






Abstract
Background. We report the first-in-human safety and immunogenicity evaluation of a highly attenuated, replication-competent recombinant vesicular stomatitis virus (rVSV) human immunodeficiency virus (HIV)-1 vaccine.
Methods. Sixty healthy, HIV-1-uninfected adults were enrolled in a randomized, double-blinded, placebo-controlled dose-escalation study. Groups of 12 participants received rVSV HIV-1 gag vaccine at 5 dose levels (4.6 × 103 to 3.4 × 107 particle forming units) (N = 10/group) or placebo (N = 2/group), delivered intramuscularly as bilateral injections at 0 and 2 months. Safety monitoring included VSV cultures from blood, urine, saliva, and swabs of oral lesions. Vesicular stomatitis virus-neutralizing antibodies, T-cell immunogenicity, and HIV-1 specific binding antibodies were assessed.
Results. Local and systemic reactogenicity symptoms were mild to moderate and increased with dose. No severe reactogenicity or product-related serious adverse events were reported, and all rVSV cultures were negative. All vaccine recipients became seropositive for VSV after 2 vaccinations. gag-specific T-cell responses were detected in 63% of participants by interferon-γ enzyme-linked immunospot at the highest dose post boost.
Conclusions. An attenuated replication-competent rVSV gag vaccine has an acceptable safety profile in healthy adults. This rVSV vector is a promising new vaccine platform for the development of vaccines to combat HIV-1 and other serious human diseases.
Attenuated, replication-competent viral vector vaccines are highly immunogenic in that they mimic natural infection. They offer the promise of eliciting a more integrated immune response and more abundant and sustained expression of the encoded viral antigens. Although a vaccine to prevent human immunodeficiency virus (HIV) infection remains an urgent global health priority, concerns about the safety of using live-attenuated HIV itself as a vaccine [1] has led the field to focus heavily on the exploration of replication-defective viral vectors to deliver HIV-1 antigens. There are limited data to suggest these vectors are able to induce an optimal, integrated immune response, which may include CD4+ T-cell help for B-cell/antibody and polyfunctional cytotoxic T-cell responses together with long-term immunologic memory [2]. To date, only 1 replication-defective viral vector, based on canarypox, administered sequentially with a gp120 subunit boost vaccine, has demonstrated partial protection from HIV acquisition in an efficacy trial [3].
Currently, only a few replicating viruses, including vaccinia (clinical trial ID NCT01705223), Sendai virus (NCT01705990), measles (NCT01320176), and adenovirus subtype 4 (NCT01989533), have advanced to clinical trials as HIV-1 vaccine vectors. Vesicular stomatitis virus (VSV) is a member of the genus Vesiculovirus in the family Rhabdoviridae. The 2 major serotypes are New Jersey and Indiana. Vesicular stomatitis virus has a single-strand, negative-sense, nonsegmented RNA genome. Similar to Sendai and measles viruses, the virus genomic organization of VSV (Figure 1A) and life cycle make it particularly attractive as a candidate vaccine vector because the genome can accommodate multiple gene inserts, is stable over many generations, and does not undergo recombination. In addition, the VSV genome replicates in the cytoplasm and is incapable of integrating within the genomes of infected host cells. Finally, VSV is a zoonosis, cycling between biting insects and livestock (cattle, horses, and swine). Human infection with VSV is rare in most regions of the world except in certain regions of Central and South America where VSV is endemic. Where VSV infection does occur, it is typically asymptomatic or is associated with an acute influenza-like illness with symptoms such as fever, muscle aches, and malaise [4, 5]. Thus, the general population is largely free of pre-existing, virus-neutralizing immunity—a factor that has limited the clinical utility of HIV-1 vaccines based on adenovirus subtype 5 [6, 7].
