67 | (69.0) | 4 | (57.1) |
| Cause of neutropenia, No. (%) | ||||
| Cancer chemotherapy | 80 | (82.5) | 5 | (71.4) |
| HSCT | 12 | (12.4) | 1 | (14.3) |
| Underlying disease | 5 | (5.2) | 1 | (14.3) |
| Primary medical condition, No. (%) | ||||
| Leukemia | 43 | (44.3) | 3 | (50.0) |
| Solid tumors | 34 | (35.1) | 2 | (33.3) |
| Bone marrow failure | 9 | (9.3) | 1 | (16.7) |
| Lymphoma | 5 | (5.2) | 0 | (0) |
| CNS tumors | 4 | (4.1) | 1 | (16.7) |
| Primary immunodeficiency | 2 | (2.1) | 0 | (0) |
| Clinical symptoms, No. (%) | ||||
| Oral | 16 | (16.5) | 1 | (14.3) |
| Skin | 5 | (5.2) | 0 | (0) |
| Gastrointestinal | 20 | (20.6) | 0 | (0) |
| Other(s) | 5 | (5.2) | 0 | (0) |
| Blood culture test positive, No. (%) | 10 | (10.3) | … | … |
| Characteristic | Febrile Neutropenia (n = 97) | Neutropenia Without Fever (n = 7) | ||
|---|---|---|---|---|
| Age, y, median | 7 | … | 6 | … |
| Age distribution, No. (%) | ||||
| ≤2 y | 17 | (17.5) | 1 | (16.7) |
| 3–5 y | 24 | (24.7) | 2 | (33.3) |
| 6–10 y | 22 | (22.7) | 3 | (50.0) |
| 11–15 y | 27 | (27.8) | 1 | (16.7) |
| ≥16 y | 7 | (7.2) | 0 | (0) |
| Sex, male, No. (%) | 67 | (69.0) | 4 | (57.1) |
| Cause of neutropenia, No. (%) | ||||
| Cancer chemotherapy | 80 | (82.5) | 5 | (71.4) |
| HSCT | 12 | (12.4) | 1 | (14.3) |
| Underlying disease | 5 | (5.2) | 1 | (14.3) |
| Primary medical condition, No. (%) | ||||
| Leukemia | 43 | (44.3) | 3 | (50.0) |
| Solid tumors | 34 | (35.1) | 2 | (33.3) |
| Bone marrow failure | 9 | (9.3) | 1 | (16.7) |
| Lymphoma | 5 | (5.2) | 0 | (0) |
| CNS tumors | 4 | (4.1) | 1 | (16.7) |
| Primary immunodeficiency | 2 | (2.1) | 0 | (0) |
| Clinical symptoms, No. (%) | ||||
| Oral | 16 | (16.5) | 1 | (14.3) |
| Skin | 5 | (5.2) | 0 | (0) |
| Gastrointestinal | 20 | (20.6) | 0 | (0) |
| Other(s) | 5 | (5.2) | 0 | (0) |
| Blood culture test positive, No. (%) | 10 | (10.3) | … | … |
Fifteen FN patients and three NE patients were excluded due to low quality of their samples for sequencing.
Abbreviations: CNS, central nervous system; HSCT, hematopoietic stem cell transplantation.
Sample Preparation and Sequencing
The NGS library was prepared from DNA extracted from 140 μL of 112 FN plasma, 10 NE plasma, and 3 distilled water used for library preparation as described previously [12]. Bioanalyzer high-sensitivity DNA assays (Agilent, Santa Clara, California) were used for sample quality control for NGS libraries. NGS libraries without the insertion sequence were excluded from sequencing except for those prepared from distilled water. Digital PCR using primers specific for the Illumina library adapter sequence by QX200 Droplet Digital PCR System (Bio-Rad, Richmond, California) was used for quantification of NGS libraries. NGS libraries of ≤12 copies/µL (equivalent to ≤0.1 nM) were excluded except for those prepared from distilled water. NGS libraries were then sequenced on the HiSeq system (Illumina, San Diego, California) with the 2 × 150-bp paired-end protocol or the MiSeq system (Illumina) with the 2 × 250-bp paired-end protocol.
Metagenomic Analysis of Obtained Sequencing Data
The NGS data generated by the Illumina sequencing platform were processed in the metagenomic pipeline PATHDET for the detection of pathogen-derived sequences [17]. The PATHDET pipeline comprises several steps conducted by different open-source software programs. First, as preprocessing, possible remaining sequencing adaptors were trimmed by Trim Galore [18] and quality filtered by PRINSEQ [19], and duplicates were identified and removed by CD-HIT [20]. Quality control was performed by FastQC [21] before and after preprocessing and summarized by MultiQC [22]. Second, as human genome subtraction, remaining sequence reads mapped to the human genome database by Bowtie2 [23] and Kraken2 [24] were removed. Third, Kraken2 and MegaBLAST [25] were used to search for highly similar species in the National Center for Biotechnology Information RefSeq database [26] from sequences that could not be classified by Kraken2. In addition, the ResFinder database [27] was used to detect acquired antibiotic resistance genes. Finally, the PATHDET pipeline integrated all the microorganisms found and reported candidates for pathogens. In detail, pathogen candidates were selected according to the following procedure. First, reads assigned Cutibacterium acnes were excluded from all analyzed data because C. acnes is likely to be a nonpathogenic/frequent contaminant of the NGS library [12]. Second, the PATHDET pipeline calculated 3 indicators: the total number of reads assigned microorganism per million total sequencing reads (PR), the number of reads assigned microorganism per million total sequencing reads (RPM), and Shannon diversity index (H′) based on each taxonomic hierarchy. Third, microorganisms satisfying PR >650, RPM >200, and H′ <3.0 were listed as pathogen candidates. These thresholds were set based on the analysis data of 7 patients with bacteremia in our previous study [12] (Supplementary Table 3 and Supplementary Figure 1).The mapping coverage in the reference genome was also calculated and used to evaluate pathogen candidates. PATHDET pipeline processed all data of sequenced samples. Samples for which the number of sequencing reads was <500 000 were excluded because of insufficient reads for analysis. We compared the results by the PATHDET pipeline with those from blood culture, DNA virus quantification, and the patient’s clinical course.
Microorganism abundance in each blood sample was described using the z score, which was normalized RPM as the number of standard deviations above or below the mean RPM for the total microorganisms in the sample at the family, genus, and species taxonomic hierarchy. Microorganisms with z scores ≥2.0 were defined as frequently detected microorganisms. The z score was calculated for microorganisms, including C. acnes, to account for contaminant microorganisms in all processes. The detection frequency of representative microorganism species of the human microbiome in the bloodstream [28, 29] was also compared between these samples. Comparison of microbiota among blood samples from patients was analyzed as follows using R packages “vegan” and “labdsv”: calculation of Morisita-Horn similarity index, hierarchical clustering by Ward method, selecting the number of clusters with silhouette analysis, and extraction of indicator species of each cluster by the IndVal method [30].
RESULTS
Pathogen Candidates Detected in Blood Samples by Metagenomic NGS
Blood samples from all 122 enrolled pediatric patients were processed into NGS libraries. Six libraries prepared from 4 FN and 2 NE patient samples were excluded due to low quality (Figure 1A). All 3 libraries prepared from distilled water were of low quality but were sequenced and analyzed as negative controls to assess contamination from the experimental procedure and environment. Total sequencing reads from distilled water were insufficient to detect putative pathogens (68 580 reads, 7104 reads, and 5874 reads). The majority of microorganisms detected in distilled water were C. acnes (82.9%, 86.5%, and 65.8%). Other microorganisms detected in distilled water did not exceed 10% of the total reads. An average of 6 573 252 reads per sample was sequenced and 12 cases (11 FN and 1 NE) were excluded because of an insufficient number of sequencing reads. The PATHDET pipeline reported 31 microorganisms as putative pathogens from 20 patients with FN (Table 2 and Figure 1B). Pathogen candidates were reported in 5 of 10 patients with positive blood cultures. The results for 3 (FN008, FN021, and FN070) of these 5 patients were consistent with the cultured bacteria (Supplementary Figure 2A). Thus, pathogen candidates by NGS were not identical to the results of blood culture in 7 of the 10 patients with positive blood cultures. However, each cultured bacterium in the 10 patients with positive blood cultures was detected by NGS for at least 1 sequencing read. Streptococcus mitis/oralis was not reported as a putative pathogen in all 3 FN patients with blood cultures positive for S. mitis/oralis. In FN patients with negative blood cultures, 15 of 87 patients (17%) had pathogen candidates detected by NGS (Supplementary Figure 2B). In 4 of these 15 patients, the pathogens were identified at the species level of taxonomic hierarchy: Acinetobacter soli, Burkholderia cepacia, and Klebsiella variicola. In addition, the pathogens were identified at the genus level in 6 patients (eg, Escherichia) and were identified at the family level in 5 patients (eg, Staphylococcaceae). In 3 of 8 patients with NE, Acinetobacter, Pseudomonas, and Brevundimonas were reported as pathogens at the genus level, even though these patients had no symptoms of infection. Forty-two FN patients had clinical symptoms other than fever (oral, skin, gastrointestinal, and other symptoms). Five of these patients were negative for blood culture and were positive for the presence of putative pathogens based on NGS. Antimicrobial resistance genes were investigated using NGS data from 104 FN and NE patients. A total of 108 resistance genes in the 45 patients are shown in Supplementary Table 4.
Identification of Putative Bacteria by Next-Generation Sequencing From Study Samples
| Patient | Category | Blood Culture Test | Taxonomic Hierarchy | NGS-Identified Pathogen | RPMa (Reads), No. | Coverage, % |
|---|---|---|---|---|---|---|
| FN006 | FN | Negative | Family | Sphingomonadaceae | 513 | … |
| Caulobacteraceae | 487 | … | ||||
| FN008 | FN | Rhizobium radiobacter | Genus | Agrobacterium | 312 | … |
| FN010 | FN | Negative | Family | Sphingomonadaceae | 282 | … |
| FN021 | FN | Acinetobacter ursingii | Species | Acinetobacter ursingii | 247 671 | 85.5 |
| FN022 | FN | Negative | Genus | Pseudomonas | 1395 | … |
| Ralstonia | 977 | … | ||||
| FN025 | FN | Negative | Genus | Pseudomonas | 290 | … |
| FN034 | FN | Negative | Family | Sphingomonadaceae | 500 | … |
| Comamonadaceae | 379 | … | ||||
| Pseudomonadaceae | 283 | … | ||||
| FN037 | FN | Negative | Species | Acinetobacter soli | 1170 | 3.6 |
| FN061 | FN | Negative | Genus | Brevundimonas | 1426 | … |
| Acinetobacter | 312 | … | ||||
| FN063 | FN | Negative | Family | Staphylococcaceae | 261 | … |
| FN070 | FN | Streptococcus mitis/oralis | Species | Acinetobacter soli | 1266 | 14.7 |
| Streptococcus mitis | 687 | 28.1 | ||||
| Brevundimonas sp DS20 | 334 | 7.5 | ||||
| Malassezia restricta | 228 | 3.4 | ||||
| FN071 | FN | Negative | Species | Burkholderia cepacia | 345 | 1.0 |
| FN073 | FN | Negative | Genus | Escherichia | 319 | … |
| Acinetobacter | 237 | … | ||||
| FN088 | FN | Streptococcus mitis/oralis | Species | Acinetobacter soli | 758 | 6.9 |
| Brevundimonas sp DS20 | 268 | 5.6 | ||||
| FN093 | FN | Negative | Genus | Roseomonas | 452 | … |
| FN099 | FN | Streptococcus mitis/oralis | Genus | Methylobacterium | 430 | … |
| FN100 | FN | Negative | Species | Acinetobacter soli | 306 | 35.3 |
| FN101 | FN | Negative | Species | Klebsiella variicola | 406 | 6.8 |
| FN104 | FN | Negative | Genus | Acinetobacter | 505 | … |
| Brevundimonas | 262 | … | ||||
| FN106 | FN | Negative | Family | Enterobacteriaceae | 551 | … |
| Staphylococcaceae | 203 | … | ||||
| NE01 | NE | … | Genus | Acinetobacter | 617 | … |
| NE04 | NE | … | Genus | Acinetobacter | 396 | … |
| NE07 | NE | … | Genus | Pseudomonas | 1590 | … |
| Brevundimonas | 205 | … |
| Patient | Category | Blood Culture Test | Taxonomic Hierarchy | NGS-Identified Pathogen | RPMa (Reads), No. | Coverage, % |
|---|---|---|---|---|---|---|
| FN006 | FN | Negative | Family | Sphingomonadaceae | 513 | … |
| Caulobacteraceae | 487 | … | ||||
| FN008 | FN | Rhizobium radiobacter | Genus | Agrobacterium | 312 | … |
| FN010 | FN | Negative | Family | Sphingomonadaceae | 282 | … |
| FN021 | FN | Acinetobacter ursingii | Species | Acinetobacter ursingii | 247 671 | 85.5 |
| FN022 | FN | Negative | Genus | Pseudomonas | 1395 | … |
| Ralstonia | 977 | … | ||||
| FN025 | FN | Negative | Genus | Pseudomonas | 290 | … |
| FN034 | FN | Negative | Family | Sphingomonadaceae | 500 | … |
| Comamonadaceae | 379 | … | ||||
| Pseudomonadaceae | 283 | … | ||||
| FN037 | FN | Negative | Species | Acinetobacter soli | 1170 | 3.6 |
| FN061 | FN | Negative | Genus | Brevundimonas | 1426 | … |
| Acinetobacter | 312 | … | ||||
| FN063 | FN | Negative | Family | Staphylococcaceae | 261 | … |
| FN070 | FN | Streptococcus mitis/oralis | Species | Acinetobacter soli | 1266 | 14.7 |
| Streptococcus mitis | 687 | 28.1 | ||||
| Brevundimonas sp DS20 | 334 | 7.5 | ||||
| Malassezia restricta | 228 | 3.4 | ||||
| FN071 | FN | Negative | Species | Burkholderia cepacia | 345 | 1.0 |
| FN073 | FN | Negative | Genus | Escherichia | 319 | … |
| Acinetobacter | 237 | … | ||||
| FN088 | FN | Streptococcus mitis/oralis | Species | Acinetobacter soli | 758 | 6.9 |
| Brevundimonas sp DS20 | 268 | 5.6 | ||||
| FN093 | FN | Negative | Genus | Roseomonas | 452 | … |
| FN099 | FN | Streptococcus mitis/oralis | Genus | Methylobacterium | 430 | … |
| FN100 | FN | Negative | Species | Acinetobacter soli | 306 | 35.3 |
| FN101 | FN | Negative | Species | Klebsiella variicola | 406 | 6.8 |
| FN104 | FN | Negative | Genus | Acinetobacter | 505 | … |
| Brevundimonas | 262 | … | ||||
| FN106 | FN | Negative | Family | Enterobacteriaceae | 551 | … |
| Staphylococcaceae | 203 | … | ||||
| NE01 | NE | … | Genus | Acinetobacter | 617 | … |
| NE04 | NE | … | Genus | Acinetobacter | 396 | … |
| NE07 | NE | … | Genus | Pseudomonas | 1590 | … |
| Brevundimonas | 205 | … |
Bacteria detected in blood cultures are shown in bold.
Abbreviations: FN, febrile neutropenia; NE, neutropenia without fever; NGS, next-generation sequencing.
aRPM indicates the number of reads assigned each pathogen per million total sequencing reads.
Identification of Putative Bacteria by Next-Generation Sequencing From Study Samples
| Patient | Category | Blood Culture Test | Taxonomic Hierarchy | NGS-Identified Pathogen | RPMa (Reads), No. | Coverage, % |
|---|---|---|---|---|---|---|
| FN006 | FN | Negative | Family | Sphingomonadaceae | 513 | … |
| Caulobacteraceae | 487 | … | ||||
| FN008 | FN | Rhizobium radiobacter | Genus | Agrobacterium | 312 | … |
| FN010 | FN | Negative | Family | Sphingomonadaceae | 282 | … |
| FN021 | FN | Acinetobacter ursingii | Species | Acinetobacter ursingii | 247 671 | 85.5 |
| FN022 | FN | Negative | Genus | Pseudomonas | 1395 | … |
| Ralstonia | 977 | … | ||||
| FN025 | FN | Negative | Genus | Pseudomonas | 290 | … |
| FN034 | FN | Negative | Family | Sphingomonadaceae | 500 | … |
| Comamonadaceae | 379 | … | ||||
| Pseudomonadaceae | 283 | … | ||||
| FN037 | FN | Negative | Species | Acinetobacter soli | 1170 | 3.6 |
| FN061 | FN | Negative | Genus | Brevundimonas | 1426 | … |
| Acinetobacter | 312 | … | ||||
| FN063 | FN | Negative | Family | Staphylococcaceae | 261 | … |
| FN070 | FN | Streptococcus mitis/oralis | Species | Acinetobacter soli | 1266 | 14.7 |
| Streptococcus mitis | 687 | 28.1 | ||||
| Brevundimonas sp DS20 | 334 | 7.5 | ||||
| Malassezia restricta | 228 | 3.4 | ||||
| FN071 | FN | Negative | Species | Burkholderia cepacia | 345 | 1.0 |
| FN073 | FN | Negative | Genus | Escherichia | 319 | … |
| Acinetobacter | 237 | … | ||||
| FN088 | FN | Streptococcus mitis/oralis | Species | Acinetobacter soli | 758 | 6.9 |
| Brevundimonas sp DS20 | 268 | 5.6 | ||||
| FN093 | FN | Negative | Genus | Roseomonas | 452 | … |
| FN099 | FN | Streptococcus mitis/oralis | Genus | Methylobacterium | 430 | … |
| FN100 | FN | Negative | Species | Acinetobacter soli | 306 | 35.3 |
| FN101 | FN | Negative | Species | Klebsiella variicola | 406 | 6.8 |
| FN104 | FN | Negative | Genus | Acinetobacter | 505 | … |
| Brevundimonas | 262 | … | ||||
| FN106 | FN | Negative | Family | Enterobacteriaceae | 551 | … |
| Staphylococcaceae | 203 | … | ||||
| NE01 | NE | … | Genus | Acinetobacter | 617 | … |
| NE04 | NE | … | Genus | Acinetobacter | 396 | … |
| NE07 | NE | … | Genus | Pseudomonas | 1590 | … |
| Brevundimonas | 205 | … |
| Patient | Category | Blood Culture Test | Taxonomic Hierarchy | NGS-Identified Pathogen | RPMa (Reads), No. | Coverage, % |
|---|---|---|---|---|---|---|
| FN006 | FN | Negative | Family | Sphingomonadaceae | 513 | … |
| Caulobacteraceae | 487 | … | ||||
| FN008 | FN | Rhizobium radiobacter | Genus | Agrobacterium | 312 | … |
| FN010 | FN | Negative | Family | Sphingomonadaceae | 282 | … |
| FN021 | FN | Acinetobacter ursingii | Species | Acinetobacter ursingii | 247 671 | 85.5 |
| FN022 | FN | Negative | Genus | Pseudomonas | 1395 | … |
| Ralstonia | 977 | … | ||||
| FN025 | FN | Negative | Genus | Pseudomonas | 290 | … |
| FN034 | FN | Negative | Family | Sphingomonadaceae | 500 | … |
| Comamonadaceae | 379 | … | ||||
| Pseudomonadaceae | 283 | … | ||||
| FN037 | FN | Negative | Species | Acinetobacter soli | 1170 | 3.6 |
| FN061 | FN | Negative | Genus | Brevundimonas | 1426 | … |
| Acinetobacter | 312 | … | ||||
| FN063 | FN | Negative | Family | Staphylococcaceae | 261 | … |
| FN070 | FN | Streptococcus mitis/oralis | Species | Acinetobacter soli | 1266 | 14.7 |
| Streptococcus mitis | 687 | 28.1 | ||||
| Brevundimonas sp DS20 | 334 | 7.5 | ||||
| Malassezia restricta | 228 | 3.4 | ||||
| FN071 | FN | Negative | Species | Burkholderia cepacia | 345 | 1.0 |
| FN073 | FN | Negative | Genus | Escherichia | 319 | … |
| Acinetobacter | 237 | … | ||||
| FN088 | FN | Streptococcus mitis/oralis | Species | Acinetobacter soli | 758 | 6.9 |
| Brevundimonas sp DS20 | 268 | 5.6 | ||||
| FN093 | FN | Negative | Genus | Roseomonas | 452 | … |
| FN099 | FN | Streptococcus mitis/oralis | Genus | Methylobacterium | 430 | … |
| FN100 | FN | Negative | Species | Acinetobacter soli | 306 | 35.3 |
| FN101 | FN | Negative | Species | Klebsiella variicola | 406 | 6.8 |
| FN104 | FN | Negative | Genus | Acinetobacter | 505 | … |
| Brevundimonas | 262 | … | ||||
| FN106 | FN | Negative | Family | Enterobacteriaceae | 551 | … |
| Staphylococcaceae | 203 | … | ||||
| NE01 | NE | … | Genus | Acinetobacter | 617 | … |
| NE04 | NE | … | Genus | Acinetobacter | 396 | … |
| NE07 | NE | … | Genus | Pseudomonas | 1590 | … |
| Brevundimonas | 205 | … |
Bacteria detected in blood cultures are shown in bold.
Abbreviations: FN, febrile neutropenia; NE, neutropenia without fever; NGS, next-generation sequencing.
aRPM indicates the number of reads assigned each pathogen per million total sequencing reads.
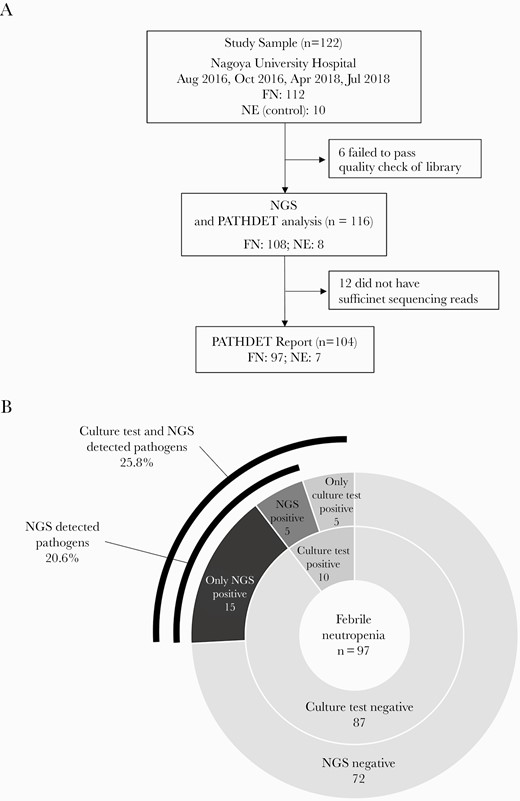
Flowchart and pie charts of the results in patients with febrile neutropenia (FN) and patients with neutropenia without fever (NE). A, Flowchart of the patient selection and comparison. B, Comparison of the results between next-generation sequencing (NGS) analysis and blood culture in patients with FN. The ratio of positive results in patients with FN is shown as a pie chart.
No viruses were reported as putative pathogens in the PATHDET pipeline. DNA viruses were detected in 19 patients (15 FN and 4 NE patients): Epstein-Barr virus in 1 case, human cytomegalovirus in 6 patients, human herpesvirus 6B in 2 patients, and torque teno virus in 13 patients. Fungus, Malassezia restricta, was detected as a pathogenic microorganism candidate in 1 case (FN070).
Commonly Detected Bacteria in Blood Samples and Translocation of the Human Microbiota to the Bloodstream
Microorganisms commonly detected in blood samples with FN are shown in Supplementary Table 5. Frequently detected microorganisms were similar among FN and NE patients, but patients with FN had more varied species compared with NE patients. In particular, C. acnes, which is frequently included in the skin microbiota, was the most abundant in blood samples of FN patients.
Translocation of human microbiota was investigated for affecting bacteremia in patients with FN. The frequency of commonly detected bacteria in blood samples was demonstrated as a heatmap in each respective group consisting of representative microorganism species in the oral cavity, skin, gut, or vagina (Figure 2). There was no difference in microorganism species between FN and NE patients. Streptococcus and Corynebacterium belonging to oral cavity flora and 3 bacteria belonging to skin flora were frequently detected by NGS.

Frequency of representative bacteria in blood samples from patients with febrile neutropenia assigned to the oral cavity, skin, gut, or vagina. The heatmap was drawn using the z score, which was normalized microbial reads as the number of standard deviations above or below the mean microbial reads for the total abundance of microorganisms in the sample at the family level of taxonomic hierarchy.
Cluster Analysis of the Microbiota in Blood Samples
The microbiota in blood samples was classified into 6 clusters by Ward method and silhouette analysis, which yielded a maximum silhouette score of 0.53 (Figure 3). After the clustering, we identified indicator species, the species that characteristically appeared in each cluster, to estimate the microorganisms associated with FN. The indicator species of each cluster were as follows: Propionibacteriaceae and Staphylococcaceae (cluster 1); Moraxellaceae (cluster 2); Enterobacteriaceae, Colwelliaceae, and Sporocadaceae (cluster 3); Streptococcaceae, Pasteurellaceae, and Alcaligenaceae (cluster 4), Pseudomonadaceae and Burkholderiaceae (cluster 5); and Bradyrhizobiaceae, Sphingomonadaceae, and Comamonadaceae (cluster 6). In 18 of the 23 cases (78.3%) where the PATHDET pipeline identified putative pathogens, the bacteria detected by NGS were consistent with the indicator species.
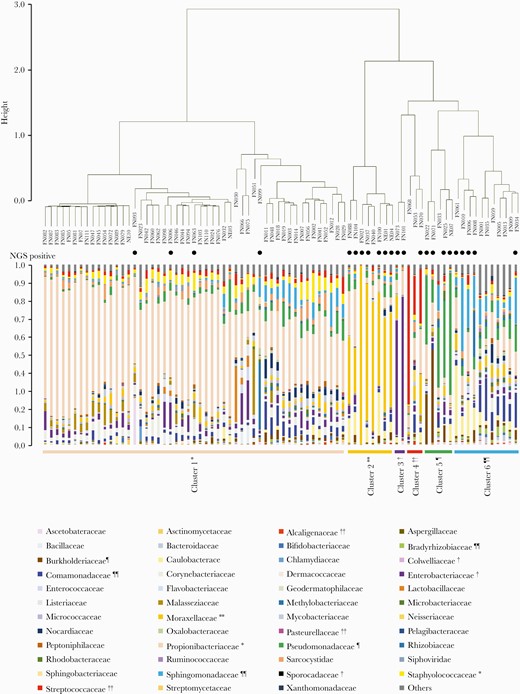
Cluster analysis of the microbiota in blood samples from patients with febrile neutropenia. Comparison of microbiota among blood samples from patients was analyzed, and selected clusters with silhouette analysis and extraction of indicator species of each cluster are shown. Closed circles indicate samples in which putative pathogens were identified by metagenomic next-generation sequencing (NGS).
DISCUSSION
To our knowledge, this is the first study to demonstrate the clinical application of metagenomic NGS for FN on a large scale. Interestingly, S. mitis was cultured in 3 of 5 FN patients who were blood culture positive but NGS negative. This may be due to poor bacterial DNA extraction of S. mitis due to the presence of a peptidoglycan layer that is difficult to destroy. DNA extraction was reported to have the largest effect on the outcome of metagenomic analysis in comparison with differences due to library preparation and sample storage [27]. In that study, 21 representative DNA extraction protocols were tested, and ascertainment biases were found to exist in estimates of community diversity and the ratio between gram-positive and gram-negative bacteria in the analyses for human fecal samples. Improvement of DNA extraction methods is required for comprehensive identification of putative pathogens by NGS. Next, previous reports have described the use of NGS methods for detecting pathogens in infectious diseases [9, 31, 32]. In these studies, threshold numbers of microbial sequencing reads were adopted for diagnosis, although there were differences between studies, such as requiring their own internal controls. However, this threshold has not yet been standardized. In our previous study [12], we considered that bacterial reads >200 were significant for diagnosis. To evaluate the microbial sequencing reads, the percentages of the reads of the dominant bacteria and microbial diversity were also calculated.
Putative pathogen candidates were found in 3 of 7 blood samples of NE patients. In these NE patients, blood collected via a central venous catheter at routine blood testing was used for NGS analysis. One explanation is that bacteria-forming colonies in the catheter lumen were detected by NGS. We previously reported that pathogenic bacteria were detected from catheters in patients who subsequently developed bloodstream infections [12]. However, NE patients whose pathogen was identified by NGS did not develop fever during the observation period.
Putative pathogens were detected in 15 of 87 FN patients (17.2%) with negative blood cultures. This detection rate in patients with negative blood cultures seems low in comparison with previous reports where pathogens were found by shotgun metagenomic sequencing in 22%–24% of patients undiagnosed for pathogens [9, 31]. This difference may be related to disease type; that is, these previous studies investigated meningitis, encephalitis, and sepsis, not FN. Different applications of NGS analysis also influence the detection rate of causative pathogens. There are 2 major applications: comprehensive metagenomic analysis and targeted 16S ribosomal RNA (rRNA) analysis. In a previous study, 16S rRNA analysis was performed using blood samples collected at onset of FN and demonstrated that the positivity rate was 13.3% (4 of 30 FN patients with negative blood cultures) [33]. This result is similar to ours using the metagenomic analysis. There is another point of comparison between these 2 applications: 16S rRNA sequencing provides only microbiome data at the family or genus level and does not include quantification of each microorganism and species-level identification. The metagenomic analysis in the present study is more useful in terms of specifying pathogenic microorganisms at the species level. In addition, all bacteria identified by metagenomic NGS from blood samples of FN patients with negative blood cultures could be cultured by blood culture procedures. The reason why these bacteria were not cultured may be that the patients were receiving prophylactic antibiotic treatment.
There were some commonly detected microorganisms in blood samples analyzed in this study. One interpretation is that environmental microorganisms may be contaminants of blood collection techniques and experimental procedures; that is, microorganisms detected by metagenomic NGS may not be derived from blood samples [34, 35]. Cutibacterium acnes was a representative bacterium that was detected by metagenomic analysis of distilled water, suggesting this was likely to be introduced by experimental procedures. Therefore, we excluded C. acnes from causative pathogen candidates. Recently, incomplete references in the database occasionally caused both false-positive and false-negative results [36]. Toxoplasma gondii, which is unlikely to exist clinically, may be a representative bioinformatics contaminant. Therefore, continued understanding of potential database contaminants and improving the reference database for infectious disease diagnosis are important. In our study, we found that the microorganisms listed in Supplementary Table 5 are potential false positives; further investigation is required. Moreover, the translocation of human microbiota plays a decisive role in bacteremia [37, 38]. There is a high risk of bloodstream infection due to human microbiota in the setting of neutropenia. As shown in Figure 2, several bacteria of oral, skin, and gut florae were commonly detected, suggesting that translocation of the human microbiota to the bloodstream may contribute to FN.
We attempted to classify metagenomic data into several clusters represented by indicator putative pathogens. These indicator pathogens were determined by comparing the composition of DNA data derived from microorganisms in blood samples. The indicators were also associated with putative pathogens and common contaminants in metagenomic composition from blood samples. This method may help to interpret complex metagenomic data and highlights pathogenic candidates in the process of bioinformatics analysis. In patient FN040, NGS was negative as defined by our diagnostic criteria and Moraxellaceae were not detected as a pathogen candidate. However, cluster analysis demonstrated that Moraxellaceae could be the pathogen candidate (Figure 3, cluster 2). Similar results were observed for Streptococcaceae in patients FN053 and FN068 in cluster 4 and Pseudomonadaceae in FN033 in cluster 5. Thus, this analytic method can be improved by accumulating metagenomic data for diagnosing causative pathogens in infectious diseases.
In conclusion, metagenomic NGS has great potential for detecting causative pathogens in patients with FN. Our data also demonstrated the probability of contamination from experimental and bioinformatic analytical processes and translocation of microbiota from other organs to the bloodstream. Finally, cluster analysis showing each indicator bacterium and other composition bacteria provides useful information to delineate pathogens in infectious diseases.
Supplementary Data
Supplementary materials are available at Open Forum Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
Notes
Data availability. The data that support the findings of this study are available from the corresponding author upon reasonable request. Sequencing reads generated in this study were deposited to the DDBJ Sequence Read Archive under experiment number DRX241793-241917.
Financial support. This work was supported by a Japan Society for the Promotion of Science Grant-in-Aid for Scientific Research (grant number 19K08298 to Y. I.).
Author contributions. K. H., T. Og., and Y. I. designed the study. Y. To., T. Ok., S. A., S. T., T. S., J. K., H. M., and Y. Ta. collected clinical samples and data. K. H., Y. To., and T. Ok. performed experiments. K. H. developed the new analysis pipeline. K. H., T. S., and Y. To. conducted bioinformatics analyses. K. H., T. Og., and Y. I. drafted the manuscript. All authors read and approved the final manuscript.
Potential conflicts of interest. All authors: No reported conflicts of interest.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments