






Abstract
Background. Successful treatment of infections caused by multidrug-resistant (MDR) Pseudomonas aeruginosa is thwarted by the emergence of antibiotic resistance and biofilm formation on prosthetic devices. Our aims were to decipher the molecular basis of resistance in a unique case of prosthetic valve endocarditis (PVE) caused by MDR P. aeruginosa.
Methods. Five sequential MDR P. aeruginosa blood isolates collected during a 7-month period were recovered from a patient suffering from PVE previously exposed to β-lactam antibiotics. Minimum inhibitory concentrations (MICs) of several classes of antibiotics were used to indicate clinical resistance characteristics; relatedness of the isolates was determined using multilocus sequence typing and repetitive sequence-based polymerase chain reaction. Amplification and sequencing of regulatory and resistance genes was performed.
Results. All isolates belonged to ST 298, possessed blaPDC-16, and were resistant to fluoroquinolones and carbapenems. In the course of therapy, we observed a >2-fold increase in cephalosporin resistance (4 µg/mL to >16 µg/mL). Sequencing of the AmpC regulator, ampR, revealed a D135N point mutation in cephalosporin-resistant isolates. Common carbapenemase genes were not identified. All isolates demonstrated a premature stop codon at amino acid 79 of the outer membrane protein OprD and mutations in the quinolone resistance-determining regions of gyrA and parC. Point mutations in nalC, an efflux pump regulator, were also observed.
Conclusions. In this analysis, we chart the molecular evolution of β-lactam resistance in a case of PVE. We show that mutations in regulatory genes controlling efflux and cephalosporinase production contributed to the MDR phenotype.
Prosthetic valve endocarditis (PVE) is a potentially life-threatening complication of cardiac valve replacement surgery. The most common pathogens found to cause infective endocarditis (IE) are Gram-positive species, primarily Staphylococcus aureus, whereas endocarditis caused by Gram-negative species is less common and often attributed to HACEK organisms, which include the following: Haemophilus species, Aggregatibacter species, Cardiobacterium hominis, Eikenella corrodens, and Kingella species [1]. Pseudomonas aeruginosa is an extremely rare cause of PVE; a large international cohort study showed that the incidence is 0.4%, or 11 of 2671 cases [2]. Biofilm formation is also a distinguishing characteristic of P. aeruginosa that complicates treatment [3].
Molecular methods are extremely valuable in determining the etiology of IE and assist in clarifying the epidemiology and course of the disease, especially when repeated episodes of infection are caused by the same strain of species and can be defined as a relapse regardless of the length of time between infections [4]. Similarly, molecular analysis serves to uncover the mechanisms of antibiotic resistance that pathogens possess and that impact therapeutic success. Thus, understanding the molecular basis of the disease (applying precision medicine) can explain the clinical course and outcome of IE.
In this report, we sought to investigate the molecular basis of a persistent case of PVE due to P. aeruginosa. The case is unique as isolates characterized were collected serially during the course of treatment, occurring throughout 2 aortic valve replacements (AVRs). The different resistance profiles between early and late isolates, in addition to the patient's history of antibiotic therapy, assisted us in the understanding of the complex relationship between medical and surgical interventions and emergent antibiotic resistance.
Case Presentation
A 58-year-old woman treated with prednisone for Takayasu's arteritis and with a previous AVR (2007) presented to a Houston, Texas tertiary hospital initially in June 2010. She was hospitalized due to worsening shortness of breath. At that time, a transthoracic echocardiograph (TTE) revealed evidence of a prosthetic valvular incompetence, corroborated by the presence of torn cusps and severe aortic regurgitation. AVR surgery was performed. On postoperative day 24, P. aeruginosa bacteremia (isolate 7166; Table 1) was detected, and Achromobacter bacteriuria was present.
Antimicrobial Susceptibility Testing (µg/mL)
| Isolate Number | Isolate ID | Date | AMK | ATM | FEP | CAZ | CIP | IMP | MEM | LVX | GEN | TZP | CST |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 7166 | 7/28/10 | ≤16 | ≤8 | ≤8 | 4 | >2 | >8 | >8 | >4 | >8 | ≤16 | 2 |
| 3 | 2961 | 1/14/11 | ≤16 | >16 | >16 | >16 | >2 | >8 | >8 | >4 | >8 | >64 | 1 |
| 4 | 7927 | 1/20/11 | ≤16 | >16 | >16 | >16 | >2 | >8 | >8 | >4 | >8 | >64 | 1 |
| 5 | 8649 | 2/8/11 | ≤16 | >16 | >16 | >16 | >2 | >8 | >8 | >4 | >8 | >64 | 12 |
| 6 | 8096 | 2/19/11 | 32 | >16 | >16 | >16 | >2 | >8 | >8 | >4 | >8 | >64 | 3 |
| Isolate Number | Isolate ID | Date | AMK | ATM | FEP | CAZ | CIP | IMP | MEM | LVX | GEN | TZP | CST |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 7166 | 7/28/10 | ≤16 | ≤8 | ≤8 | 4 | >2 | >8 | >8 | >4 | >8 | ≤16 | 2 |
| 3 | 2961 | 1/14/11 | ≤16 | >16 | >16 | >16 | >2 | >8 | >8 | >4 | >8 | >64 | 1 |
| 4 | 7927 | 1/20/11 | ≤16 | >16 | >16 | >16 | >2 | >8 | >8 | >4 | >8 | >64 | 1 |
| 5 | 8649 | 2/8/11 | ≤16 | >16 | >16 | >16 | >2 | >8 | >8 | >4 | >8 | >64 | 12 |
| 6 | 8096 | 2/19/11 | 32 | >16 | >16 | >16 | >2 | >8 | >8 | >4 | >8 | >64 | 3 |
Antimicrobial susceptibility tests were interpreted according to 2015 Clinical Laboratory Standards Institute (CLSI) criteria for Pseudomonas aeruginosa: for aztreonam (ATM), cefepime (FEP), and ceftazidime (CAZ), minimum inhibitory concentration (MIC) ≤ 8 is susceptible (S), MIC = 16 is intermediate (I), and MIC ≥ 32 is resistant (R); for meropenem (MEM), imipenem (IMP), levofloxacin (LVX), and colistin (CST), MIC ≤ 2 is S, MIC = 4 is I, MIC ≥ 8 is R; for amikacin (AMK), MIC ≤ 16 is S, MIC = 32 is I, and MIC ≥ 64 is R; for ciprofloxacin (CIP), MIC ≤ 1 is S, MIC = 2 is I, MIC ≥ 4 is R MIC; for gentamicin (GEN), MIC ≤ 4 is S, MIC = 8 is I, MIC ≥ 16 is R; for piperacillin/tazobactam (TZP), MIC ≤ 16 is S, MIC = 32–64 is I, MIC ≥ 128 is R.
Antimicrobial Susceptibility Testing (µg/mL)
| Isolate Number | Isolate ID | Date | AMK | ATM | FEP | CAZ | CIP | IMP | MEM | LVX | GEN | TZP | CST |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 7166 | 7/28/10 | ≤16 | ≤8 | ≤8 | 4 | >2 | >8 | >8 | >4 | >8 | ≤16 | 2 |
| 3 | 2961 | 1/14/11 | ≤16 | >16 | >16 | >16 | >2 | >8 | >8 | >4 | >8 | >64 | 1 |
| 4 | 7927 | 1/20/11 | ≤16 | >16 | >16 | >16 | >2 | >8 | >8 | >4 | >8 | >64 | 1 |
| 5 | 8649 | 2/8/11 | ≤16 | >16 | >16 | >16 | >2 | >8 | >8 | >4 | >8 | >64 | 12 |
| 6 | 8096 | 2/19/11 | 32 | >16 | >16 | >16 | >2 | >8 | >8 | >4 | >8 | >64 | 3 |
| Isolate Number | Isolate ID | Date | AMK | ATM | FEP | CAZ | CIP | IMP | MEM | LVX | GEN | TZP | CST |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 7166 | 7/28/10 | ≤16 | ≤8 | ≤8 | 4 | >2 | >8 | >8 | >4 | >8 | ≤16 | 2 |
| 3 | 2961 | 1/14/11 | ≤16 | >16 | >16 | >16 | >2 | >8 | >8 | >4 | >8 | >64 | 1 |
| 4 | 7927 | 1/20/11 | ≤16 | >16 | >16 | >16 | >2 | >8 | >8 | >4 | >8 | >64 | 1 |
| 5 | 8649 | 2/8/11 | ≤16 | >16 | >16 | >16 | >2 | >8 | >8 | >4 | >8 | >64 | 12 |
| 6 | 8096 | 2/19/11 | 32 | >16 | >16 | >16 | >2 | >8 | >8 | >4 | >8 | >64 | 3 |
Antimicrobial susceptibility tests were interpreted according to 2015 Clinical Laboratory Standards Institute (CLSI) criteria for Pseudomonas aeruginosa: for aztreonam (ATM), cefepime (FEP), and ceftazidime (CAZ), minimum inhibitory concentration (MIC) ≤ 8 is susceptible (S), MIC = 16 is intermediate (I), and MIC ≥ 32 is resistant (R); for meropenem (MEM), imipenem (IMP), levofloxacin (LVX), and colistin (CST), MIC ≤ 2 is S, MIC = 4 is I, MIC ≥ 8 is R; for amikacin (AMK), MIC ≤ 16 is S, MIC = 32 is I, and MIC ≥ 64 is R; for ciprofloxacin (CIP), MIC ≤ 1 is S, MIC = 2 is I, MIC ≥ 4 is R MIC; for gentamicin (GEN), MIC ≤ 4 is S, MIC = 8 is I, MIC ≥ 16 is R; for piperacillin/tazobactam (TZP), MIC ≤ 16 is S, MIC = 32–64 is I, MIC ≥ 128 is R.
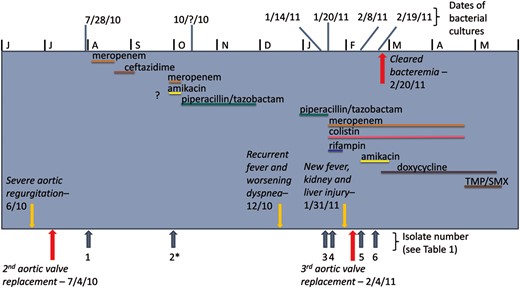
Timeline of antibiotic therapy, bacterial cultures, and patient health information. ?, Treated at outside hospital, unclear details; *, Isolate 2 recoved at outside hospital, not included in analysis.
The patient returned to the tertiary hospital in Houston, Texas, in January 2011 with recurrent fever and dyspnea of 1 month duration. Her cultures again grew P. aeruginosa (isolate 2961; Table 1). Piperacillin/tazobactam therapy was started based upon previous susceptibilities. This regimen changed to colistin, rifampin, and high-dose, prolonged infusion meropenem once susceptibility data were determined during admission. The patient's blood cultures cleared, but TTE showed evidence of aortic regurgitation. Subsequently, the patient was found to have dehiscence of the new valve and underwent a third aortic valve replacement with homograft placed 215 days after the second AVR. Despite ongoing antibiotics (including the addition of amikacin), the bacteremia persisted until postoperative day 16 after the addition of doxycycline to meropenem and colistin. Antibiotics were stopped after 8 weeks of PVE treatment, which lead to microbiological resolution.
During the course of treatment, 6 sequential P. aeruginosa blood isolates were collected, 1 of which was not included in this analysis because it was obtained while the patient was in an outside care facility and was not available for further analysis. The date of each isolate collection, relevant to the timeline of antibiotic therapy and patient health information, is detailed in Figure 1. The 5 isolates of interest were analyzed to understand the molecular mechanisms behind the observed clinical presentation.
METHODS AND RESULTS
Genotypic Analysis
Herein, we analyze a complicated case of PVE caused by MDR P. aeruginosa, where resistance to cephalosporins and aztreonam emerged while on therapy. This is one of a few studies where the genetic backgrounds of multiple isolates of P. aeruginosa collected from the same patient during a period of 7 months were analyzed to explain the resistance phenotype and correlate with the course of infection.
Susceptibility testing to antibiotics was performed using the MicroScan System (Siemens Healthcare Diagnostics, Tarrytown, NY) and interpreted according to the 2015 Clinical and Laboratory Standards Institute guidelines for P. aeruginosa. American Type Culture Collection control strains P. aeruginosa 27853 and E. coli 25922 were used as control strains. Colistin minimum inhibitory concentrations (MICs) were determined by Etest. All isolates were resistant to carbapenems and fluoroquinolones [ciprofloxacin (MIC >2 µg/mL), meropenem, and imipenem (MICs >8 µg/mL)]. Whereas the initial isolate was susceptible to aztreonam (ATM; MIC of ≤8 mg/mL), cefepime (FEP; MIC of 8 µg/mL), and ceftazidime (CAZ; MIC of 4 µg/mL), subsequent isolates were resistant (MIC >16 µg/mL) (Table 1). There was a clinically important change in this susceptibility (ie, CAZ, ATM, and FEP, from susceptible to resistant) during the 7 months isolates were collected.
β-lactamase genetic amplification and detection was performed using the CT103 kit produced by Check-Points B.V. (Wageningen, The Netherlands) This is a polymerase chain reaction (PCR)–based DNA microarray method for detecting the following β-lactamase (bla) genes: blaKPC, blaNDM, blaOXA-48, blaVIM, and blaIMP (carbapenemases). Additionally, this method detects extended-spectrum β-lactamase (ESBL) genes and AmpC genes: blaTEM, blaSHV, blaCTX-M, blaCMY, blaDHA, blaFOX, blaMOX, blaACC, blaMIR, and blaACT. For blaTEM and blaSHV, the assay method allows for distinction between ESBL and non-ESBL variants that exist within an isolate. Briefly, genomic DNA was extracted from bacterial colonies grown overnight using the DNeasy Blood and Tissue Kit (Qiagen, Inc., Valencia, CA). Microarray assays were performed generating templates of the target DNA sequence that were amplified and hybridized, detected by the microarray reader, and interpreted using software supplied by the manufacturer. The panel of β-lactamases that are detected by the microarray are optimized for Enterobacteriaceae. The performance of this kit using P. aeruginosa is being explored and tested. We emphasize that carbapenemase genes were not detected.
PCR amplifications of genes involved in resistance to cephalosporins (ie, ampD, ampR, blaPDC, and dacB), resistance to fluoroquinolones (ie, gyrA, gyrB, parC, parE), other mechanisms of resistance to carbapenems (oprD), and regulation of efflux pumps (ie, nalB (mexR), nalC, nalD, PA5471, and mexZ) were also performed using the primers and methodology described by Cabot et al [5]. Amplicons obtained using primers for the above resistance genes were sequenced at a commercial sequencing facility (MCLAB, South San Francisco, CA). Generated sequences were analyzed using DNAstar SeqMan Pro software (Madison, WI). Generated sequences were then compared with BLAST online software (https://blast.ncbi.nlm.nih.gov), or translated in ExPASy (http://web.expasy.org/translate/), then aligned using ClustalX software (www.clustal.org) to determine DNA mutations that resulted in amino acid mutations.
Cephalosporin Resistance
Summary of Mutations
| Isolate Number | Isolate ID | AmpC | PDC Substitutions | AmpC Regulators | QRDRs | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| AmpD and DacB | AmpR | GyrA | GyrB | ParC | ParE | |||||
| PA01 | ST Type | PDC-1 | WT | WT | WT | WT | WT | WT | WT | |
| 1 | 7166 | 298 | PDC-16 | G27D, T105A, V205L, G391A | WT | M1L, G238E, M288R | T83I | WT | S87L | WT |
| 3 | 2961 | 298 | PDC-16 | G27D, T105A, V205L, G391A | WT | M1L, D135N, G238E, M288R | T83I | WT | S87L | WT |
| 4 | 7927 | 298 | PDC-16 | G27D, T105A, V205L, G391A | WT | M1L, D135N, G238E, M288R | T83I | WT | S87L | WT |
| 5 | 8649 | 298 | PDC-16 | G27D, T105A, V205L, G391A | WT | M1L, D135N, G238E, M288R | T83I | WT | S87L | WT |
| 6 | 8096 | 298 | PDC-16 | G27D, T105A, V205L, G391A | WT | M1L, D135N, G238E, M288R | T83I | WT | S87L | WT |
| Isolate Number | Isolate ID | AmpC | PDC Substitutions | AmpC Regulators | QRDRs | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| AmpD and DacB | AmpR | GyrA | GyrB | ParC | ParE | |||||
| PA01 | ST Type | PDC-1 | WT | WT | WT | WT | WT | WT | WT | |
| 1 | 7166 | 298 | PDC-16 | G27D, T105A, V205L, G391A | WT | M1L, G238E, M288R | T83I | WT | S87L | WT |
| 3 | 2961 | 298 | PDC-16 | G27D, T105A, V205L, G391A | WT | M1L, D135N, G238E, M288R | T83I | WT | S87L | WT |
| 4 | 7927 | 298 | PDC-16 | G27D, T105A, V205L, G391A | WT | M1L, D135N, G238E, M288R | T83I | WT | S87L | WT |
| 5 | 8649 | 298 | PDC-16 | G27D, T105A, V205L, G391A | WT | M1L, D135N, G238E, M288R | T83I | WT | S87L | WT |
| 6 | 8096 | 298 | PDC-16 | G27D, T105A, V205L, G391A | WT | M1L, D135N, G238E, M288R | T83I | WT | S87L | WT |
| OMP | MexAB-OprM regulators | MexXY-OprM regulators | |||||
|---|---|---|---|---|---|---|---|
| Isolate ID | OprD | NalB (MexR) | NalC | NalD | PA5471 | MexZ | |
| Isolate Number | PA01 | WT | WT | WT | WT | WT | WT |
| 1 | 7166 | Premature stop codon at aa 79 | V126E | G71E, A145V, S209R | WT | Won't amplify | WT |
| 3 | 2961 | Premature stop codon at aa 79 | V126E | G71E, A145V, S209R | WT | Won't amplify | WT |
| 4 | 7927 | Premature stop codon at aa 79 | V126E | G71E, A145V, S209R | WT | Won't amplify | WT |
| 5 | 8649 | Premature stop codon at aa 79 | V126E | G71E, A145V, S209R | WT | Won't amplify | WT |
| 6 | 8096 | Premature stop codon at aa 79 | V126E | G71E, A145V, S209R | WT | Won't amplify | WT |
| OMP | MexAB-OprM regulators | MexXY-OprM regulators | |||||
|---|---|---|---|---|---|---|---|
| Isolate ID | OprD | NalB (MexR) | NalC | NalD | PA5471 | MexZ | |
| Isolate Number | PA01 | WT | WT | WT | WT | WT | WT |
| 1 | 7166 | Premature stop codon at aa 79 | V126E | G71E, A145V, S209R | WT | Won't amplify | WT |
| 3 | 2961 | Premature stop codon at aa 79 | V126E | G71E, A145V, S209R | WT | Won't amplify | WT |
| 4 | 7927 | Premature stop codon at aa 79 | V126E | G71E, A145V, S209R | WT | Won't amplify | WT |
| 5 | 8649 | Premature stop codon at aa 79 | V126E | G71E, A145V, S209R | WT | Won't amplify | WT |
| 6 | 8096 | Premature stop codon at aa 79 | V126E | G71E, A145V, S209R | WT | Won't amplify | WT |
Reference wild-type comparator amino acid sequences from Pseudomonas aeruginosa PA01 isolate listed in Table 2 are as follows:
PDC-1, ACQ82807; AmpD, AAG07910; DacB, NP_251737; AmpR, AAG07496; GyrA, AAG06556; GyrB, AAG03394; ParC, NP_253651; ParE, AAG08352; OprD, NP_249649; NalB (MexR) AAG03813; NalC, AAG07108; NalD, AAG06962; PA5471, AAG08856; MexZ, AF073776.
Abbreviations: PDC, Pseudomonas-derived cephalosporinase; QRDR, quinolone resistance-determining region; ST, sequence type; WT, wild-type or no mutation.
Summary of Mutations
| Isolate Number | Isolate ID | AmpC | PDC Substitutions | AmpC Regulators | QRDRs | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| AmpD and DacB | AmpR | GyrA | GyrB | ParC | ParE | |||||
| PA01 | ST Type | PDC-1 | WT | WT | WT | WT | WT | WT | WT | |
| 1 | 7166 | 298 | PDC-16 | G27D, T105A, V205L, G391A | WT | M1L, G238E, M288R | T83I | WT | S87L | WT |
| 3 | 2961 | 298 | PDC-16 | G27D, T105A, V205L, G391A | WT | M1L, D135N, G238E, M288R | T83I | WT | S87L | WT |
| 4 | 7927 | 298 | PDC-16 | G27D, T105A, V205L, G391A | WT | M1L, D135N, G238E, M288R | T83I | WT | S87L | WT |
| 5 | 8649 | 298 | PDC-16 | G27D, T105A, V205L, G391A | WT | M1L, D135N, G238E, M288R | T83I | WT | S87L | WT |
| 6 | 8096 | 298 | PDC-16 | G27D, T105A, V205L, G391A | WT | M1L, D135N, G238E, M288R | T83I | WT | S87L | WT |
| Isolate Number | Isolate ID | AmpC | PDC Substitutions | AmpC Regulators | QRDRs | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| AmpD and DacB | AmpR | GyrA | GyrB | ParC | ParE | |||||
| PA01 | ST Type | PDC-1 | WT | WT | WT | WT | WT | WT | WT | |
| 1 | 7166 | 298 | PDC-16 | G27D, T105A, V205L, G391A | WT | M1L, G238E, M288R | T83I | WT | S87L | WT |
| 3 | 2961 | 298 | PDC-16 | G27D, T105A, V205L, G391A | WT | M1L, D135N, G238E, M288R | T83I | WT | S87L | WT |
| 4 | 7927 | 298 | PDC-16 | G27D, T105A, V205L, G391A | WT | M1L, D135N, G238E, M288R | T83I | WT | S87L | WT |
| 5 | 8649 | 298 | PDC-16 | G27D, T105A, V205L, G391A | WT | M1L, D135N, G238E, M288R | T83I | WT | S87L | WT |
| 6 | 8096 | 298 | PDC-16 | G27D, T105A, V205L, G391A | WT | M1L, D135N, G238E, M288R | T83I | WT | S87L | WT |
| OMP | MexAB-OprM regulators | MexXY-OprM regulators | |||||
|---|---|---|---|---|---|---|---|
| Isolate ID | OprD | NalB (MexR) | NalC | NalD | PA5471 | MexZ | |
| Isolate Number | PA01 | WT | WT | WT | WT | WT | WT |
| 1 | 7166 | Premature stop codon at aa 79 | V126E | G71E, A145V, S209R | WT | Won't amplify | WT |
| 3 | 2961 | Premature stop codon at aa 79 | V126E | G71E, A145V, S209R | WT | Won't amplify | WT |
| 4 | 7927 | Premature stop codon at aa 79 | V126E | G71E, A145V, S209R | WT | Won't amplify | WT |
| 5 | 8649 | Premature stop codon at aa 79 | V126E | G71E, A145V, S209R | WT | Won't amplify | WT |
| 6 | 8096 | Premature stop codon at aa 79 | V126E | G71E, A145V, S209R | WT | Won't amplify | WT |
| OMP | MexAB-OprM regulators | MexXY-OprM regulators | |||||
|---|---|---|---|---|---|---|---|
| Isolate ID | OprD | NalB (MexR) | NalC | NalD | PA5471 | MexZ | |
| Isolate Number | PA01 | WT | WT | WT | WT | WT | WT |
| 1 | 7166 | Premature stop codon at aa 79 | V126E | G71E, A145V, S209R | WT | Won't amplify | WT |
| 3 | 2961 | Premature stop codon at aa 79 | V126E | G71E, A145V, S209R | WT | Won't amplify | WT |
| 4 | 7927 | Premature stop codon at aa 79 | V126E | G71E, A145V, S209R | WT | Won't amplify | WT |
| 5 | 8649 | Premature stop codon at aa 79 | V126E | G71E, A145V, S209R | WT | Won't amplify | WT |
| 6 | 8096 | Premature stop codon at aa 79 | V126E | G71E, A145V, S209R | WT | Won't amplify | WT |
Reference wild-type comparator amino acid sequences from Pseudomonas aeruginosa PA01 isolate listed in Table 2 are as follows:
PDC-1, ACQ82807; AmpD, AAG07910; DacB, NP_251737; AmpR, AAG07496; GyrA, AAG06556; GyrB, AAG03394; ParC, NP_253651; ParE, AAG08352; OprD, NP_249649; NalB (MexR) AAG03813; NalC, AAG07108; NalD, AAG06962; PA5471, AAG08856; MexZ, AF073776.
Abbreviations: PDC, Pseudomonas-derived cephalosporinase; QRDR, quinolone resistance-determining region; ST, sequence type; WT, wild-type or no mutation.

Immunoblot expression of Pseudomonas-derived cephalosporinase (PDC) β-lactamse, PDC-16. Lanes: 1 – Isolate 2961, 2 – Isolate 7927, 3 – Isolate 8649, 4 – Isolate 7166, 5 – Isolate 8096, 6 – Marker. Probed with 0.1 µg/mL anti-PDC antibody; 1:10 000 dilution of Protein G-HRP conjugate.
Quinolone Resistance
T83I in gyrA and S87L in parC were found in all isolates. These mutations confer high-level fluoroquinolone resistance [8].
Carbapenem Resistance
All isolates also had a premature stop codon at amino acid 79 in OprD, resulting in inactivation of the main porin for imipenem uptake [9].
Mutations in Efflux Pump Regulators
Three point mutations (G71E, A145V, and S209R) were observed in nalC; however, their role in an MDR phenotype in P. aeruginosa is unclear at the present time. Also of note, isolate 6 developed moderate amikacin resistance (32 µg/mL). In this case, MexXY overexpression might be expected, although specific mutations in mexZ were not seen. It could be that additional MexXY regulators were involved, or the presence of an aminoglycoside modifying enzyme, in that resistance phenotype. Additionally, the gene PA5471 would not amplify in any of the isolates. Deletions of the coding sequence in PA5471 can lead to MexXY-OprM overexpression [5, 10].
To determine whether recurrent P. aeruginosa bacteremia represented relapse of infection or acquisition of a new strain, we performed multilocus sequence typing on all P. aeruginosa isolates following the method of Curran et al [11]. All isolates belonged to sequence type 298 and displayed resistance to ciprofloxacin (MIC >2 µg/mL), meropenem, and imipenem (MICs >8 µg/mL) by Microscan; only isolate 5 appeared to be resistant to colistin. The cause of that colistin resistance is undetermined.

Genetic typing with repetitive sequence-based PCR (rep-PCR). Dendrogram of rep-PCR illustrating >98% similarity in band patterns, suggesting persistence of the same Pseudomonas aeruginosa strain, rather than re-infection with a different strain.
Although much information was uncovered in this investigation it should be noted that certain limitations to this study exist, such as: (1) MICs were reported by breakpoints only (ie, endpoints were not determined), (2) expression of efflux pumps was not examined, and (3) the relevance of nalC mutations to efflux pump expression still remains unknown.
CONCLUSION
We have chronicled the molecular basis of evolving antibiotic resistance in a case of MDR P. aeruginosa in a patient with PVE, an infection that is very difficult to eradicate. Serial analysis shows that the occurrence of point mutations contributed to the MDR phenotype. Likely, changes in the efflux pump regulators and reduced porin expression contribute to the carbapenem-resistant phenotype. Our molecular analysis also uncovered point mutations in quinolone resistance-determining regions. However, the key finding during the course of treatment was the mutation in ampR leading to the emergence of cephalosporin and aztreonam resistance. This was due to the D135N AmpR amino acid change, which causes a conformational change in the Amp R regulator, leading to overexpression of the PDC-16 β-lactamase that was present in these isolates.
We stress that ampR is a global transcriptional regulator also associated with alginate production and biofilm formation. Prolonged exposure to β-lactams before surgical intervention may have contributed to the selection of ampR regulator mutants. This should serve as a reminder of the complexity of acquired resistance in P. aeruginosa PVE and the therapeutic challenges it entails.
Acknowledgments
The authors would like to acknowledge the contributions of Dr Joseph Coselli and his cardiothoracic surgery team.
Financial support. Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award numbers R01AI100560, R01AI063517, and R01AI072219. This study was also supported in part by funds and/or facilities provided by the Cleveland Department of Veterans Affairs (award number 1I01BX001974 to R. A. B. from the Biomedical Laboratory Research & Development Service of the VA Office of Research and Development and the Geriatric Research Education and Clinical Center VISN 10 to R. A. B.) and the National Center for Research Resources (Clinical and Translational Science Award UL1 RR024989 to F. P.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the Department of Veterans Affairs.
Potential conflicts of interest. All authors: No reported conflicts. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
Author notes
Correspondence: R. A. Bonomo, Medical Service, Louis Stokes Cleveland Department of Veterans Affairs Medical Center, 10701 East Boulevard, Cleveland, OH 44106 ([email protected]).


{kind=link}
{kind=link}
{kind=link}
Comments