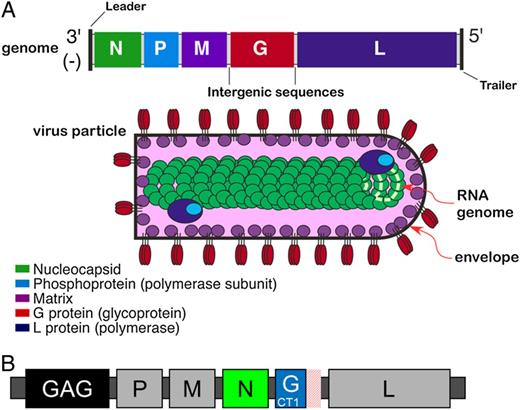
(A) Wild-type vesicular stomatitis virus (VSV) genome organization and virion structure. Viral transcription is polar, initiating at the single 3′ promoter and proceeding to the 5′ end of the genome, producing a steep 3′ to 5′ gradient of gene expression. The N protein encapsidates viral genomic RNA forming viral nucleocapsid; the M protein drives virus assembly and budding from infected cells, and the G protein forms trimers in host cell plasma membrane, which becomes the envelope of budding virus particles. The P and L proteins associate to form the viral RNA polymerase that copackages into virus particles with nucleocapsid, providing the components needed to initiate the next infectious cycle. The mature virus particle is bullet shaped (60 nm × 180 nm) with a lipid envelope and underlying M protein surrounding a dense nucleocapsid core. (B) The recombinant VSV human immunodeficiency virus (HIV)-1 vaccine vector (rVSVN4CT1gag1). The vaccine construct encodes the HIV-1 gag transgene (strain HXB-2 p55 gag) in position one of the genome (gag1) to maximize gene expression, has the N gene translocated from first to fourth position (N4), and encodes a G protein that has the cytoplasmic tail truncated from 29 to 1 amino acid (CT1).
Recombinant VSV (rVSV) has been extensively studied as a potential HIV vaccine vector in preclinical, nonhuman primate (NHP) models [8–11]. After 2 sequential immunizations, rhesus macaques that received rVSV expressing HIV-1 env and simian immunodeficiency virus (SIV) gag were all infected but were protected from disease progression for 4 years or more after infection with SHIV 89.6P, maintaining low or undetectable viral load set points and preserved CD4+ T-cell counts [11]. This encouraging level of postchallenge protection from disease suggested that rVSV vectors expressing HIV genes might be an effective HIV vaccine in humans. To maximize safety for first-in-human clinical evaluation, a highly attenuated form of rVSV Indiana was developed. This involved downregulating VSV N protein expression by translocating the N gene further away from the 3′ transcription promoter and truncating the VSV G protein cytoplasmic tail from 29 amino acids found in wild-type virus to a single amino acid [12]. In addition, HIV-1 gag expression was achieved by insertion of the gag gene adjacent to the 3′ transcription promoter (Figure 1B). The resulting vector (rVSVN4CT1gag1) was shown to be safe, well tolerated, and immunogenic in several murine and NHP studies [13, 14]. In the work described here, the HIV Vaccine Trials Network (HVTN) evaluated the safety and immunogenicity of this novel vaccine vector in a phase 1a dose-escalation, first-in-human trial, HVTN 090.
METHODS
Vaccine
The vaccine research virus seed was prepared under carefully controlled (compliant) laboratory conditions starting with plasmids encoding viral cDNA in a process known as virus rescue [15]. Research virus seed was amplified and then transferred to Novasep (Charleroi, Belgium) for production of master virus seed and clinical trial material (CTM) at 10 L scale under good manufacturing practices. Clinical trial material lots were formulated in 7.5% w/v sucrose, 3.8 mM KH2PO4, 7.2 mM K2HPO4, 5.5 mM l-glutamate, pH 7.1 and 0.2% w/v hydrolyzed gelatin as a stabilizer, and vialed at 2.3 × 104 particle-forming units (PFU)/mL, 2.4 × 105 PFU/mL, 2.1 × 106 PFU/mL, and 1.7 × 107 PFU/mL. The CTM was stored at −80°C until administered to trial participants. The placebo was Sodium Chloride Injection USP, 0.9%.
Study Design and Procedures
HVTN 090 was a randomized, double-blind, placebo-controlled dose-escalation phase 1a trial to evaluate the safety and immunogenicity of a 2-immunization regimen (0 and 8 weeks) of rVSV gag at 4.6 × 103 PFU (T1), 4.6 × 104 PFU (T2), 4.8 × 105 PFU (T3), 4.2 × 106 PFU (T4), and 3.4 × 107 PFU (T5) in HIV-uninfected volunteers at 4 clinical sites in the United States Table 1. The protocol was approved by the institutional review boards of all participating centers and registered at ClinicalTrials.gov (NCT01438606). Sixty adults aged 18–50 years who reported low behavioral risk for HIV infection and tested HIV-1-seronegative and were determined to be healthy based on medical history, physical exam, and laboratory tests were enrolled after providing written informed consent. Due to a potential concern for rare cases of neurovirulence associated with wild-type VSV infection [16] and to avoid misclassification of commonly experienced adverse events during trials (eg, headaches), individuals were excluded if they had a history of neurologic or neuropsychiatric disorders. In addition, given that this was a novel replicating vector vaccine, precautions were taken to exclude individuals in close contact with infants, the elderly, immunocompromised individuals, or persons with chronic lung disease.
Protocol Schema
| Treatment Group | Vaccine/Placebo | rVSV gag Dose (PFU) | Injection Schedule, Months | |
|---|---|---|---|---|
| Month 0 (Prime) | Month 2 (Boost) | |||
| T1 | 10/2 | 4.6 × 103 | rVSV gag or placebo | rVSV gag or placebo |
| T2 | 10/2 | 4.6 × 104 | rVSV gag or placebo | rVSV gag or placebo |
| T3 | 10/2 | 4.8 × 105 | rVSV gag or placebo | rVSV gag or placebo |
| T4 | 10/2 | 4.2 × 106 | rVSV gag or placebo | rVSV gag or placebo |
| T5 | 10/2 | 3.4 × 107 | rVSV gag or placebo | rVSV gag or placebo |
| Total | 60 (50/10) | |||
| Treatment Group | Vaccine/Placebo | rVSV gag Dose (PFU) | Injection Schedule, Months | |
|---|---|---|---|---|
| Month 0 (Prime) | Month 2 (Boost) | |||
| T1 | 10/2 | 4.6 × 103 | rVSV gag or placebo | rVSV gag or placebo |
| T2 | 10/2 | 4.6 × 104 | rVSV gag or placebo | rVSV gag or placebo |
| T3 | 10/2 | 4.8 × 105 | rVSV gag or placebo | rVSV gag or placebo |
| T4 | 10/2 | 4.2 × 106 | rVSV gag or placebo | rVSV gag or placebo |
| T5 | 10/2 | 3.4 × 107 | rVSV gag or placebo | rVSV gag or placebo |
| Total | 60 (50/10) | |||
Abbreviations: PFU, particle-forming unit; rVSV, recombinant vesicular stomatitis virus.
Protocol Schema
| Treatment Group | Vaccine/Placebo | rVSV gag Dose (PFU) | Injection Schedule, Months | |
|---|---|---|---|---|
| Month 0 (Prime) | Month 2 (Boost) | |||
| T1 | 10/2 | 4.6 × 103 | rVSV gag or placebo | rVSV gag or placebo |
| T2 | 10/2 | 4.6 × 104 | rVSV gag or placebo | rVSV gag or placebo |
| T3 | 10/2 | 4.8 × 105 | rVSV gag or placebo | rVSV gag or placebo |
| T4 | 10/2 | 4.2 × 106 | rVSV gag or placebo | rVSV gag or placebo |
| T5 | 10/2 | 3.4 × 107 | rVSV gag or placebo | rVSV gag or placebo |
| Total | 60 (50/10) | |||
| Treatment Group | Vaccine/Placebo | rVSV gag Dose (PFU) | Injection Schedule, Months | |
|---|---|---|---|---|
| Month 0 (Prime) | Month 2 (Boost) | |||
| T1 | 10/2 | 4.6 × 103 | rVSV gag or placebo | rVSV gag or placebo |
| T2 | 10/2 | 4.6 × 104 | rVSV gag or placebo | rVSV gag or placebo |
| T3 | 10/2 | 4.8 × 105 | rVSV gag or placebo | rVSV gag or placebo |
| T4 | 10/2 | 4.2 × 106 | rVSV gag or placebo | rVSV gag or placebo |
| T5 | 10/2 | 3.4 × 107 | rVSV gag or placebo | rVSV gag or placebo |
| Total | 60 (50/10) | |||
Abbreviations: PFU, particle-forming unit; rVSV, recombinant vesicular stomatitis virus.
Participants received vaccinations as 1 mL injections given intramuscularly (IM) into both deltoids, at 0 and 8 weeks. Safety evaluations included physical examinations and standard clinical chemistry and hematological tests. Local injection site (pain, tenderness, redness, erythema, and induration) and systemic reactogenicity symptoms (malaise, headache, fever, chills, myalgias, arthralgias, nausea, vomiting, and fatigue) were assessed daily for 7 days following each vaccination or until resolution. Adverse events were graded based on the HVTN Table for Grading Severity of Adverse Experiences (http://rsc.tech-res.com/Document/safetyandpharmacovigilance/Table_For_Grading_Severity_of_Adult_Pediatric_Adverse_Events.pdf). Participants were observed for 6 months after the last vaccination, and then they were contacted annually to complete 3 years of long-term follow-up. Additional safety monitoring included mini-mental state examinations at baseline and at each visit. Participants were also evaluated for any oral lesions that might be indicative of rVSV-induced pathology. To assess virus dissemination and shedding, blood, urine, and saliva samples were collected at days 3 and 7 postvaccination and swabs were taken from any oral lesions observed up to 2 weeks postvaccination. Test samples were thawed rapidly in a 37°C water bath, and duplicate 100 µL samples were then adsorbed to freshly confluent Vero cell monolayers, containing 100 µL Dulbecco's modified Eagles medium (DMEM) supplemented with 200 IU of penicillin, 200 µg/mL streptomycin, and 5 µg/mL fungizone in 24-well plates, for 15 minutes at room temperature followed by 30 minutes at 37°C. The inoculum was then replaced with 1 mL virus growth medium (DMEM supplemented with 5% fetal bovine serum, 100 IU of penicillin, 100 µg/mL streptomycin, 2.5 µg/mL fungizone, and 10 µg/mL gentamycin), and cells were incubated at 32°C for 7 days to allow VSV-induced cytopathic effects typified by cell rounding and detachment to be observed by microscopy. The detection limit of the infectivity assay used in these studies, determined by spiking prevaccination samples with defined amounts of vaccine virus, was 5 PFU/100 µL of sample.
Blood samples for assessment of immunogenicity were collected at 1 and 2 weeks after each vaccination (days 7, 14, 63, and 70). Several licensed diagnostic HIV enzyme-linked immunosorbent assays (Abbott HIVAB HIV 1/2 [rDNA], Abbott Architect HIV Ag/Ab Combo, Bio-Rad Genetic System HIV 1/2 Plus O EIA, Bio-Rad Genetic System HIV 1/2 rLAV, and Bio-Rad Multispot HIV-1/HIV-2 Rapid Test) were performed on sera from all participants at the end of study (day 364) to assess vaccine-induced seroreactivity.
Immune Response Assays
Vesicular Stomatitis Virus-Neutralizing Antibodies
Serum VSV-neutralizing antibody (nAb) titers were determined by standard neutralization assay [17]. In brief, duplicate serial 2-fold serum dilutions were prepared from 2 independent 10-fold dilutions of each serum sample in virus growth medium, and then each dilution was mixed with 100 PFU of VSV Indiana followed by a 1-hour incubation at 37°C, 5% CO2 in a 96-well plate format to allow virus neutralization. An aliquot of 1 × 104 Vero cells suspended in 100 µL cell growth medium was then added to each well followed by incubation at 37°C, 5% CO2 for 4–6 days to allow development of viral cytopathic effects (CPEs). The neutralization titer was recorded as the reciprocal of the lowest serum dilution providing complete protection from viral-induced CPEs.
HIV-1-Specific Cellular Responses
Intracellular cytokine staining (ICS) was performed on cryopreserved peripheral blood mononuclear cells (PBMCs) by flow cytometry to examine HIV-1-specific vaccine-induced CD4+ and CD8+ T-cell responses at 1 and 2 weeks after each vaccination. Cytokine production was assessed after stimulation with a Gag Consensus B peptide pool (15-mers overlapping by 11 amino acids, 1 µg/mL for each peptide) as previously described [18]. The 6-hour stimulation included brefeldin A (10 μg/mL; Sigma-Aldrich, St. Louis, MO) and anti-CD28/anti-CD49d (each at 1 μg/mL; BD Biosciences, San Jose, CA). Staphylococcus enterotoxin B (Sigma-Aldrich) was used as a positive control, and peptide diluent (dimethyl sulfoxide [DMSO] at a final concentration of 1%) was used as a negative control. Data were acquired on an LSRII and analyzed using FlowJo (Treestar, Inc., Ashland, OR). Positivity of the ICS responses of individual cytokines or cytokine combination was determined by a 1-sided Fisher's exact test applied to each peptide pool-specific response versus the negative control response with a discrete Bonferroni adjustment for the multiple comparisons due to testing against multiple peptide pools. Peptide pools with adjusted P values less than α = 0.00001 were considered positive. If at least 1 peptide pool for a specific HIV-1 protein is positive, then the overall response to the protein is considered positive [19]. Data were filtered if background responses (DMSO control) were >0.1% cytokine secretion or if <5000 events were acquired within the CD4+ or CD8+ T-cell subpopulations. In addition, HIV-specific T-cell responses were assessed by interferon-γ (IFN-γ) enzyme-linked immunospot (ELISPOT) on cryopreserved PBMCs stimulated overnight with a Gag Consensus B peptide pool as previously described [20, 21].
HIV-1-Specific Antibodies
Serum HIV-1-specific immunoglobulin G responses (1:50 dilution) against Gag p24 were measured at 2 weeks after each vaccination by a validated HIV-1-binding antibody multiplex assay as previously described [22]. Antibody measurements were acquired on a Bio-Plex instrument (Bio-Rad), and the readout was expressed as mean fluorescent intensity (MFI). The positive control in each assay was HIV-positive sera, and the negative control was HIV-negative human sera and blank beads. Samples with blank bead MFI >5000 after 1 retesting were excluded. Samples were determined to be positive if both the MFI and blank-subtracted MFI were greater than 3-fold over the baseline MFI and blank-subtracted MFI, respectively, and if the blank-subtracted baseline MFI values were above an antigen-specific cutoff that equals the antigen-specific average MFI plus 3 standard deviations of 60 seronegative plasma samples.
Statistical Analysis
Data were analyzed according to a modified intention-to-treat approach, wherein all participants who received the first vaccination were included in the analysis, and analyzed based on initial randomization assignment regardless of the number of injections received. Five vaccine arms and a pooled placebo group comprising placebo recipients from all 5 treatment groups were evaluated and compared for safety and immunogenicity. All statistical tests were 2-sided and were deemed statistically significant if P was ≤ .05. All statistical analyses were performed using SAS or R statistical software, and detailed descriptions of methods can be found in the Supplementary data.
RESULTS
Demographics and Accrual
The median age of the 60 participants enrolled was 24 years; 45% were female, and 37% were nonwhite and/or mixed race or ethnicity. Overall retention in the study was 92%; 3 individuals did not receive the second vaccination—1 at the 4.8 × 105 PFU dose (refused) and 2 at the 3.4 × 107 PFU dose (1 following a protocol safety team decision in a participant with injection site reactogenicity and hypoesthesia after the first dose; the second was unable to be contacted). Five individuals discontinued participation early; 1 due to HIV infection approximately 5 months after his second vaccination; the 4 others were lost to follow-up. All had received active vaccine. One participant, who received the 4.6 × 103 PFU dose, reported a pregnancy 168 days after her second vaccination, resulting in a healthy full-term birth. Another pregnancy, reported after the study ended, during long-term follow-up, was associated with fetal anomalies and was electively terminated. The pregnancy occurred approximately 18 months after the second vaccination of the 4.6 × 104 PFU dose.
Safety and Tolerability
Overall, local injection site and systemic reactogenicity symptoms including injection site tenderness, chills, and fever, were mild to moderate and increased with dose (see Supplementary Appendix, Figure 1). Eleven (22%) vaccine recipients reported mild or moderate fever (37.7–39.3°C) in the 7-day reactogenicity period (including 50% of those receiving the highest dose). No severe reactogenicity, changes in mental status, encephalitis, or product-related serious adverse events (SAEs) were reported. Viral infectivity cultures of blood (n = 179), urine (n = 180), saliva (n = 205), and oral swabs/saliva (n = 42) obtained after vaccinations did not recover VSV. There were no significant differences in the occurrence of oral lesions comparing vaccines and placebo recipients. One vaccinee (2%) had vaccine-induced HIV seroreactivity at the end of study.
Immunogenicity
Vesicular Stomatitis Virus-Neutralizing Antibody Responses
The VSV G protein forms trimeric spikes on the surface of the virus particle and is the target of VSV-nAb responses. A VSV-nAb response was detected in all vaccine recipients after 2 vaccinations at all doses tested (Figure 2). Induction of a neutralizing response after a single vaccine dose in many participants indicated significant antivirus response (“take”) in these vaccine recipients. The dose response was linear after 1 vaccination, and it achieved saturation after 2 vaccinations at higher dose levels.
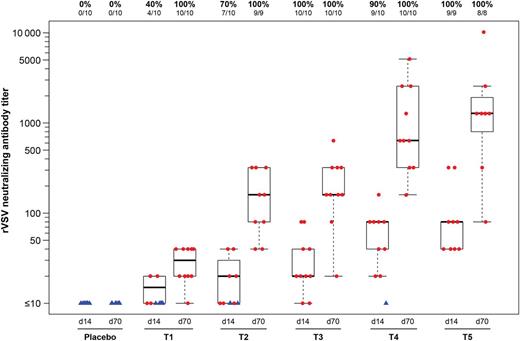
Vesicular stomatitis virus (VSV)-neutralizing antibody titers. Neutralizing antibody titer responses are represented as the reciprocal of the lowest serum dilution, demonstrating complete protection of Vero cell monolayers from VSV-induced cytopathic effects using serum from 2 weeks after the first vaccination (d14) and 2 weeks after the second vaccination (d70), by group. Responders are shown as red dots, and nonresponders are shown as blue triangles. Box plots show the distribution of titers among positive responders only. The box indicates the median and interquartile range (IQR); whiskers extend to the furthest point within 1.5 times the IQR from the upper or lower quartile. Numbers above the panel show the number of responders with an assay result out of the evaluable subjects and the percentage with positive responses. Abbreviations: T1, 4.6 × 103 PFU; T2, 4.6 × 104 PFU ; T3, 4.8 × 105 PFU; T4, 4.2 × 106 PFU; and T5, 3.4 × 107 PFU. d, day; PFU, particle-forming units.
HIV-1-Specific T-Cell Responses
Human immunodeficiency virus Gag-specific T-cell responses were measured in all participants with available samples at 1 and 2 weeks after each immunization. The primary readout for the validated ICS assay is expression of IFN-γ and/or interleukin (IL)-2 (Figure 3A). Responses are absent in the lowest dose groups (T1–T3). At 4.2 × 106 PFU (T4), there is 1 responder (1 of 10) after the prime and a different responder after the boost. At the highest dose (T5), responses are detected in 1 participant after the prime and in 3 participants (2 new responders) after the boost, leading to a CD4+ T-cell response rate of 38% (3 of 8) 2 weeks after vaccination with the highest dose. The magnitude of the CD4+ T-cell response to Gag was correlated with VSV nAb titer after 2 doses (day 70, Pearson correlation coefficient, r2 = 0.46, P = .001) but not after a single dose (day 14, r2 = 0.21, P = .14). CD8+ T cells were only found in a single individual in the 4.2 × 106 PFU dose group at 2 weeks after the prime (data not shown).
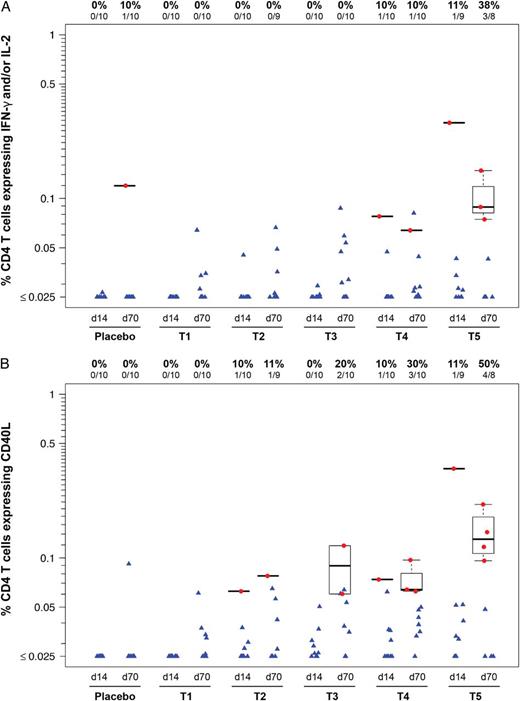
Intracellular cytokine staining assay. CD4+ T-cell responses to Gag Consensus (Con) B peptides. The percentage of CD4+ T cells expressing interleukin (IL)-2 or interferon (IFN)-γ (A) or expressing CD40 ligand (CD154) (B), in response to Gag Con B peptide pools. Human immunodeficiency virus-1-specific CD4+ and CD8+ T-cell responses were measured using a validated Intracellular cytokine staining assay. Results shown are from samples obtained 2 weeks after the prime (d14) and 2 weeks after boost vaccination (d70) for each treatment group. Responders are shown as red dots, and nonresponders are shown as blue triangles. Box plots show the distribution of the magnitude of response in positive responders only. The box indicates the median and interquartile range (IQR); whiskers extend to the furthest point within 1.5 times the IQR from the upper or lower quartile. Numbers at the top of each panel show the number and percentage of responders from each group. One Group 4 participant had a CD8+ T-cell response to Gag Con B at day 14 (data not shown). Abbreviations: T1, 4.6 × 103 PFU; T2, 4.6 × 104 PFU; T3, 4.8 × 105 PFU; T4, 4.2 × 106 PFU; and T5, 3.4 × 107 PFU. d, day; PFU, particle-forming units.
In addition to IFN-γ and IL-2, we measured expression of tumor necrosis factor-α, CD40L, CD107, macrophage-inflammatory protein-1β, and IL-4. CD40L provided a more sensitive readout for vaccine-induced CD4+ T-cell responses in this trial, and expression of this marker was detected in 4 of 8 (50%) of vaccinees after the second vaccination at the highest dose (Figure 3B). Responses measured 1 week and 2 weeks after vaccination were not significantly different regardless of the marker analyzed (data not shown).
To further characterize the cellular immune response, we also tested PBMC from participants in the 2 highest dose groups 2 weeks postvaccination, by IFN-γ ELISPOT. Consistent with the observations by ICS, Figure 4 shows a dose-response relationship and higher responses after the boost. An additional responder was ide.jpegied using this assay, leading to an overall response rate of 63% (5 of 8 participants) at the highest dose of 3.4 × 107 PFU.
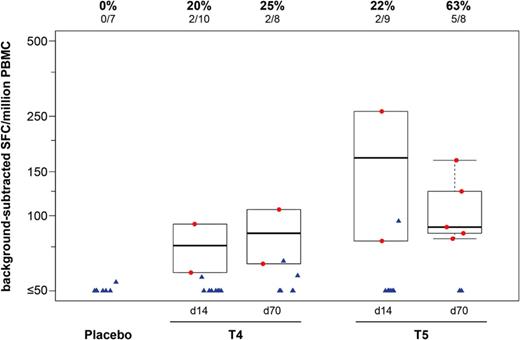
T-cell responses to Gag Consensus B peptides by interferon (IFN)-γ enzyme-linked immunospot (ELISPOT). The validated IFN-γ ELISPOT assay demonstrates a dose-response relationship, with a higher response rate and magnitude with a 10-fold higher dose and a second vaccination. Results are shown from 2 weeks after the first vaccination (d14) and 2 weeks after the second vaccination (d70), for study groups T4, 4.2 × 106 PFU and T5, 3.4 × 107 PFU. Responses are expressed as the number of spot-forming units (SFUs) per 106 cells. Responders are shown as red dots, and nonresponders are shown as blue triangles. Box plots show the distribution of the magnitude of response in positive responders only. The box indicates the median and interquartile range (IQR); whiskers extend to the furthest point within 1.5 times the IQR from the upper or lower quartile. Numbers above the panel show the number and percentage of responders from each group. Abbreviations: d, day; PBMC, peripheral blood mononuclear cells.
Binding Antibody to Gag p24
There were no positive responders 2 weeks after the first vaccination (day 14) or 2 weeks after the second vaccination (day 70) in Groups 1–3. At day 70, positive responses were detected in 1 of 10 vaccine recipients at the 4.2 × 106 dose and 3 of 8 recipients at the 3.4 × 107 PFU dose (data not shown).
DISCUSSION
In this first-in-human clinical trial of a highly attenuated VSV-based HIV-1 vaccine, we found that it had an acceptable safety and tolerability profile, with mostly mild-to-moderate reactogenicity, and a dose-dependent increase in fever and chills. There were no product-related SAEs, and no VSV was detected from samples of blood, urine, and saliva or from swabs of oral lesions. In contrast, a clinical study of a less attenuated form of rVSV, developed as an Ebola virus vaccine [23], demonstrated some severe reactogenicity events including arthritis, and vaccine virus was detected in blood, joint fluid, and skin vesicles. That vaccine, rVSV-ZEBOV, was attenuated by replacing the VSV G gene with the glycoprotein (GP) gene of Ebola virus generating an rVSV-GP pseudotype, which may alter virus tropism but does not constrain intracellular VSV replication. However, the rVSVN4CT1 vaccine platform described herein is attenuated by 2 different, stable genome modifications [12–14], resulting in significant growth attenuation in vitro. In HVTN 090, we found that VSV seroconversion was 100% after 2 vaccinations, with a dose-dependent increase in VSV nAb. More than 60% of vaccinees developed HIV-1-specific T-cell responses after the prime and boost at the highest dose tested (3.4 × 107 PFU), mediated almost entirely by CD4+ T cells and the magnitude of CD4+ T-cell responses to Gag correlated with VSV nAb titers. In addition, Gag p24 antibody was detected in one third of participants after 2 vaccinations at the highest dose.
Although some trials of vector-based HIV vaccines in humans have demonstrated lower levels of immunogenicity compared with their SIV-based homologs tested in preclinical NHP models [24, 25], we found that the frequency and magnitude of HIV-1-specific T-cell immune responses were only modestly lower than predicted with the rVSV vector. In preclinical NHP immunogenicity studies, the rVSVN4CT1gag1 vector elicited a Gag-specific cellular immune response in a majority of animals, with an average of 200 IFN-γ spot-forming cells (SFC)/million PBMCs after a single IM injection of 107 PFU of virus and over 500 SFC/million PBMCs after a booster dose with rVSVN4CT1gag1 serotype New Jersey, intended to minimize effects of antivector immunity [14, 17]. HVTN 090 showed comparable frequency, albeit somewhat lower magnitude ELISPOT responses (median of approximately 100 SFC/million PBMCs after 2 doses of rVSVIN), with a homologous serotype boost. It is notable that the observed boosting of T-cell responses after a second dose of the same rVSVIN vector indicates that future clinical development should consider regimens with homologous rVSV boosting.
There were a few limitations of this study. Two individuals did not receive their second doses of rVSV gag at 3.4 × 107 PFU, with consequent loss of data at this dose. Based on available data, this highest dose was taken forward as a boost for a DNA prime (HIV-1 multiantigen vaccine; Profectus BioSciences, Inc.) given with IL-12 pDNA adjuvant and delivered IM with electroporation in another study, HVTN 087. Preliminary evidence suggests that this prime-boost regimen substantially enhanced HIV Gag-specific T-cell responses after vaccination with rVSV HIV-1 gag [26]. The rVSV prototype vaccine evaluated in both HVTN 087 and HVTN 090 encoded gag only, and, as expected, there were limited HIV-specific antibody responses generated. Given the correlation of vaccine-induced Env-specific responses with lower risk of HIV infection [27], Profectus has moved forward with development of a clinical candidate that encodes the env gene from an HIV-1 clade C transmitted/founder strain, 1086.C. This vaccine elicited a robust Env-specific humoral response in guinea pigs (unpublished data) and has been manufactured in preparation for clinical evaluation in a DNA/rVSV prime-boost study.
CONCLUSIONS
In summary, these first-in-human data demonstrate that an attenuated replication-competent rVSV vaccine is both safe and immunogenic, and it is a promising candidate for future HIV vaccine development. These data also support the use of this attenuated replication-competent rVSV vaccine vector platform to combat other infectious diseases, such as Ebola and Marburg viruses, for which promising preclinical [28–31] and clinical data exist [23, 32] and effective vaccines are urgently needed.
Acknowledgments
We thank the HVTN 090 study staff, the sites' community advisory boards, and the clinical trial participants. We also acknowledge the longstanding support of Dr. John Rose who first developed the recombinant vesicular stomatitis virus (rVSV) vector system and performed many of the early proof of concept studies. In addition, we acknowledge the late Dr. Steve Udem for his commitment, support, and stewardship of the rVSV vector program at Wyeth Pharmaceuticals. Finally, we thank the following HVTN 090 Study Group and Clinical Research Site Investigators: Mark Mulligan and Nadine Rouphael (Atlanta, GA); Scharla Estep (Bethesda, MD); Kyle Rybczyk (Memphis, TN); Deb Dunbar (Philadelphia, PA); Susan Buchbinder, Theresa Wagner, and Reese Isbell (San Francisco, CA); Victoria Chinnell, Jin Bae, Gina Escamilla, Jenny Tseng, Ramey Fair, Shelly Ramirez, Gail Broder, Liz Briesemeister, and Adi Ferrara (Seattle, WA).
Financial support. HVTN 090 was conducted by the HIV Vaccine Trials Network (HVTN) and funded by the National Institutes of Allergy and Infectious Diseases. This work was also funded by the following grants: HHSN272200800061C (Profectus); UM1 AI068614 (HVTN Core Fred Hutchinson Cancer Research Center); UM1 AI068635 (Statistical Center for HIV/AIDS Research and Prevention); UM1 AI068618 (HVTN Laboratory Program); UM1 AI069467 and P30 AI450008 (University of Pennsylvania); UM1 AI069418 (Emory); UM1 AI069439 (Vanderbilt); and UM1 AI069496 (San Francisco Department of Public Health).
Potential conflicts of interest. M. A. E., D. K. C., T. L., M. A. T., T. J. H., and J. H. E. are employees of Profectus BioSciences, Inc.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
Author notes
Presented in part: AIDS Vaccine Conference, September 2013, Barcelona, Spain; Keystone Symposium, HIV Vaccines, March 2014, Banff, Canada.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments